1. Introduction
Today, the most environmentally short-term issue is mandatory CO2 emission reduction to limit global warming. One way to move towards carbon neutrality relies on the use of renewable carbon sources to produce liquid fuels for transportation or domestic uses or for chemical and material synthesis. Lignocellulosic biomass is abundant and nonedible, which makes it the best alternative to moving towards carbon neutrality thanks to its short carbon cycle conversely to fossil resources. Cellulose, the main component of lignocellulose, represents 35–50% depending on the type of biomass. Cellulose is made of linear chains of D-glucose linked by β-1-4 glycosidic bonds which are strongly associated by hydrogen bonds in cellulose micro-fibers. This is at the origin of cellulose crystallinity. The native cellulose contains highly crystalline domains and less ordered ones as well.
Cellulose is seen as a sugar reserve. Its robust hydrogen bond network makes it insoluble in most solvents and very resistant against depolymerization into glucose or other derived small molecules. Methods exist among biological and thermochemical processes, and both approaches are the subjects of considerable efforts of research. With the aim to selectively and rapidly depolymerize cellulose into glucose, the use of solid acid catalysts gathers the advantages of robustness, easy recovery, and absence of safety or corrosion issues, the conditions to pave the way towards cost effective and sustainable processes.
To date, carbon-based solid acids were reported as one of the most efficient catalytic systems for cellulose hydrolysis into glucose.
Porous acid materials obtained via acidification of active carbon or via partial carbonization of biomass and subsequent sulfonation were first reported for this application [
1,
2,
3,
4,
5]. In earlier studies, in 2008, Onda et al. [
1] reported that sulfonated activated carbon-converted selectively ball-milled cellulose at 150 °C with a high glucose yield of approximately 40%. The significant higher catalytic activity and selectivity of sulfonated activated carbon with regards to usual mineral solid acids (zeolites, sulfated zirconia, etc.) was ascribed to its hydrophobicity combined with the strong acidity of the SO
3H group. In the same year, Hara et al. [
2] investigated crystalline cellulose hydrolysis at low temperatures, 100 °C, with amorphous carbon prepared by sulfonation of partial carbonization of cellulose, leading to a material bearing various oxygenated groups: SO
3H, COOH, and OH. In these soft hydrolysis conditions, only the carbon materials were able to hydrolyze the microcrystalline cellulose into soluble β1-4 glucans and glucose. Based on cellobiose and glucose adsorption experiments, the authors proposed that the specificity of this functionalized carbon material relies on its ability to interact strongly with β1-4 glucans and with the presence of hydrophobic graphene sheets which make SO
3H groups water tolerant. Later on, in 2015, Sievers et al. [
3] investigated active carbon chemically modified with H
2O
2 and sulfuric acid for cellulose hydrolysis. He concluded that, despite the relative weakness of the acid functionalized group, their impact on the β1-4 glucan adsorption would induce conformational changes assumed to be at the origin of the hydrolysis activity. Thereafter, the hydrolysis rate of oligosaccharides of different polymerizations (DPs) catalyzed by functionalized carbon materials was studied by Fukuoka et al. [
5]. The detailed kinetic study revealed an enhanced hydrolysis rate of longer oligosaccharides explained by their higher affinity with the carbon material surface assumed to cause β1-4 glycosidic bond angle changes and to reduce the activation energy required for their cleavage. The authors concluded that this would justify the unique selectivity of the carbon materials for cellulose hydrolysis in glucose despite their low acidity. One can retain that these works agree on the role of favored β1-4 glucan adsorption on functionalized amorphous active carbon while the nature and the acid strength of the active sites remain less documented.
