One-Pot Synthesis of B-Aryl Carboranes with Sensitive Functional Groups Using Sequential Cobalt- and Palladium-Catalyzed Reactions



Abstract
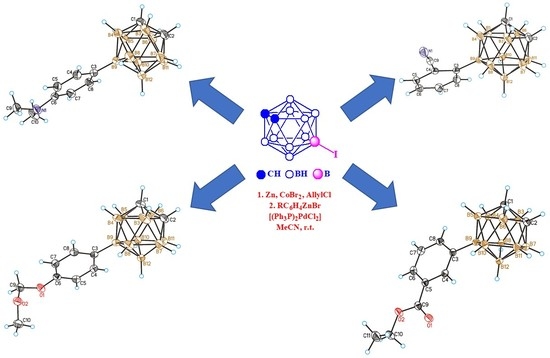
{kind=link}
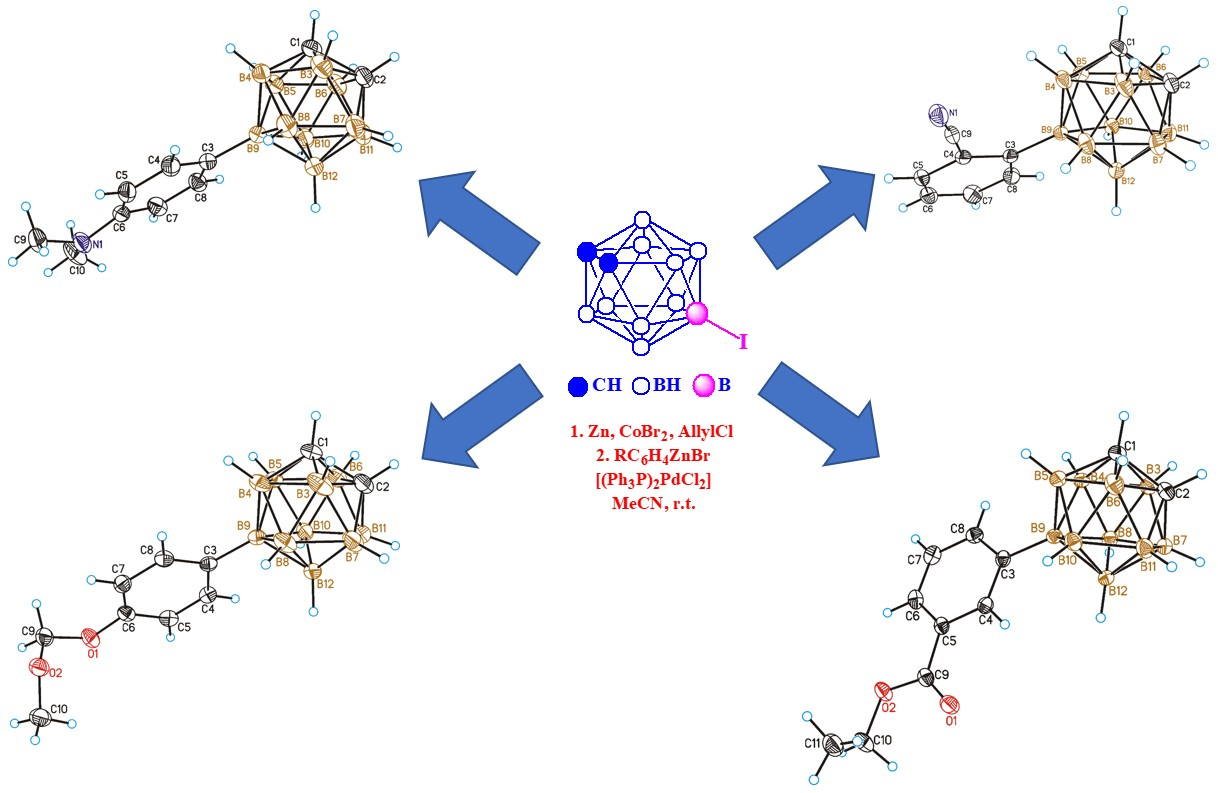
{kind=link}
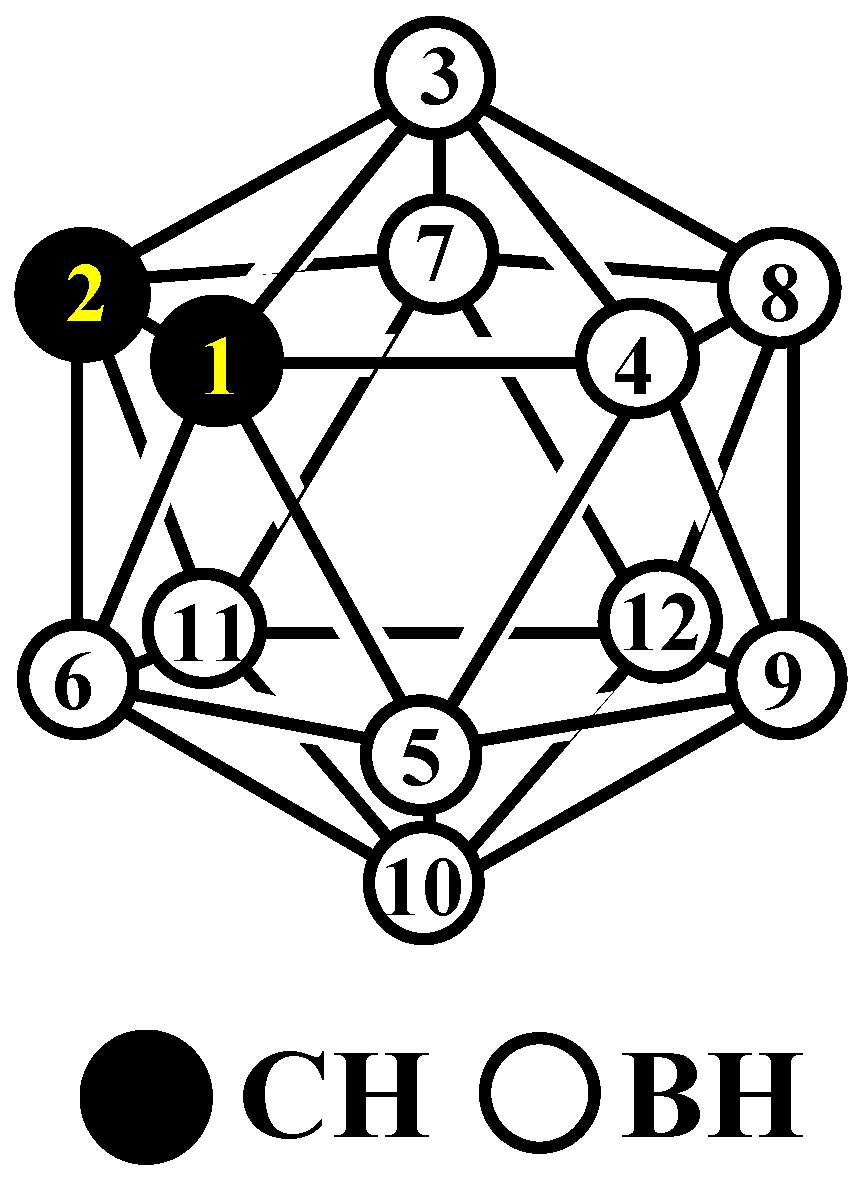
{kind=link}
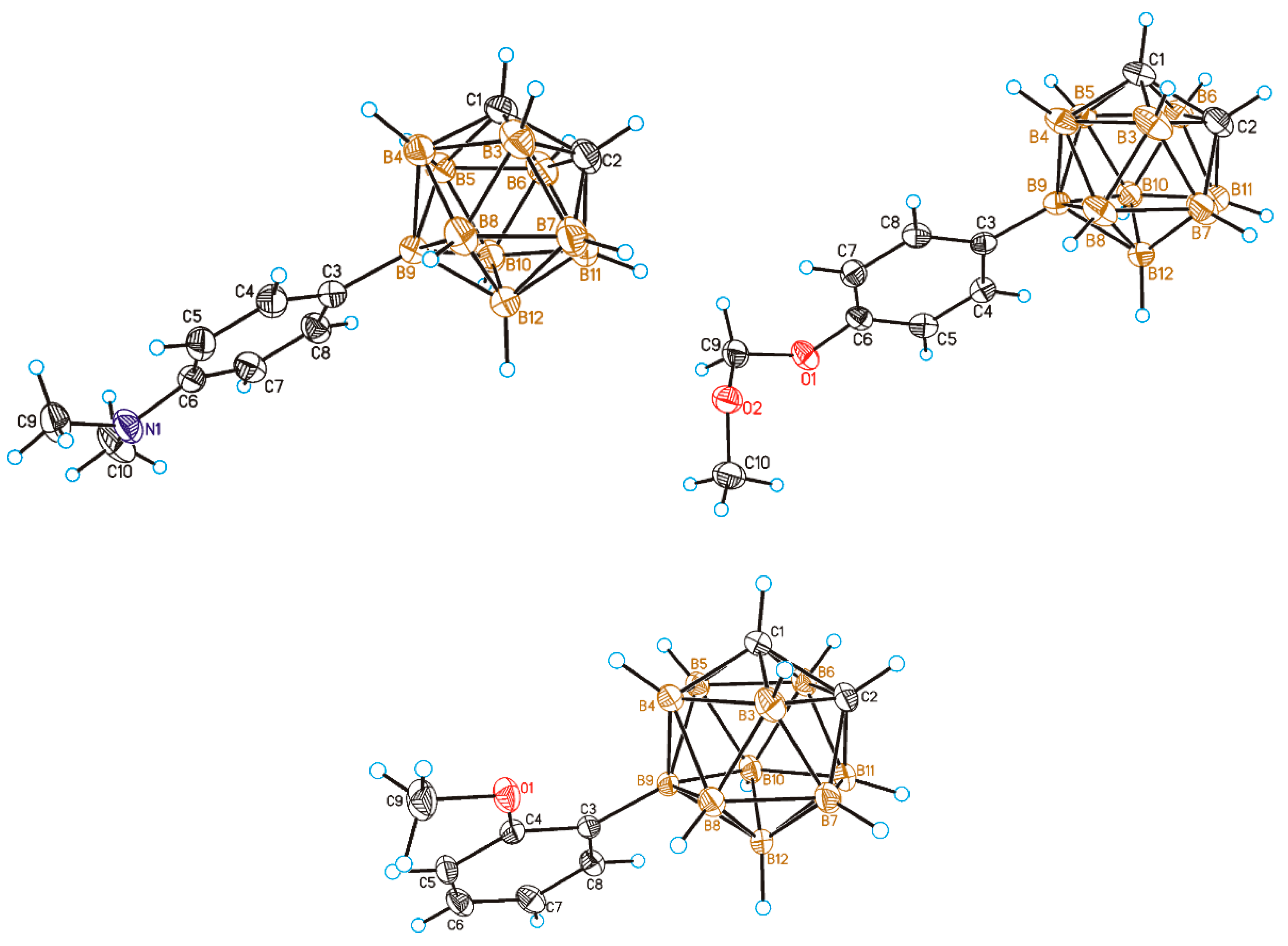
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
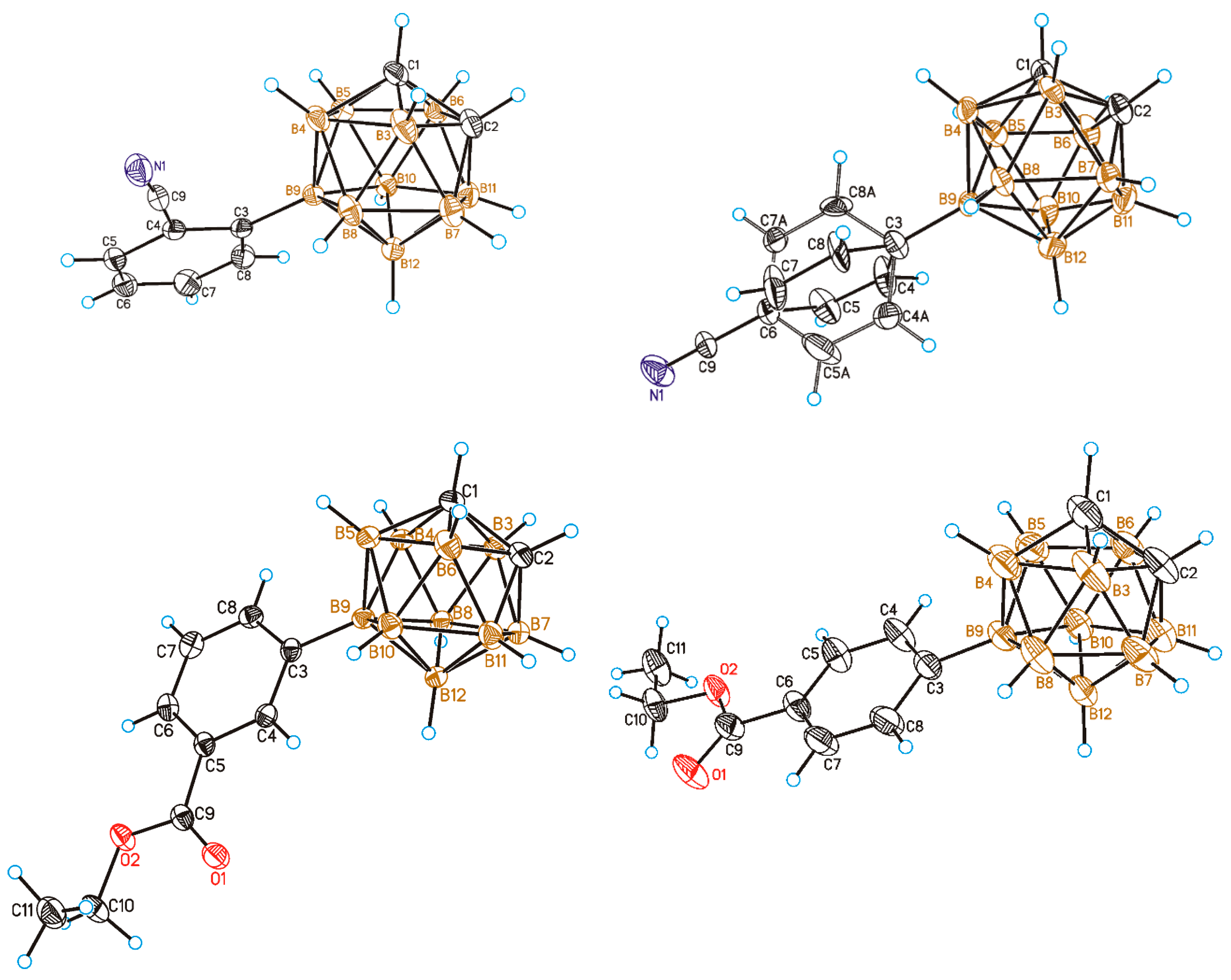
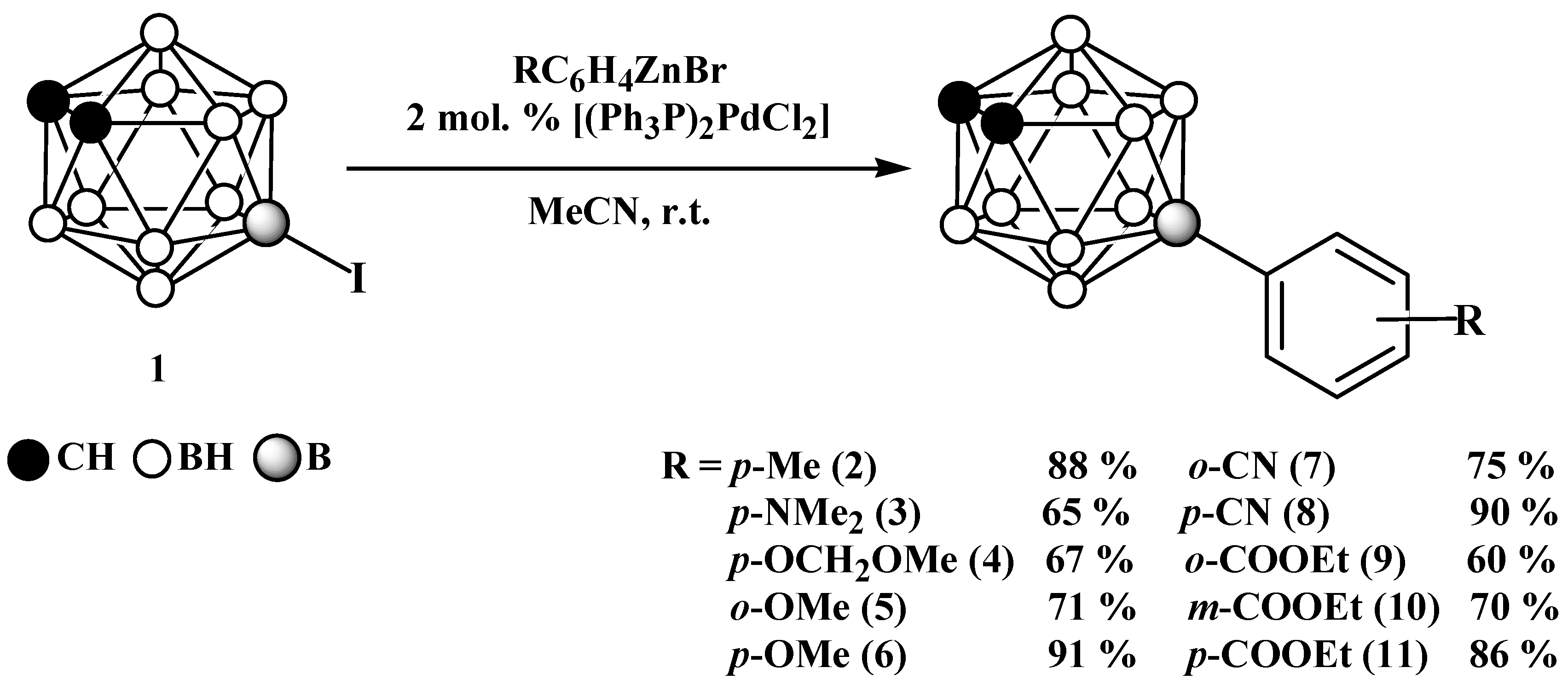
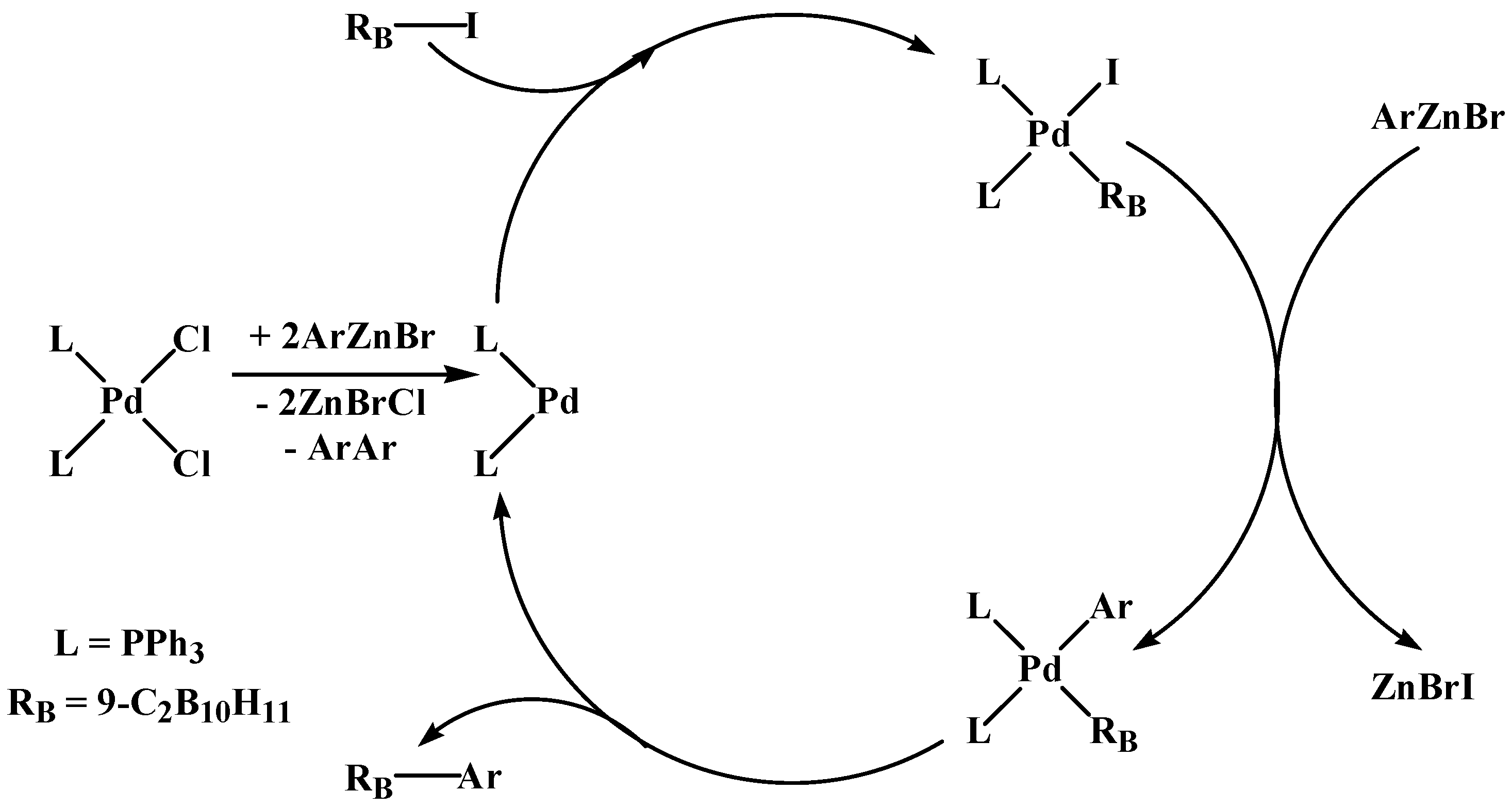
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. General Synthetic Procedure and Characterization of Monosubstituted B-Aryl Derivatives of Ortho-Carborane
3.3. General Synthetic Procedure and Characterization of Disubstituted B-Aryl Derivatives of Ortho-Carborane
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yamamoto, K.; Endo, Y. Utility of boron clusters for drug design. Hansch-Fujita hydrophobic parameters π of dicarba-closo-dodecaboranyl groups. Bioorg. Med. Chem. Lett. 2001, 11, 2389–2392. [Google Scholar] [CrossRef]
- Issa, F.; Kassiou, M.; Rendina, L.M. Boron in drug discovery: Carboranes as unique pharmacophores in biologically active compounds. Chem. Rev. 2011, 111, 5701–5722. [Google Scholar] [CrossRef] [PubMed]
- Scholz, M.; Hey-Hawkins, E. Carbaboranes as pharmacophores: Properties, synthesis, and application strategies. Chem. Rev. 2011, 111, 7035–7062. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y. Carboranes as hydrophobic pharmacophores: Applications for design of nuclear receptor ligands. In Boron-Based Compounds: Potential and Emerging Applications in Medicine; Hey-Hawkins, E., Viñas Teixidor, C., Eds.; John Wiley & Sons Ltd.: Oxford, UK, 2018; pp. 3–19. [Google Scholar] [CrossRef]
