Conformational Landscapes of Halohydrin Dehalogenases and Their Accessible Active Site Tunnels



Abstract
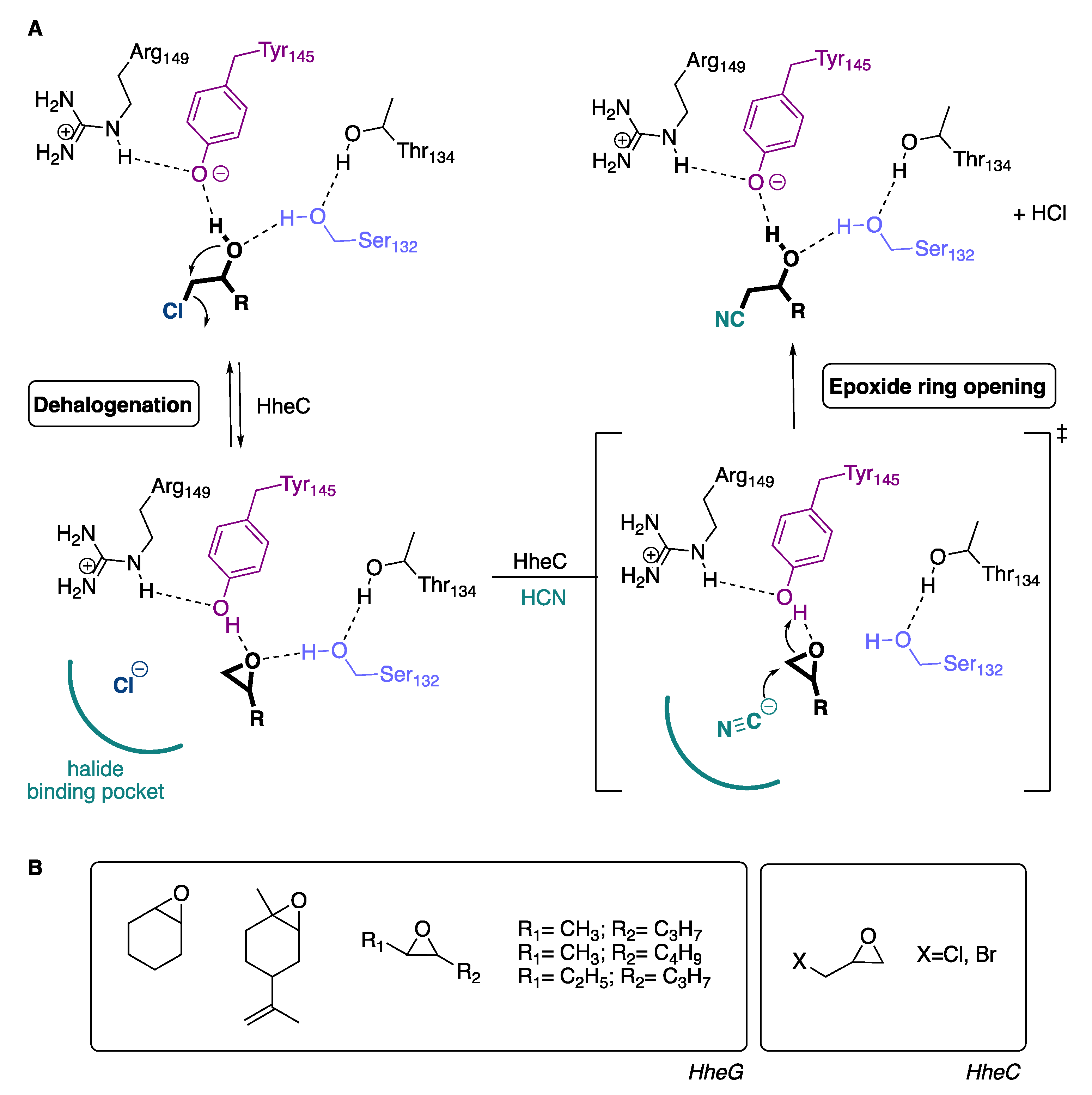
1. Introduction
2. Results
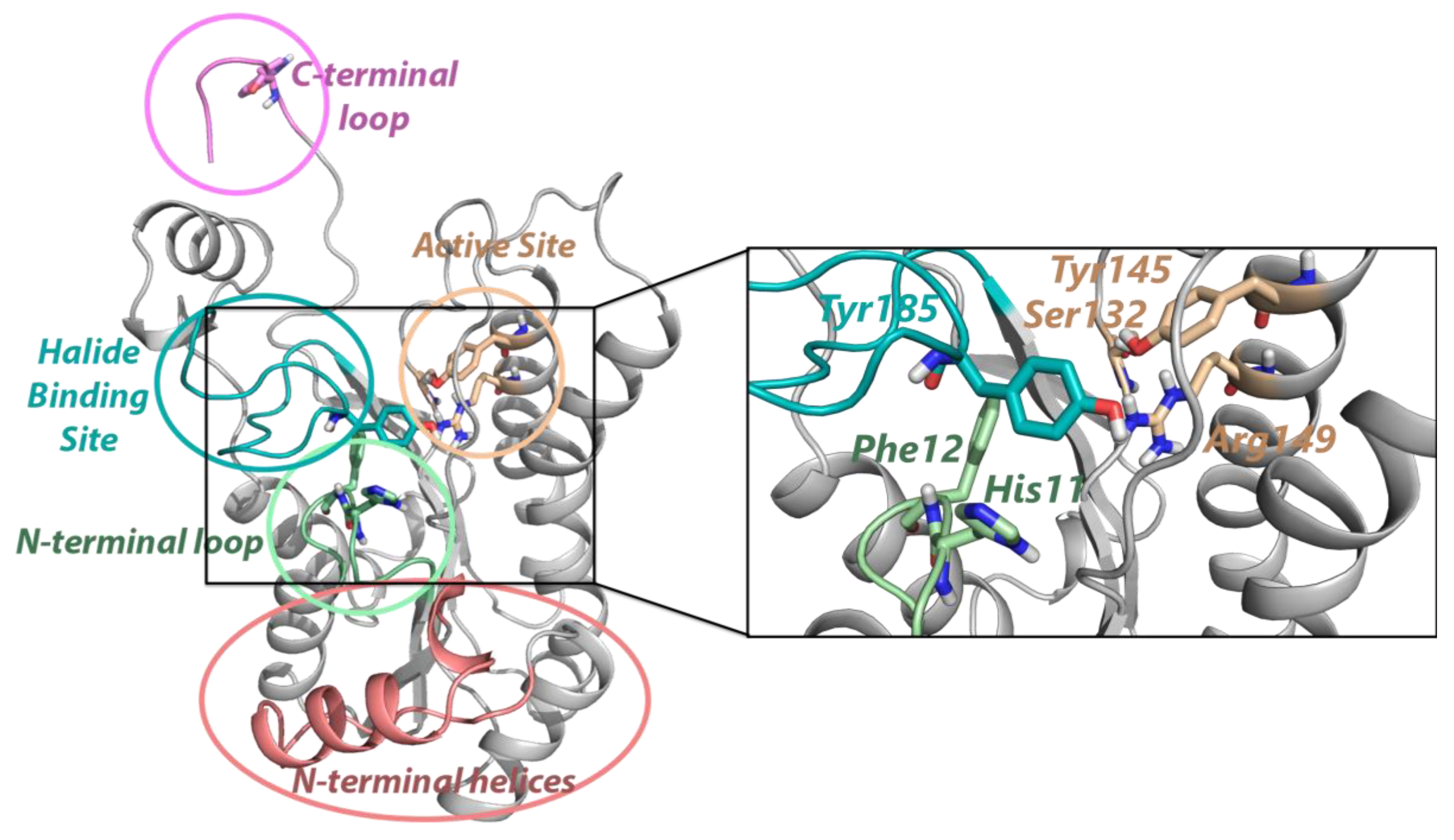
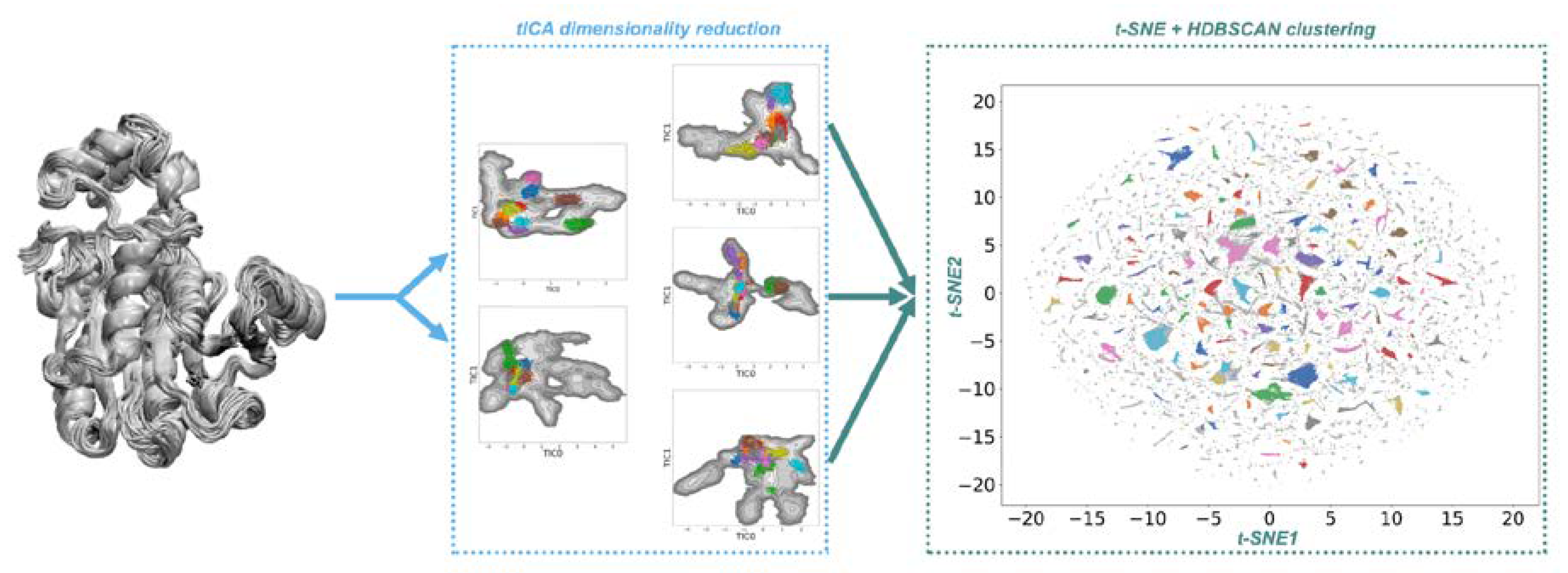
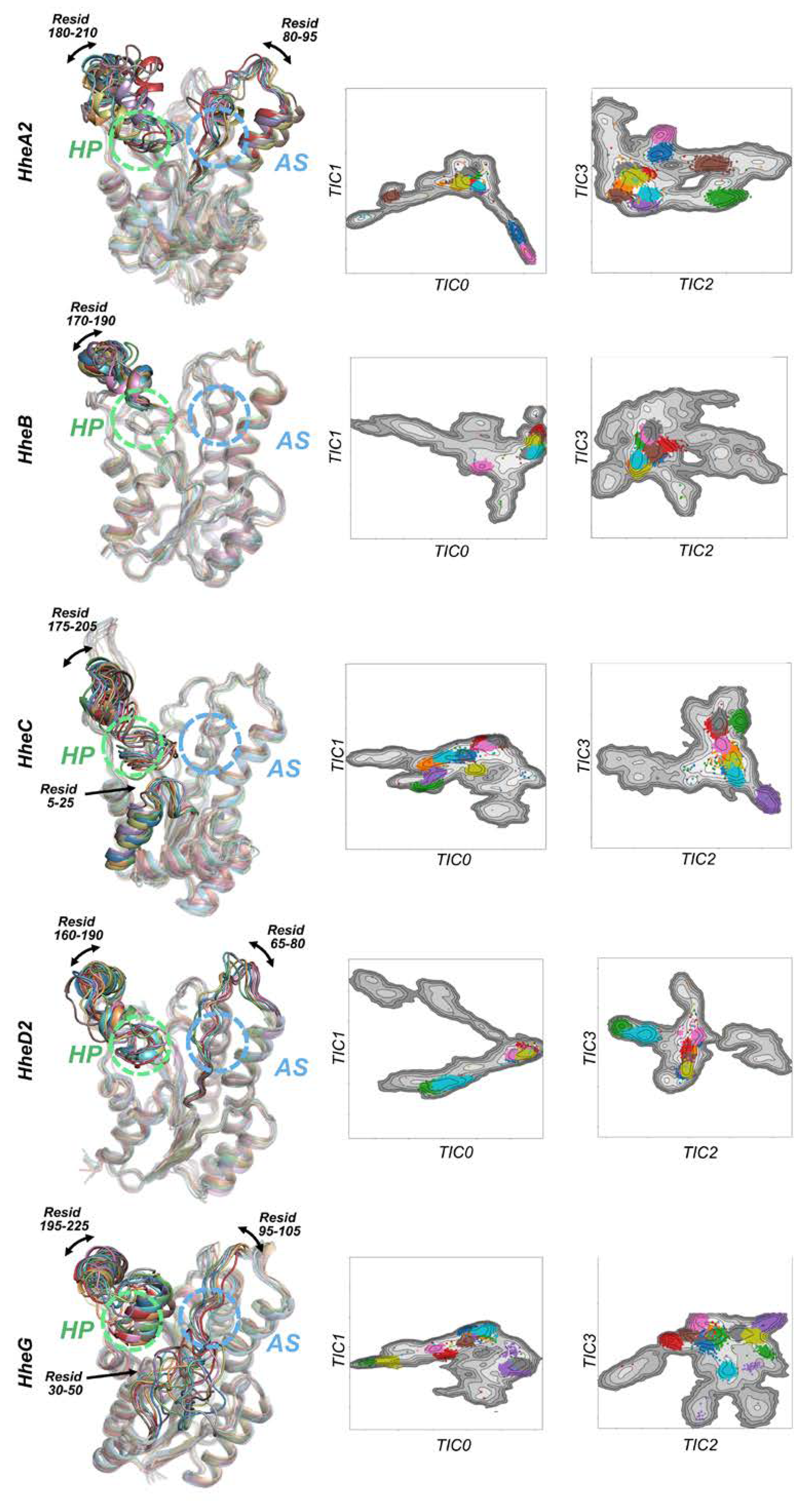
2.1. Conformational Landscapes of Halohydrin Dehalogenases (HHDHs)
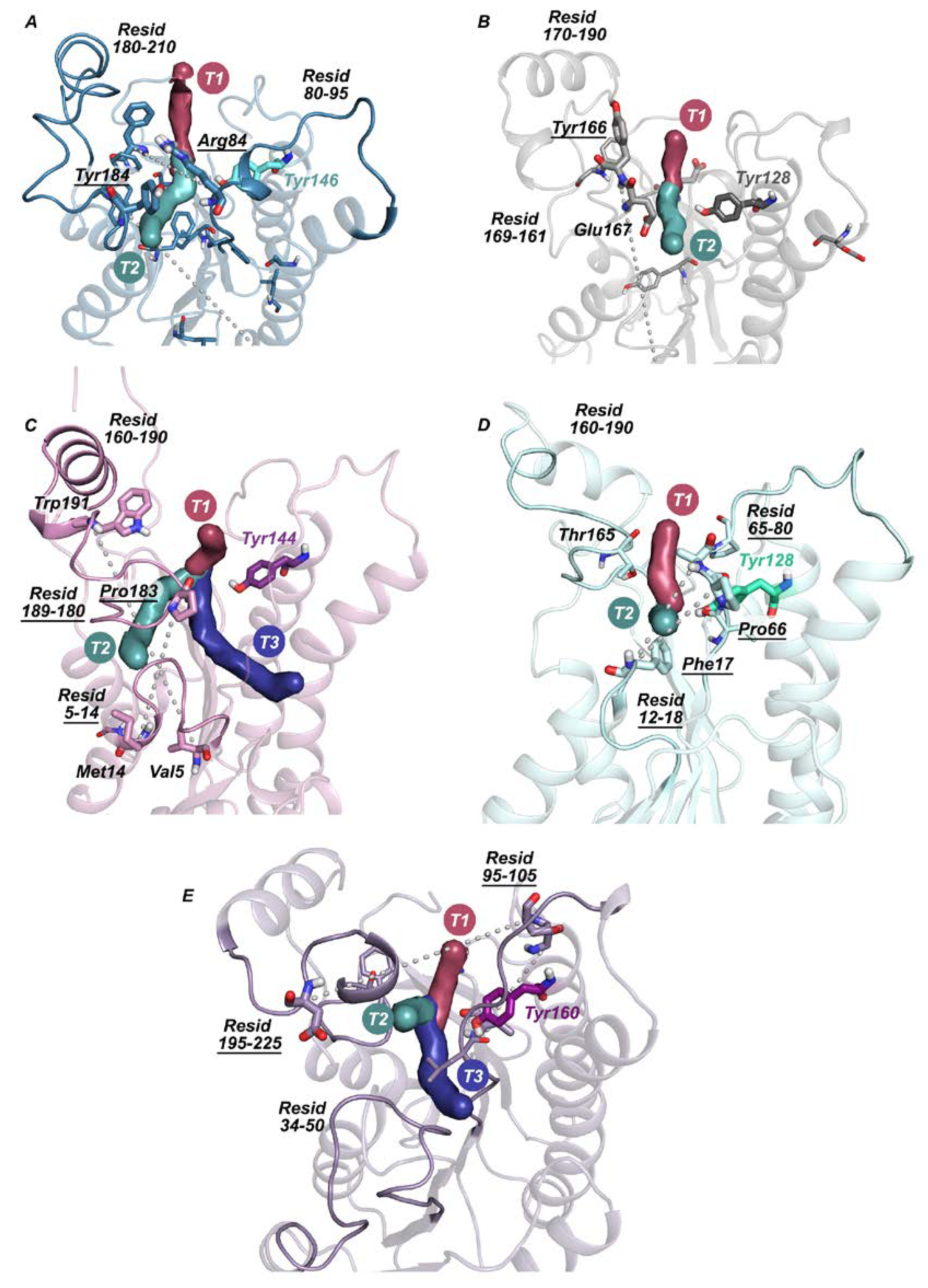
2.2. Active Site Accessibility Tunnels of Halohydrin Dehalogenases (HHDHs)
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Tokuriki, N.; Tawfik, D.S. Protein Dynamism and Evolvability. Science 2009, 324, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, O.K.; Dan, S. Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annu. Rev. Biochem. 2010, 79, 471–505. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.C.; Correy, G.J.; Mabbitt, P.D.; Buckle, A.M.; Tokuriki, N.; Jackson, C.J. Laboratory evolution of protein conformational dynamics. Curr. Opin. Struct. Biol. 2018, 50, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Maria-Solano, M.A.; Serrano-Hervás, E.; Romero-Rivera, A.; Iglesias-Fernández, J.; Osuna, S. Role of conformational dynamics in the evolution of novel enzyme function. Chem. Commun. 2018, 54, 6622–6634. [Google Scholar] [CrossRef]
- Petrović, D.; Risso, V.A.; Kamerlin, S.C.L.; Sanchez-Ruiz, J.M. Conformational dynamics and enzyme evolution. J. R. Soc. Interface 2018, 15. [Google Scholar] [CrossRef]