Recently, graphene oxide (GO) and sulfonated GO are objects of substantial interest for cellulose hydrolysis into glucose [
6,
7,
8,
9,
10,
11,
12]. GO, obtained from graphite exfoliation/oxidation, is constituted of single or few flat nanolayers of 2D sheets constituted of sp
2 hybridized carbons with in-plane and edge defects made of sp
3 hybridized carbons. These defects in sp
3 carbons gave rise to various oxygenated functional groups: OH, epoxy, and COOH. SO
3H groups are also present when GO is prepared via Hummer’s method, which involves H
2SO
4 among the reactants [
13,
14]. These oxygenated groups expand the layer separation and are the origin of GO hydrophilicity, which makes it highly dispersed in water. The chemical nature of the functional groups is similar to those described previously on functionalized active carbons. GO is also reported to be highly active and selective for cellulose hydrolysis into glucose. Earlier studies from Zhang’s group, in 2014 [
7], have shown that GO hydrolyzed cellulose to glucose up to 50% yield at 150 °C, after 24 h, using a quantitative catalyst/cellulose weight ratio. It was proposed that carboxylic and phenolic groups of the GO layered structure would act in synergy with the strong acid active sites, SO
3H, for efficient activation of the β1-4 glycosidic bond. In 2014, Deng et al. [
6] also found that GOs have superior activity compared to other conventional solid acids and even H
2SO
4 for cellobiose hydrolysis into glucose. They proposed that the GO sheets would work as enzyme-like activation with hydrophobic graphene sheet decorated with oxygenated groups capable of activating the reactants. Later on, in 2015, Yang et al. concluded the outstanding catalytic performances of reduced graphene oxide to hydrolyze crystalline Avicel celluloses in glucose, leading to 30% glucose yield after 6 h at 150 °C using a quantitative cellulose to catalyst weight ratio. A value twice higher that obtained with Amberlyst-15 while zeolites were inactive. The authors explained the activity of their graphene-based material by the catalytic action of the acidic –COOH and SO
3H groups while the –OH groups participated in the adsorption of the β1-4 glucans and therefore made it possible to access cellulose to the solid catalyst active sites. Quite similar explanations were also proposed to explain the efficiency for cellulose hydrolysis in glucose over a GO-based material obtained from cellulose carbonization done in the presence of GO [
10]. Note that only post-sulfonated carbon materials were active. The determining role of solid–solid contact for graphene-based catalyzed cellulose hydrolysis was recently highlighted by Huang et al. [