- Spokoyny, A.M.; Farha, O.K.; Mulfort, K.L.; Hupp, J.T.; Mirkin, C.A. Porosity tuning of carborane-based metal–organic frameworks (MOFs) via coordination chemistry and ligand design. Inorg. Chim. Acta 2010, 364, 266–271. [Google Scholar] [CrossRef]
- Kennedy, R.D.; Krungleviciute, V.; Clingerman, D.J.; Mondloch, J.E.; Peng, Y.; Wilmer, C.E.; Sarjeant, A.A.; Snurr, R.Q.; Hupp, J.T.; Yildirim, T.; et al. A carborane-based metal-organic framework with high methane and hydrogen storage capacities. Chem. Mater. 2013, 25, 3539–3543. [Google Scholar] [CrossRef]
- Qi, S.-C.; Han, G.; Wang, H.-R.; Li, N.; Zhang, X.; Jiang, S.-L.; Lu, Y.-F. Synthesis and characterization of carborane bisphenol resol phenolic resins with ultrahigh char yield. Chin. J. Polym. Sci. 2015, 33, 1606–1617. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, G.; Feng, C.; Yang, J. High Tg and thermo-oxidatively stable thermosetting polyimides derived from a carborane-containing diamine. Macromol. Rapid Commun. 2018, 39, 1800484. [Google Scholar] [CrossRef]
- Cui, M.; Zhang, L.; Lou, P.; Zhang, X.; Han, X.; Zhang, Z.; Zhu, S. Study on thermal degradation mechanism of heat-resistant epoxy resin modified with carboranes. Polym. Degrad. Stabil. 2020, 109143. [Google Scholar] [CrossRef]
- So, H.; Kim, J.H.; Lee, J.H.; Hwang, H.; An, D.K.; Lee, K.M. Planarity of terphenyl rings possessing o-carborane cages: Turning on intramolecular-charge-transfer-based emission. Chem. Commun. 2019, 55, 14518–14521. [Google Scholar] [CrossRef]
- Ochi, J.; Tanaka, K.; Chujo, Y. Recent progress in the development of solid-state luminescent o-carboranes with stimuli responsivity. Angew. Chem. Int. Ed. 2020, 59. [Google Scholar] [CrossRef]
