Organocatalyzed Michael Addition to Nitroalkenes via Masked Acetaldehyde

 ,
,  ,
,  and
and 
Abstract
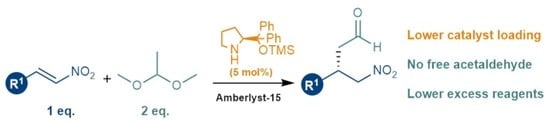
:
1. Introduction
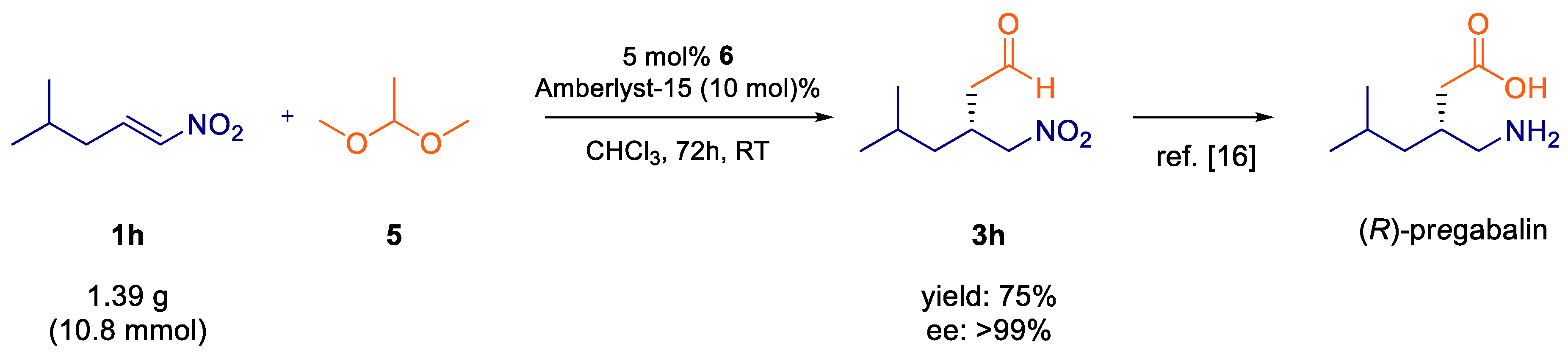
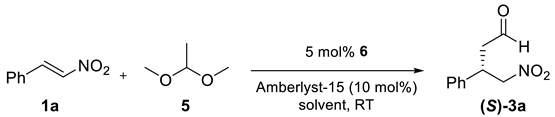
2. Results and Discussion
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carlone, A.; Bernardi, L. Enantioselective organocatalytic approaches to active pharmaceutical—Selected industrial examples. Phys. Sci. Rev. 2019, 4, 20180097. [Google Scholar] [CrossRef]
- Blaser, H.U.; Federsel, H.J. Asymmetric Catalysis on Industrial Scale: Challenges, Approaches and Solutions, 2nd ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; ISBN 9783527324897. [Google Scholar]
- Wallace, D.; Challenger, S.; Ding, Z.D.; Osminski, W.E.G.; Ren, H.; Wulff, W.D.; Desai, A.A.; Munmun, M.; Scott, J.P.; Alam, M.; et al. Special feature section: Asymmetric synthesis on large scale. Org. Process. Res. Dev. 2011, 15, 1088–1211. Available online: https://pubs.acs.org/toc/oprdfk/15/5 (accessed on 9 November 2020). [CrossRef]
- Dunn, P.J.; Hii, K.K.; Krische, M.J.; Williams, M.T. Sustainable Catalysis: Challenges and Practices for the Pharmaceutical and Fine Chemical Industries; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; ISBN 9781118155424. [Google Scholar]
- Adamo, M.F.A.; Kelly, B.G.; Moccia, M. Recent advances in the preparation of active pharmaceutical ingredient (S)-Pregabalin. Chim. Oggi Chem. Today 2016, 34, 54–57. [Google Scholar]
- Dalko, P.I. Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Applications; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; Volume 1–3, ISBN 9783527658862. [Google Scholar]
- List, B. Asymmetric Organocatalysis in Topics in Current Chemistry; Springer: Berlin, Germany; New York, NY, USA, 2009; ISBN 978-3-642-02815-1. [Google Scholar]
- Torres, R.R. Stereoselective Organocatalysis: Bond Formation Methodologies and Activation Modes; Wiley: Hoboken, NJ, USA, 2013; ISBN 978-1-118-60470-0. [Google Scholar]
- Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Asymmetric aminocatalysis—Gold rush in organic chemistry. Angew. Chem. Int. Ed. 2008, 47, 6138–6171. [Google Scholar] [CrossRef] [PubMed]
- Córdova, A.; Notz, W.; Barbas, C.F. Proline-catalyzed one-step asymmetric synthesis of 5-hydroxy-(2E)-hexenal from acetaldehyde. J. Org. Chem. 2002, 67, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Bøgevig, A.; Kumaragurubaran, N.; Jørgensen, K.A. Direct catalytic asymmetric aldol reactions of aldehydes. Chem. Commun. 2002, 2, 620–621. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Itoh, T.; Aratake, S.; Ishikawa, H. A diarylprolinol in an asymmetric, catalytic, and direct crossed-aldol reaction of acetaldehyde. Angew. Chem. Int. Ed. 2008, 47, 2082–2084. [Google Scholar] [CrossRef]
- Hayashi, Y.; Itoh, T.; Ohkubo, M.; Ishikawa, H. Asymmetric Michael reaction of acetaldehyde catalyzed by diphenylprolinol silyl ether. Angew. Chem. Int. Ed. 2008, 47, 4722–4724. [Google Scholar] [CrossRef] [PubMed]