- Jiménez-Osés, G.; Osuna, S.; Gao, X.; Sawaya, M.R.; Gilson, L.; Collier, S.J.; Huisman, G.W.; Yeates, T.O.; Tang, Y.; Houk, K.N. The role of distant mutations and allosteric regulation on LovD active site dynamics. Nat. Chem. Biol. 2014, 10, 431–436. [Google Scholar] [CrossRef]
- Romero-Rivera, A.; Garcia-Borràs, M.; Osuna, S. Role of Conformational Dynamics in the Evolution of Retro-Aldolase Activity. ACS Catal. 2017, 7, 8524–8532. [Google Scholar] [CrossRef]
- Nestl, B.M.; Hauer, B. Engineering of Flexible Loops in Enzymes. ACS Catal. 2014, 4, 3201–3211. [Google Scholar] [CrossRef]
- Pavlova, M.; Klvana, M.; Prokop, Z.; Chaloupkova, R.; Banas, P.; Otyepka, M.; Wade, R.C.; Tsuda, M.; Nagata, Y.; Damborsky, J. Redesigning dehalogenase access tunnels as a strategy for degrading an anthropogenic substrate. Nat. Chem. Biol. 2009, 5, 727. [Google Scholar] [CrossRef]
- de Vries, E.J.; Janssen, D.B. Biocatalytic conversion of epoxides. Curr. Opin. Biotechnol. 2003, 14, 414–420. [Google Scholar] [CrossRef]
- Hasnaoui-Dijoux, G.; Majerić Elenkov, M.; Lutje Spelberg, J.H.; Hauer, B.; Janssen, D.B. Catalytic Promiscuity of Halohydrin Dehalogenase and its Application in Enantioselective Epoxide Ring Opening. ChemBioChem 2008, 9, 1048–1051. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.J.; Davis, S.C.; Mundorff, E.C.; Newman, L.M.; Gavrilovic, V.; Ma, S.K.; Chung, L.M.; Ching, C.; Tam, S.; Muley, S.; et al. Improving catalytic function by ProSAR-driven enzyme evolution. Nat. Biotechnol. 2007, 25, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Schallmey, A.; Schallmey, M. Recent advances on halohydrin dehalogenases-from enzyme identification to novel biocatalytic applications. Appl. Microbiol. Biotechnol. 2016, 100, 7827–7839. [Google Scholar] [CrossRef] [PubMed]
- Schallmey, M.; Floor, R.J.; Hauer, B.; Breuer, M.; Jekel, P.A.; Wijma, H.J.; Dijkstra, B.W.; Janssen, D.B. Biocatalytic and Structural Properties of a Highly Engineered Halohydrin Dehalogenase. ChemBioChem 2013, 14, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Wan, N.-W.; Liu, Z.-Q.; Huang, K.; Shen, Z.-Y.; Xue, F.; Zheng, Y.-G.; Shen, Y.-C. Synthesis of ethyl (R)-4-cyano-3-hydroxybutyrate in high concentration using a novel halohydrin dehalogenase HHDH-PL from Parvibaculum lavamentivorans DS-1. RSC Adv. 2014, 4, 64027–64031. [Google Scholar] [CrossRef]
- Assis, H.M.S.; Bull, A.T.; Hardman, D.J. Synthesis of Chiral Epihalohydrins Using Haloalcohol Dehalogenase A from Arthrobacter Erithii H10a. Enzyme Microb. Technol. 1998, 22, 545–551. [Google Scholar] [CrossRef]