11]. Adding N,N-dimethylacetamide resulted in improved dispersibility of a sulfonated GO and in high glucose yield of 45% after 8 h at 130 °C with an Avicel cellulose/catalyst weight ratio of 1. One can underline that all the cited studies dealing with GO-based catalysts underlined the unique role of the functionalized 2D carbon sheet in adsorbing soluble β 1-4 glucan and even cellulose. Again, one can remark that the nature and the acid strength of the active sites are not clearly addressed.
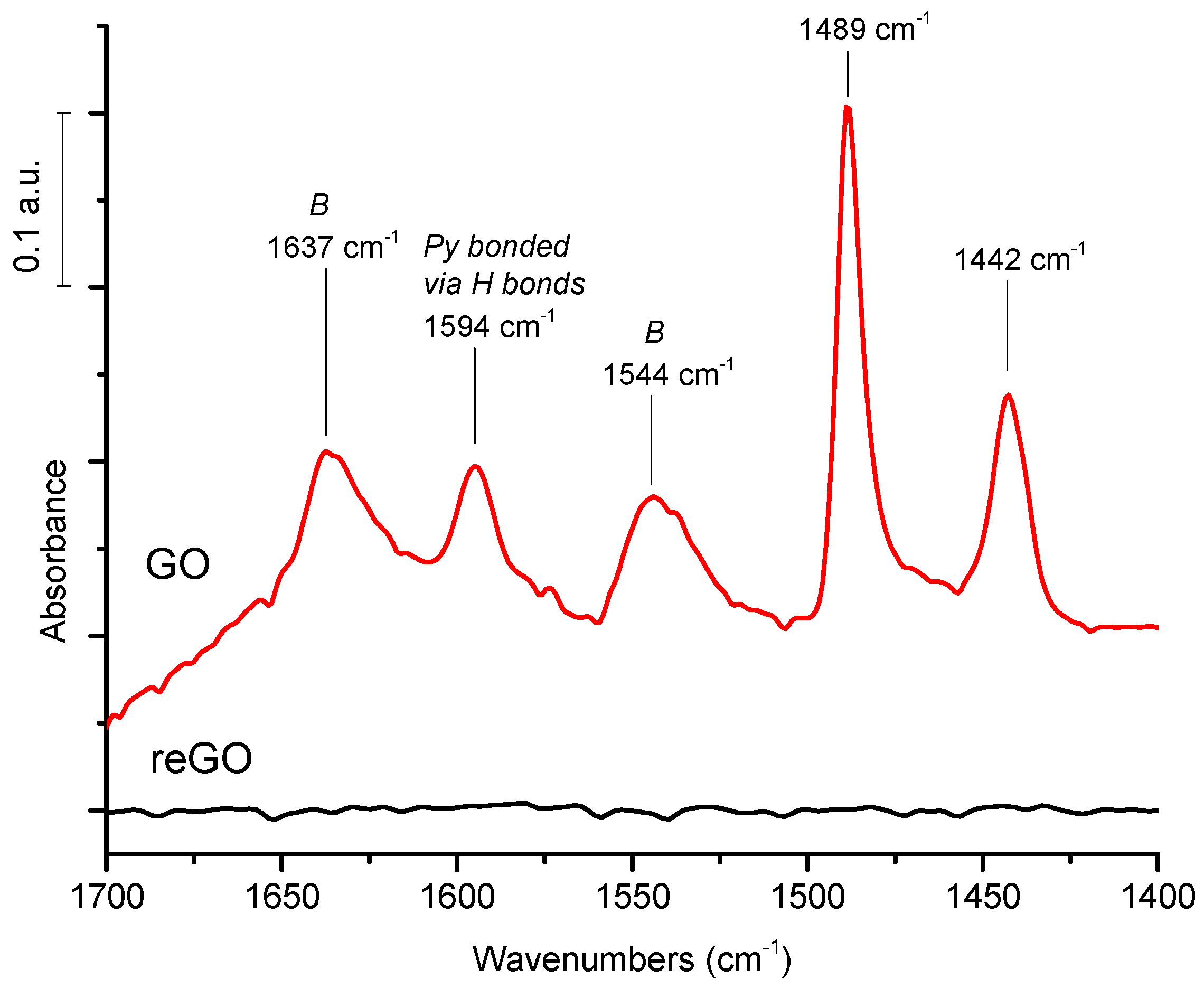
In the above reported publications, qualitative information on the chemical nature of the potential acid sites are provided by FTIR of GO in KBr pellets and/or by XPS. With regards to quantitative data, they are deduced from S chemical analysis and titrations after Na+ exchanges. Surprisingly, one can note the absence of data obtained from the usual methods based on the adsorption of probe basic molecules (pyridine/ammonia) monitored by FTIR or calorimetry to characterize the nature (Br∅nsted/Lewis), the strength, and the amount of acid sites. Indisputably, this would contribute to clarifying the role of oxygenated groups as effective acid sites with regards to their accepted role in β 1-4 glucan adsorption.
In this article, we measured the acid strength of graphene-based materials, GO prepared by Hummer’s method, and reduced GO prepared by calorimetry of NH3 adsorption. The nature of the acid sites is studied by FTIR of pyridine adsorption/desorption. In parallel, their catalytic performances for cellulose hydrolysis and hydrogenolysis using GO combined with Pt were evaluated. Very strong acid sites are probed on graphene oxides by calorimetry with heats of NH3 adsorption as high as 200 kJ/mole, while reduced graphene oxide has a very weak acid surface. The FTIR of pyridine adsorption shows the prevailing presence of Br∅nsted acid sites and a peculiar feature: the presence of pyridine coordinated by hydrogen bonds. This exceptionally strong Br∅nsted acidity is tentatively explained by the presence of graphene domains made of intact sp2 carbon decorated by hydroxyl, carboxylic, or sulfonated groups in the GO sheets, resulting in high mobility of the negative charges borne by the oxygenated groups within the graphene sheet of high conductivity, which makes the proton weakly retained, explaining its strong acidity. In parallel, we examined the catalytic activity of GO and reduced GO. In agreement with their acidic features, only GO is active and selective for native cellulose hydrolysis in glucose conversely to reduced GO. Finally, we failed to directly produce sugar alcohols from cellulose using a combination of GO with Pt/reduced GO under hydrogen pressure, explained by the reduction of oxygenated functions of GO which cancel out its acidic properties.