- Wu, X.; Guo, J.; Lv, Y.; Jia, D.; Zhao, J.; Shan, H.; Jin, X.; Ma, Y. Aggregation-induced emission characteristics of o-carborane-functionalized fluorene and its heteroanalogs: The influence of heteroatoms on photoluminescence. Mater. Chem. Front. 2020, 4, 257–267. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes, 3rd ed.; Academic Press: London, UK, 2016; pp. 283–502. [Google Scholar] [CrossRef]
- Heying, T.L.; Ager, J.W.; Clark, S.L.; Mangold, D.J.; Goldstein, H.L.; Hillman, M.; Polak, R.J.; Szymanski, J.W. A new series of organoboranes. I. Carboranes from the reaction of decaborane with acetylenic compounds. Inorg. Chem. 1963, 2, 1089–1092. [Google Scholar] [CrossRef]
- Ohta, K.; Goto, T.; Endo, Y. 1,2-Dicarba-closo-dodecaboran-1-yl naphthalene derivatives. Inorg. Chem. 2005, 44, 8569–8573. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Morisaki, Y.; Chujo, Y. o-Carborane-based anthracene: A variety of emission behaviors. Angew. Chem. Int. Ed. 2015, 54, 5084–5087. [Google Scholar] [CrossRef] [PubMed]
- Coult, R.; Fox, M.A.; Gill, W.R.; Herbertson, P.L.; MacBride, J.A.H.; Wade, K. C-arylation and C-heteroarylation of icosahedral carboranes via their copper(I) derivatives. J. Organomet. Chem. 1993, 462, 19–29. [Google Scholar] [CrossRef]
- Colquhoun, H.M.; Herbertson, P.L.; Wade, K.; Baxter, I.; Williams, D.J. A carborane-based analogue of poly(p-phenylene). Macromolecules 1998, 31, 1694–1696. [Google Scholar] [CrossRef]
- Fujii, S.; Yamada, A.; Nakano, E.; Takeuchi, Y.; Mori, S.; Masuno, H.; Kagechika, H. Design and synthesis of nonsteroidal progesterone receptor antagonists based on C,C′-diphenylcarborane scaffold as a hydrophobic pharmacophore. Eur. J. Med. Chem. 2014, 84, 264–277. [Google Scholar] [CrossRef]
- Naito, H.; Nishino, K.; Morisaki, Y.; Tanaka, K.; Chujo, Y. Solid-state emission of the anthracene-o-carborane dyad from the twisted-intramolecular charge transfer in the crystalline state. Angew. Chem. Int. Ed. 2017, 56, 254–259. [Google Scholar] [CrossRef]