- Elenkov, M.M.; Tang, L.; Meetsma, A.; Hauer, B.; Janssen, D.B. Formation of Enantiopure 5-Substituted Oxazolidinones through Enzyme-Catalysed Kinetic Resolution of Epoxides. Org. Lett. 2008, 10, 2417–2420. [Google Scholar] [CrossRef]
- Molinaro, C.; Guilbault, A.-A.; Kosjek, B. Resolution of 2,2-Disubstituted Epoxides via Biocatalytic Azidolysis. Org. Lett. 2010, 12, 3772–3775. [Google Scholar] [CrossRef]
- de Jong, R.M.; Tiesinga, J.J.W.; Rozeboom, H.J.; Kalk, K.H.; Tang, L.; Janssen, D.B.; Dijkstra, B.W. Structure and mechanism of a bacterial haloalcohol dehalogenase: A new variation of the short-chain dehydrogenase/reductase fold without an NAD(P)H binding site. EMBO J. 2003, 22, 4933–4944. [Google Scholar] [CrossRef]
- de Jong, R.M.; Tiesinga, J.J.W.; Villa, A.; Tang, L.; Janssen, D.B.; Dijkstra, B.W. Structural Basis for the Enantioselectivity of an Epoxide Ring Opening Reaction Catalyzed by Halo Alcohol Dehalogenase HheC. J. Am. Chem. Soc. 2005, 127, 13338–13343. [Google Scholar] [CrossRef]
- Elenkov, M.M.; Hauer, B.; Janssen, D.B. Enantioselective Ring Opening of Epoxides with Cyanide Catalysed by Halohydrin Dehalogenases: A New Approach to Non-Racemic β-Hydroxy Nitriles. Adv. Synth. Catal. 2006, 348, 579–585. [Google Scholar] [CrossRef]
- Koopmeiners, J.; Diederich, C.; Solarczek, J.; Voß, H.; Mayer, J.; Blankenfeldt, W.; Schallmey, A. HheG, a Halohydrin Dehalogenase with Activity on Cyclic Epoxides. ACS Catal. 2017, 7, 6877–6886. [Google Scholar] [CrossRef]