3. Materials and Methods
3.1. Materials
Sulfuric acid 95–97%, Potassium permanganate (≥99.0%), sodium nitrate (≥99.0%), hydrogen peroxide (30% in water), hydrochloric acid (37% in water), hydrazine hydrate (80% in water), H2PtCl6, 6H2O (99%), and ethylene glycol (99.8%) were purchased from Sigma-Aldrich. Expanded graphite, used as carbon source for graphene oxide, was purchased from SGL Group.
Commercial cellulose MN 301, at standard grade, 95% of fibers with lengths 2–20 μm, and average degree of polymerization 400–500 and used as model substrate for hydrolysis reaction, was purchased from Macherey-Nagel.
3.2. Graphene Oxide and Reduced Graphene Oxide Preparation
Graphene oxide was synthesized following the modified Hummers’ method [
15]; 50 mL of sulfuric acid 95% was added to a mixture of 1 g expanded graphite and 0.5 g sodium nitrate under constant stirring at room temperature. After 1 h, the temperature of the solution was cooled to less than 20 °C by ice bath; then, 3 g KMnO
4 was added gradually to the solution. The mixture was stirred at 35 °C for 2 h, followed by the slow addition of 50 mL of deionized (DI) water. After stirring at 90 °C for 2 h, 5 mL of H
2O
2 30% was added to the mixture to ensure completion of the reaction with KMnO
4. The resulting mixture was washed with 100 mL HCl 3.7% once and with DI water using centrifugation until pH 7, followed by freeze drying. The result powder was graphene oxide.
Reduced graphene oxide (reGO) was achieved from the reduction of graphene oxide by using ethylene glycol; 0.3 g of GO was dispersed in 300 mL DI water by ultrasonication for 30 min to obtain homogeneous dispersion. Then, 150 mL of ethylene glycol was added into the solution and heated in an oil bath under reflux for 24 h. After the reaction, the obtained reGO was separated by filtration and washed three times with ethanol and DI water. reGO were dried at 60 °C for 24 h to remove water content.
Platinum dispersed on reduced graphene oxide (1% Pt/reGO) was achieved from a reduction of the platinum precursor and graphene oxide using ethylene glycol; 0.3 g of GO was dispersed in 300 mL DI water by ultrasonication for 30 min to obtain homogeneous dispersion before 3 mL of H2PtCl6 solution (1 mg Pt/mL) was added to obtain 1%Pt/reGO. Then, 150 mL of ethylene glycol was added into the solution and heated in oil bath under reflux for 24 h. After the reaction, the obtained 1% Pt/reGO was filtrated and washed three times with ethanol and DI water. Pt/reGO were dried at 60 °C for 24 h to remove water content.
3.3. Physicochemical Characterization of Catalysts
The FTIR spectra were recorded with a Brucker Vector 22 spectrometer in the absorption mode with a resolution of 2 cm−1. GO and reGO samples were dispersed in water and spread on an inert infrared transparent silicon holder. For pyridine adsorption monitored by FTIR, the GO and reGO samples were also dispersed on a silicon plate and placed in an IR cell equipped with CaF2 windows. The samples were treated at ambient temperature or at 150 °C under vacuum. Pyridine was adsorbed under saturation vapor pressure at ambient temperature and then desorbed at ambient temperature or at 150 °C in order to remove the physisorbed Py species.
The acidic strength and the number of acid sites of the catalyst were characterized by NH3 adsorption at 80 °C using a TianCalvet calorimeter. The samples were placed in the glass cell and pretreated at 150 °C for 3 h under secondary vacuum and then, placed into the calorimeter up to the stabilization of the temperature (80 °C, one night). After that, the sample was contacted with successive small doses of NH3 vapor while the differential enthalpies of NH3 adsorption were recorded.