- Tang, C.; Xie, Z. Nickel-catalyzed cross-coupling reactions of o-carboranyl with aryl iodides: Facile synthesis of 1-aryl-o-carboranes and 1,2-diaryl-o-carboranes. Angew. Chem. Int. Ed. 2015, 54, 7662–7665. [Google Scholar] [CrossRef]
- Wu, X.; Guo, J.; Cao, Y.; Zhao, J.; Jia, W.; Chen, Y.; Jia, D. Mechanically triggered reversible stepwise tricolor switching and thermochromism of anthracene-o-carborane dyad. Chem. Sci. 2018, 9, 5270–5277. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Lebedev, V.N. Reaction of lithium 1-methyl-o-carborane and lithium 1-methyl-m-carborane with hexafluorobenzene and pentafluorochlorobenzene. Bull. Acad. Sci. USSR Div. Chem. Sci. 1972, 21, 2273–2275. [Google Scholar] [CrossRef]
- Thomas, R.L.; Welch, A.J. Haloaryl carbaboranes: 2. Synthesis and characterisation of haloaryl-closo-carbaboranes. Crystal structures of 1-(4-BrC6F4)-2-R-1,2-closo-C2B10H10 (R = Me, Ph, But). Polyhedron 1999, 18, 1961–1968. [Google Scholar] [CrossRef]
- Batsanov, A.S.; Fox, M.A.; Howard, J.A.K.; Wade, K. Syntheses and reactions of some new C-pentafluorophenyl and tetrafluorophenylene carborane systems. J. Organomet. Chem. 2000, 597, 157–163. [Google Scholar] [CrossRef]
- Ohta, K.; Goto, T.; Endo, Y. New synthetic method of 1,2-diaryl-1,2-dicarba-closo-dodecaboranes employing aromatic nucleophilic substitution (SNAr) reaction. Tetrahedron Lett. 2005, 46, 483–485. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Koveredov, A.I.; Ol’shevskaya, V.A.; Shaugumbekova, S. Synthesis of B-organo-substituted 1,2-, 1,7-, and 1,12-Dicarba-closo-dodecarboranes(12). J. Organomet. Chem. 1982, 226, 217–226. [Google Scholar] [CrossRef]