- Schallmey, M.; Koopmeiners, J.; Wells, E.; Wardenga, R.; Schallmey, A. Expanding the Halohydrin Dehalogenase Enzyme Family: Identification of Novel Enzymes by Database Mining. Appl. Environ. Microbiol. 2014, 80, 7303–7315. [Google Scholar] [CrossRef] [PubMed]
- Calderini, E.; Wessel, J.; Süss, P.; Schrepfer, P.; Wardenga, R.; Schallmey, A. Selective Ring-Opening of Di-Substituted Epoxides Catalysed by Halohydrin Dehalogenases. ChemCatChem 2019, 11, 2099–2106. [Google Scholar] [CrossRef]
- Orozco, M. A theoretical view of protein dynamics. Chem. Soc. Rev. 2014, 43, 5051–5066. [Google Scholar] [CrossRef]
- Osuna, S. The challenge of predicting distal active site mutations in computational enzyme design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, e1502. [Google Scholar] [CrossRef]
- Wang, L.; Marciello, M.; Estévez-Gay, M.; Rodriguez, P.E.D.S.; Morato, Y.L.; Iglesias-Fernández, J.; Huang, X.; Osuna, S.; Filice, M.; Sanchez, S. Enzyme Conformation Influences the Performance of Lipase-powered Nanomotors. Angew. Chem. Int. Ed. 2020, 59, 21080–21087. [Google Scholar] [CrossRef]
- Mu, Y.; Nguyen, P.H.; Stock, G. Energy landscape of a small peptide revealed by dihedral angle principal component analysis. Proteins 2005, 58, 45–52. [Google Scholar] [CrossRef]