3.4. Catalytic Tests
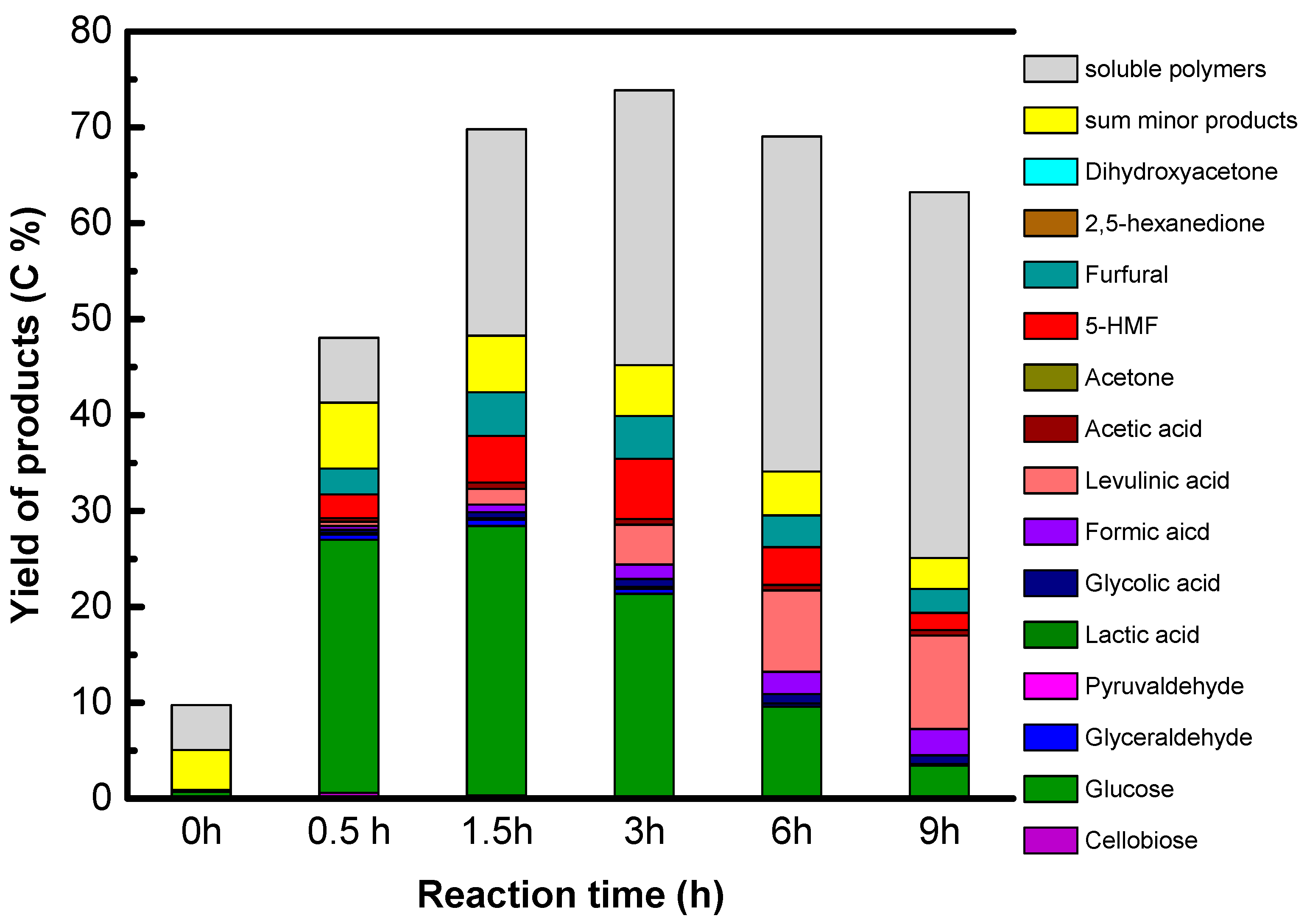
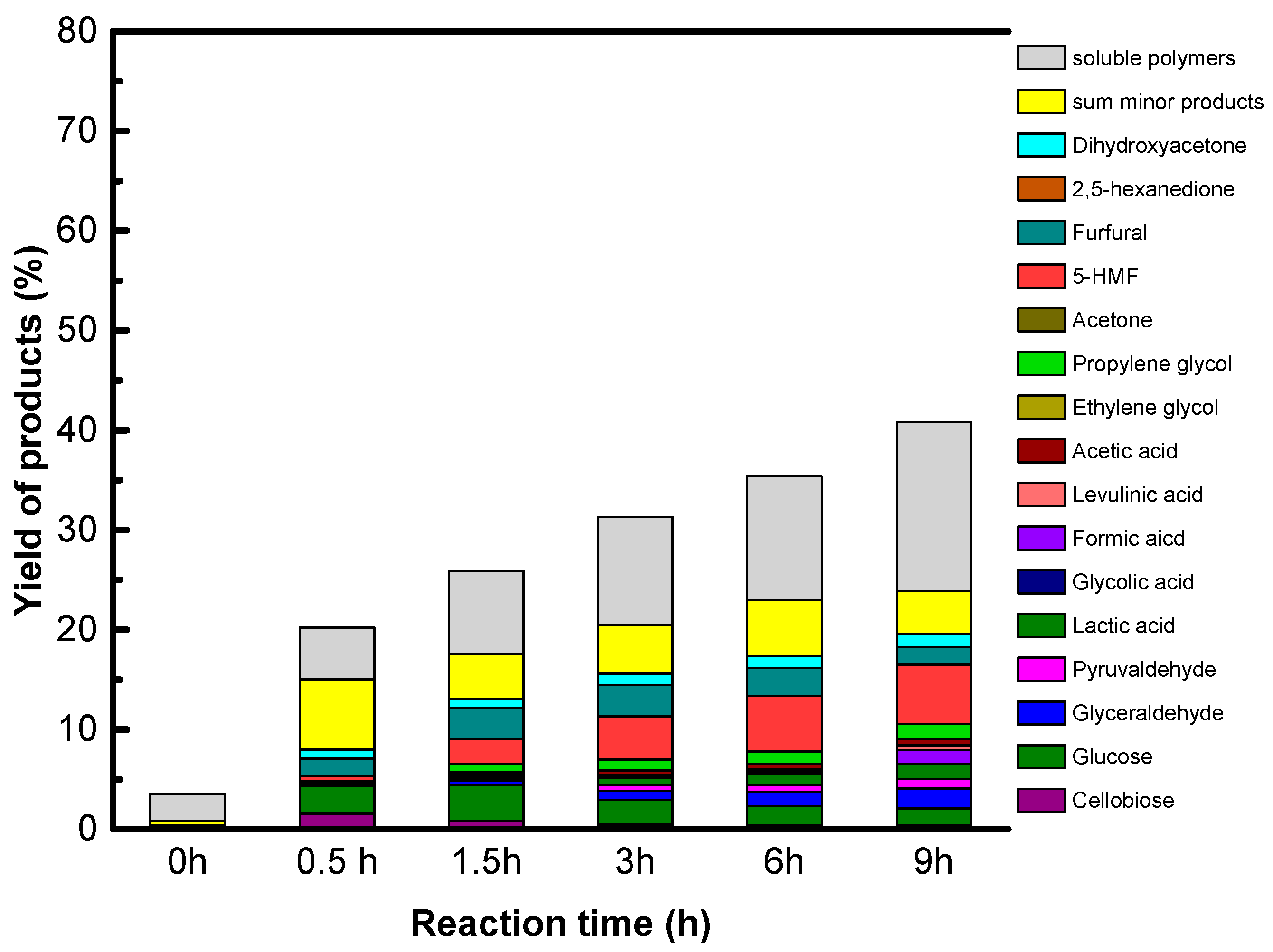
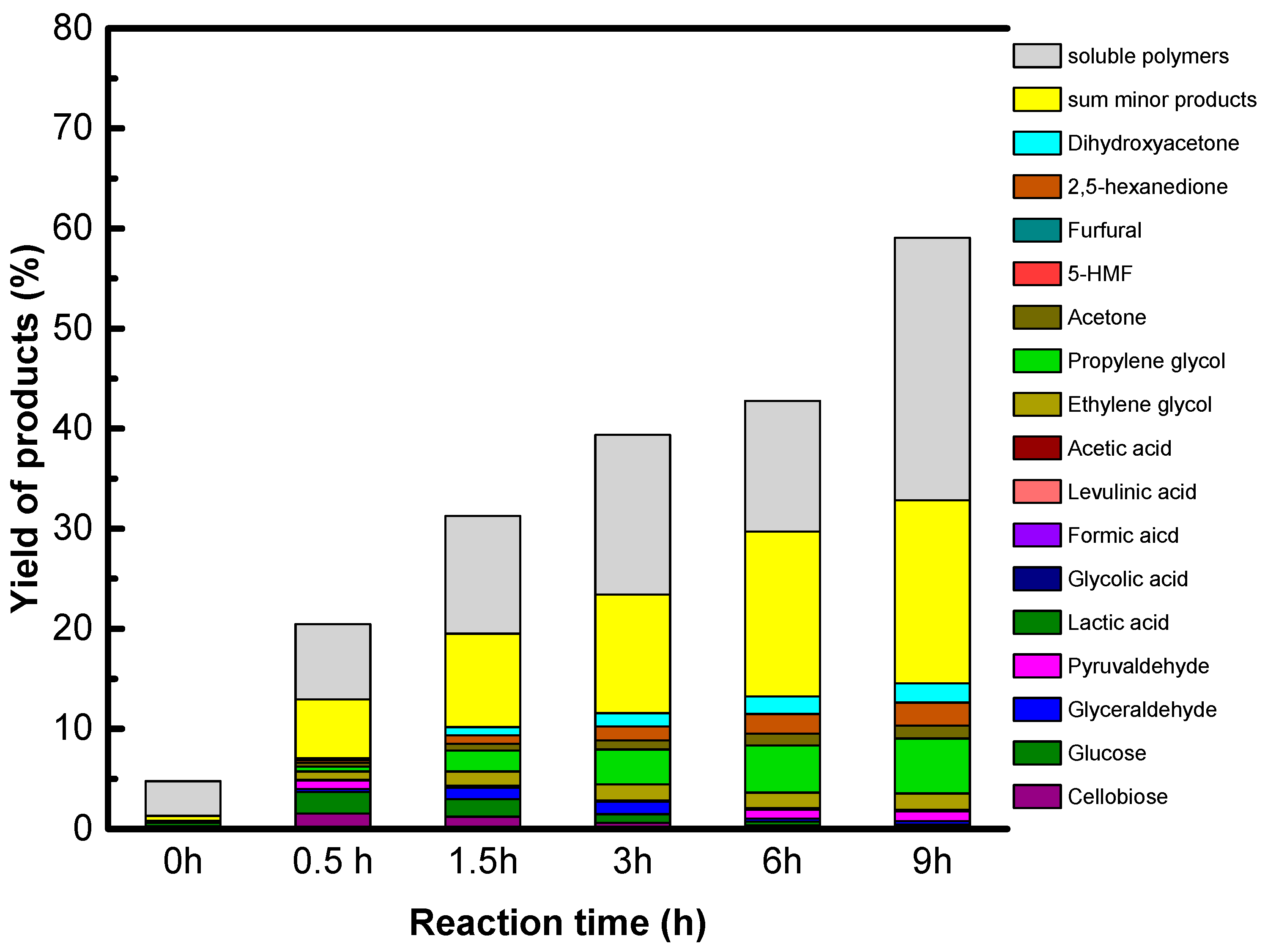
The reaction conditions are as follow: 1.6 g of native cellulose, 0.34 g or 0.68 g of catalysts and 65 mL of water were introduced in a 100 mL Paar Hastelloy autoclave equipped with a Rushton turbine under auto pressure at 190 °C. The autoclave was purged with He and heated. The starting time is the time when temperature reached 190 °C. After the period reaction time, 2 mL of samples was taken out for kinetic analysis. The samples were filtrated over polyvinylidene difluoride (PVDF) filter (0.45 μm). All products in liquid phase were analyzed by a HPLC-RID system using COREGEL 107H column.