- Zheng, Z.; Jiang, W.; Zinn, A.A.; Knobler, C.B.; Hawthorne, M.F. Facile electrophilic iodination of icosahedral carboranes. Synthesis of carborane derivatives with boron-carbon bonds via the palladium- catalyzed reaction of diiodocarboranes with Grignard reagents. Inorg. Chem. 1995, 34, 2095–2100. [Google Scholar] [CrossRef]
- Jiang, W.; Knobler, C.B.; Curtis, C.E.; Mortimer, M.D.; Hawthorne, M.F. Iodination reactions of icosahedral para-carborane and the synthesis of carborane derivatives with boron-carbon bonds. Inorg. Chem. 1995, 34, 3491–3498. [Google Scholar] [CrossRef]
- Lee, H.; Knobler, C.B.; Hawthorne, M.F. Supramolecular self-assembly directed by carborane C–H…F interactions. Chem. Commun. 2000, 2485–2486. [Google Scholar] [CrossRef]
- Fox, M.A.; Wade, K. Model compounds and monomers for phenylene ether carboranylene ketone (PECK) polymer synthesis: Preparation and characterization of boron-arylated ortho-carboranes bearing carboxyphenyl, phenoxyphenyl or benzoylphenyl substituents. J. Mater. Chem. 2002, 12, 1301–1306. [Google Scholar] [CrossRef]
- Bayer, M.J.; Herzog, A.; Diaz, M.; Harakas, G.A.; Lee, H.; Knobler, C.B.; Hawthorne, M.F. The synthesis of carboracycles derived from B,B’-bis(aryl) derivatives of icosahedral ortho-carborane. Chem. Eur. J. 2003, 9, 2732–2744. [Google Scholar] [CrossRef]
- Viñas, C.; Barbera, G.; Oliva, J.M.; Teixidor, F.; Welch, A.J.; Rosair, G.M. Are halocarboranes suitable for substitution reactions? The case for 3-I-1,2-closo-C2B10H11: Molecular orbital calculations, aryldehalogenation reactions, 11B NMR interpretation of closo-carboranes, and molecular structures of 1-Ph-3-Br-1,2-closo-C2B10H10 and 3-Ph-1,2-closo-C2B10H11. Inorg. Chem. 2001, 40, 6555–6562. [Google Scholar] [CrossRef] [PubMed]
- Spokoyny, A.M.; Li, T.C.; Farha, O.K.; Machan, C.W.; She, C.; Stern, C.L.; Marks, T.J.; Hupp, J.T.; Mirkin, C.A. Electronic tuning of nickel-based bis(dicarbollide) redox shuttles in dye-sensitized solar cells. Angew. Chem. Int. Ed. 2010, 49, 5339–5343. [Google Scholar] [CrossRef] [PubMed]