- Ferguson, A.L.; Panagiotopoulos, A.Z.; Kevrekidis, I.G.; Debenedetti, P.G. Nonlinear dimensionality reduction in molecular simulation: The diffusion map approach. Chem. Phys. Lett. 2011, 509, 1–11. [Google Scholar] [CrossRef]
- Ceriotti, M.; Tribello, G.A.; Parrinello, M. Simplifying the representation of complex free-energy landscapes using sketch-map. Proc. Natl. Acad. Sci. USA 2011, 108, 13023–13028. [Google Scholar] [CrossRef]
- Hernández, C.X.; Wayment-Steele, H.K.; Sultan, M.M.; Husic, B.E.; Pande, V.S. Variational encoding of complex dynamics. Phys. Rev. E 2018, 97, 062412. [Google Scholar] [CrossRef] [PubMed]
- Mardt, A.; Pasquali, L.; Wu, H.; Noé, F. VAMPnets for deep learning of molecular kinetics. Nat. Commun. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wang, F.; Tao, P. t-Distributed Stochastic Neighbor Embedding Method with the Least Information Loss for Macromolecular Simulations. J. Chem. Theory Comput. 2018, 14, 5499–5510. [Google Scholar] [CrossRef] [PubMed]
- Spiwok, V.; Kříž, P. Time-Lagged t-Distributed Stochastic Neighbor Embedding (t-SNE) of Molecular Simulation Trajectories. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Hernández, G.; Paul, F.; Giorgino, T.; De Fabritiis, G.; Noé, F. Identification of slow molecular order parameters for Markov model construction. J. Chem. Phys. 2013, 139, 015102. [Google Scholar] [CrossRef]