The total mass of carbon in the liquid phase (solubility) was determined by a Shimadzu TOC-VSCH analyzer [
17,
18]:
in which mg C
liquid phase: mg of carbon contents in liquid phase, obtained from the total soluble organic carbon (TOC) analysis, in mg of carbon and mg C
initial cellulose: mg of carbon contents in the initial cellulose
The carbon yields of the products were calculated as the moles of the product i detected by HPLC and the initial glucosyl units in the initial cellulose, corrected by the number of carbon atoms:
in which nC
i: number of carbon atoms in the product i; n
i: number of moles of the product i determined by the HPLC analysis; and n
glucosyl units: initial number of moles of glucosyl units in the cellulose sample = m
cellulose/162.
The yield of soluble oligosaccharides and polymers (SP) was determined from subtraction from the solubility analyzed by TOC to the sum of products analyzed by HPLC:
4. Discussion and Conclusions
The catalytic results reported here confirm that GO acts as an excellent solid acid catalyst for cellulose hydrolysis into glucose. As recalled in the Introduction section, there is a general agreement to correlate the GO unique catalytic performances to its ability to adsorb carbohydrates but the origin of its acidity remains unclear. We believe that this article is a contribution which brings new insights into the origin of the acidity of GO. The specificity of GO prepared by Hummer’s method implies that sulfonated groups are present in the as-synthesized GO in addition to carboxylic and hydroxyl groups or lactones since sulfuric acid is one of the reactants used to oxidize the graphite precursor. In this paper, we disclose by calorimetry of NH
3 adsorption the presence of very strong acid sites on GO with heat of NH
3 adsorption as high as 200 kJ/mol, a value similar to that measured on heteropolyacids, H
3PW
12O
40 or Cs
2HPW
12O
40, reported as superacids [
18,
19]. It is clear that the acidity of GO is significantly higher than that developed by sulfonic groups borne by a classical carbonous matrix such as sulfonated acidic resins (Amberlyst-15) or usual sulfonated carbons characterized by the lower heat of ammonia adsorption of approximately 140 kJ/mol [
15,
20]. Besides, it is shown here that reGO has a weak acidic surface capable of retaining NH
3 rather by physisorption than chemisorption. FTIR of pyridine adsorption shows the prevailing presence of Br∅nsted acid sites and a peculiar feature of GO, the presence of pyridine retained on the surface by hydrogen bonding. Since reGO is not capable of retaining pyridine species in good agreement with the calorimetric results and since the surface of reGO is shown by FTIR to be depleted from most of the functional groups, it seems reasonable to propose that the strong acidity of GO originated from a synergy between the 2D carbon sheet of GO and the oxygenated functional groups. We tentatively explain the exceptional strong Br∅nsted acidity of GO by drawing inspiration from what we know about the origin of the superacidity of heteropolyacids [
21,
22], i.e., the existence of negative charge delocalization on the surface of large Kegging anions which makes the protons weakly retained. By similitude, we propose that, within the GO sheet, graphene sp2 carbon domains decorated with hydroxyl, carboxylic, or sulfonated groups exist and result in high mobility of the negative charges of the functional groups within the graphene domain, which makes the protons free and explains their strong acidity (
Scheme 2).
Accordingly, only GO is active and selective for native cellulose hydrolysis leading selectively to glucose in high yield, 27%. However, we show that sugar alcohols cannot be formed directly from cellulose using GO combined with a metallic function under hydrogen, explained by the reduction of the oxygenated functions of GO which are essential for GO acidity. The instability of the acid functions of GO in a reducing atmosphere is the weak point of this singular solid acid.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