- Anufriev, S.A.; Sivaev, I.B.; Bregadze, V.I. Synthesis of 9,9´,12,12´-substituted cobalt bis(dicarbollide) derivatives. Russ. Chem. Bull. 2015, 64, 712–717. [Google Scholar] [CrossRef]
- Anderson, K.P.; Mills, H.A.; Mao, C.; Kirlikovali, K.O.; Axtell, J.C.; Rheingold, A.L.; Spokoyny, A.M. Improved synthesis of icosahedral carboranes containing exopolyhedral B-C and C-C bonds. Tetrahedron 2019, 75, 187–191. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Balagurova, E.V.; Lebedev, V.N. Suzuki cross-coupling in the carborane series. Russ. J. Gen. Chem. 1998, 68, 922–924. [Google Scholar]
- Eriksson, L.; Beletskaya, I.P.; Bregadze, V.I.; Sivaev, I.B.; Sjöberg, S. Palladium-catalyzed cross-coupling reactions of arylboronic acids and 2-I-p-carborane. J. Organomet. Chem. 2002, 657, 267–272. [Google Scholar] [CrossRef]
- Aizawa, K.; Ohta, K.; Endo, Y. Synthesis of 3-aryl-1,2-dicarba-closo-dodecaboranes by Suzuki-Miyaura coupling reaction. Heterocycles 2010, 80, 369–377. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Ol’shevskaya, V.A. Synthesis of 9-organyl-1,2 and 1,7-dicarba-closo-dodecaboranes(12) via the cross-coupling reactions between organozinc compounds and 9-iodo-1,2- or 1,7-dicarba-closo-dodecaboranes. Synth. React. Inorg. Met. Org. Chem. 1991, 21, 1041–1046. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Ol’shevskaya, V.A.; Zhigareva, G.G. Synthesis of B-organyl-ortho-carboranes and meta-carboranes via cross-correlation reaction of B-iodine-ortho-carboranes and meta-carboranes with organozinc compounds during catalysis by palladium complexes. Russ. J. Gen. Chem. 1998, 68, 925–927. [Google Scholar]
- Cao, K.; Huang, Y.; Yang, J.; Wu, J. Palladium catalyzed selective mono-arylation of o-carboranes via B–H activation. Chem. Commun. 2015, 51, 7257–7260. [Google Scholar] [CrossRef]