- Case, D.A.; Darden, T.A.; Cheatham, T.E.; Simmerling, C.L.; Wang, J.; Duke, R.E.; Luo, R.; Crowley, M.; Walker, R.C.; Zhang, W.; et al. AMBER 16, University of California, San Francisco; United States of America: Washington, DC, USA, 2016. [Google Scholar]
- Campello, R.J.G.B.; Moulavi, D.; Sander, J. Density-Based Clustering Based on Hierarchical Density Estimates; Springer: Berlin/Heidelberg, Germany, 2013; pp. 160–172. [Google Scholar]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlikova, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P.; et al. CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 2012, 8, e1002708. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Scherer, M.K.; Trendelkamp-Schroer, B.; Paul, F.; Pérez-Hernández, G.; Hoffmann, M.; Plattner, N.; Wehmeyer, C.; Prinz, J.-H.; Noé, F. PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J. Chem. Theory Comput. 2015, 11, 5525–5542. [Google Scholar] [CrossRef] [PubMed]
- Jurcik, A.; Bednar, D.; Byska, J.; Marques, S.M.; Furmanova, K.; Daniel, L.; Kokkonen, P.; Brezovsky, J.; Strnad, O.; Stourac, J.; et al. CAVER Analyst 2.0: Analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories. Bioinformatics 2018, 34, 3586–3588. [Google Scholar] [CrossRef] [PubMed]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.-P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HHDH | Tunnel T1 | Tunnel T2 | Tunnel T3 |
|---|---|---|---|
| HheA2 | 1.8 ± 0.4 | 1.6 ± 0.6 | n.d. 1 |