- Quan, Y.; Xie, Z. Controlled functionalization of o-carborane via transition metal catalyzed B–H activation. Chem. Soc. Rev. 2019, 48, 3660–3673. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Zhang, J.; Zhang, J.; Xie, Z. Regioselective nucleophilic alkylation/arylation of B−H bonds in. o‑carboranes: An alternative method for selective cage boron functionalization. J. Am. Chem. Soc. 2018, 140, 16423–16427. [Google Scholar] [CrossRef]
- Klatt, T.; Markiewicz, J.T.; Sämann, C.; Knochel, P. Strategies to prepare and use functionalized organometallic reagents. J. Org. Chem. 2014, 79, 4253–4269. [Google Scholar] [CrossRef] [PubMed]
- Balkenhohl, M.; Knochel, P. Recent advances of the halogen-zinc exchange reactions. Chem. Eur. J. 2020, 26, 3688–3697. [Google Scholar] [CrossRef] [PubMed]
- Fillon, H.; Gosmini, C.; Périchon, J. New chemical synthesis of functionalized arylzinc compounds from aromatic or thienyl bromides under mild conditions using a simple cobalt catalyst and zinc dust. J. Am. Chem. Soc. 2003, 125, 3867–3870. [Google Scholar] [CrossRef]
- Kazmierski, I.; Gosmini, C.; Paris, J.-M.; Périchon, J. New progress in the cobalt-catalysed synthesis of aromatic organozinc compounds by reduction of aromatic halides by zinc dust. Tetrahedron Lett. 2003, 44, 6417–6420. [Google Scholar] [CrossRef]
- Gosmini, C.; Moncomble, A. Cobalt-catalyzed cross-coupling reactions of aryl halides. Isr. J. Chem. 2010, 50, 568–576. [Google Scholar] [CrossRef]
- Claudel, S.; Gosmini, C.; Paris, J.M.; Périchon, J. A novel transmetallation of arylzinc species into arylboronates from aryl halides in a barbier procedure. Chem. Commun. 2007, 3667–3669. [Google Scholar] [CrossRef]
- Safronov, A.V.; Sevryugina, Y.V.; Pichaandi, K.R.; Jalisatgi, S.S.; Hawthorne, M.F. Synthesis of closo- and nido-biscarboranes with rigid unsaturated linkers as precursors to linear metallacarborane-based molecular rods. Dalton Trans. 2014, 43, 4969–4977. [Google Scholar] [CrossRef]
- Andrews, J.S.; Zayas, J.; Jones, M. 9-Iodo-o-carborane. Inorg. Chem. 1985, 24, 3715–3716. [Google Scholar] [CrossRef]
- Brauer, G. Handbuch der Präparativen Anorganischen Chemie, 3rd ed.; Ferdinand Enke Verlag: Stuttgart, Germany, 1981. [Google Scholar]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 6th ed.; Butterworth-Heinemann: Burlington, NJ, USA, 2009. [Google Scholar] [CrossRef]
- APEX2 and SAINT; Bruker AXS Inc.: Madison, WI, USA, 2014.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anufriev, S.A.; Shmal’ko, A.V.; Suponitsky, K.Y.; Sivaev, I.B. One-Pot Synthesis of B-Aryl Carboranes with Sensitive Functional Groups Using Sequential Cobalt- and Palladium-Catalyzed Reactions. Catalysts 2020, 10, 1348. https://doi.org/10.3390/catal10111348
Anufriev SA, Shmal’ko AV, Suponitsky KY, Sivaev IB. One-Pot Synthesis of B-Aryl Carboranes with Sensitive Functional Groups Using Sequential Cobalt- and Palladium-Catalyzed Reactions. Catalysts. 2020; 10(11):1348. https://doi.org/10.3390/catal10111348
Chicago/Turabian StyleAnufriev, Sergey A., Akim V. Shmal’ko, Kyrill Yu. Suponitsky, and Igor B. Sivaev. 2020. "One-Pot Synthesis of B-Aryl Carboranes with Sensitive Functional Groups Using Sequential Cobalt- and Palladium-Catalyzed Reactions" Catalysts 10, no. 11: 1348. https://doi.org/10.3390/catal10111348
APA StyleAnufriev, S. A., Shmal’ko, A. V., Suponitsky, K. Y., & Sivaev, I. B. (2020). One-Pot Synthesis of B-Aryl Carboranes with Sensitive Functional Groups Using Sequential Cobalt- and Palladium-Catalyzed Reactions. Catalysts, 10(11), 1348. https://doi.org/10.3390/catal10111348