| HheB | 1.9 ± 0.6 | 1.8 ± 0.8 | n.d. 1 |
| HheC | 2.0 ± 0.3 | 1.3 ± 0.2 | 1.0 ± 0.02 |
| HheD2 | 1.8 ± 0.5 | 1.7 ± 0.4 | n.d. 1 |
| HheG | 2.2 ± 0.4 | 1.9 ± 0.5 | 1.8 ± 0.5 Å |
| HHDH | Tunnel T1 | Tunnel T2 | Tunnel T3 |
|---|---|---|---|
| HheA2 | 92.4% | 12.3% | n.d. 1 |
| HheB | 97.6% | 25.7% | n.d. 1 |
| HheC | 96.9% | 77.5% | 36.2% |
| HheD2 | 88.0% | 71.1% | n.d. 1 |
| HheG | 97.6% | 91.8% | 65.8% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estévez-Gay, M.; Iglesias-Fernández, J.; Osuna, S. Conformational Landscapes of Halohydrin Dehalogenases and Their Accessible Active Site Tunnels. Catalysts 2020, 10, 1403. https://doi.org/10.3390/catal10121403
Estévez-Gay M, Iglesias-Fernández J, Osuna S. Conformational Landscapes of Halohydrin Dehalogenases and Their Accessible Active Site Tunnels. Catalysts. 2020; 10(12):1403. https://doi.org/10.3390/catal10121403
Chicago/Turabian StyleEstévez-Gay, Miquel, Javier Iglesias-Fernández, and Sílvia Osuna. 2020. "Conformational Landscapes of Halohydrin Dehalogenases and Their Accessible Active Site Tunnels" Catalysts 10, no. 12: 1403. https://doi.org/10.3390/catal10121403
APA StyleEstévez-Gay, M., Iglesias-Fernández, J., & Osuna, S. (2020). Conformational Landscapes of Halohydrin Dehalogenases and Their Accessible Active Site Tunnels. Catalysts, 10(12), 1403. https://doi.org/10.3390/catal10121403