- García-García, P.; Ladépêche, A.; Halder, R.; List, B. Catalytic asymmetric michael reactions of acetaldehyde. Angew. Chem. Int. Ed. 2008, 47, 4719–4721. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Rodríguez-Escrich, C.; Sayalero, S.; Pericàs, M.A. Paraldehyde as an acetaldehyde precursor in asymmetric michael reactions promoted by site-isolated incompatible catalysts. Chem. Eur. J. 2013, 19, 10814–10817. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, H.; Ishikawa, H.; Hayashi, Y. Diphenylprolinol silyl ether as catalyst of an asymmetric, catalytic, and direct michael reaction of nitroalkanes with α,β-unsaturated aldehydes. Org. Lett. 2007, 9, 5307–5309. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
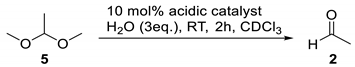
| Entry | Acid | Conv. (%) 2 |
|---|---|---|
| 1 | Benzoic Acid | 0 |
| 2 | AcOH | 1 |
| 3 | p-NO2-C6H4CO2H | 0 |
| 4 | pTSA | 0 |
| 5 | HCl | 14 |
| 6 | H2SO4 | 14 |
| 7 | Amberlyst-36 | 12 |
| 8 | Nafion NRE 212 | 15 |
| 9 | TFA | 16 |
| 10 | Amberlyst-15 | 18 |
| Entry | 5 (eq) | H2O (eq) | Solvent | t (h) | Conv. [%] 2 | ee (%) 3 |
|---|---|---|---|---|---|---|
| 1 | 5 | 0 | CHCl3 | 72 | 46 | 96 |
| 2 | 5 | 10 | CHCl3 | 72 | 94 | 93 |
| 3 | 5 | 10 | AcOEt | 72 | 32 | 88 |
| 4 | 5 | 10 | MeCN | 72 | 5 | 44 |
| 5 | 5 | 10 | Acetone | 72 | 75 | 87 |
| 6 | 5 | 10 | Toluene | 72 | 85 | 94 |
| 7 | 5 | 10 | Et2O | 72 | 71 | 94 |
| 8 | 5 | 10 | CH2Cl2 | 72 | 61 | 92 |
| 9 | 5 | 10 | Dioxane | 72 | 93 | 94 |
| 10 | 5 | 10 | Dioxane | 24 | 71 | 93 |
| 11 | 5 | 10 | CHCl3 | 24 | 93 | 93 |
| 12 | 5 | 15 | Dioxane | 24 | 84 | 95 |
| 13 | 5 | 15 | CHCl3 | 24 | 97 | 93 |
| 14 | 1.2 | 3.6 | CHCl3 | 24 | 57 | 91 |
| 15 | 2 | 6 | CHCl3 | 24 | 74 | 90 |
| 16 | 3 | 9 | CHCl3 | 24 | 81 | 92 |
| 17 4 | 3 | 9 | CHCl3 | 24 | >99 | 94 |
| 18 4 | 2 | 6 | CHCl3 | 24 | 81 | 92 |
| 19 5 | 2 | 6 | CHCl3 | 24 | 94 | 90 |
| 20 5,6 | 2 | 6 | CHCl3 | 24 | 91 | 90 |
| 21 5,7 | 2 | 6 | CHCl3 | 24 | 10 | 93 |
| 22 5,8 | 2 | 6 | CHCl3 | 24 | 0 | n.d |
| 23 9 | 2 | 6 | CHCl3 | 24 | 48 | 91 |
| 24 5 | 2 | 6 | Dioxane | 24 | 65 | 92 |
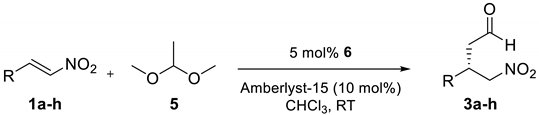
| Entry | R | 3 | t (h) | Yield (Conv.) (%) 2 | ee (%) 3 |
|---|---|---|---|---|---|
| 1 | 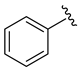 | 3a | 24 | 90 (94) | 95 |
| 2 | 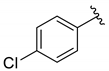 | 3b | 72 | 62 (65) | 94 |
| 3 | 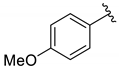 | 3c | 72 | 76 (80) | 92 |
| 4 | 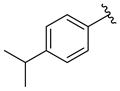 | 3d | 48 | 83 (85) | 93 |
| 5 |  | 3e | 48 | 75 (78) | 96 |
| 6 4 | 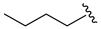 | 3f | 48 | 89 (92) | −95 5 |
| 7 4 | 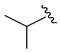 | 3g | 72 | 93 (94) | 93 |
| 8 | 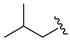 | 3h | 72 | 83 (86) | 94 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giorgianni, G.; Nori, V.; Baschieri, A.; Palombi, L.; Carlone, A. Organocatalyzed Michael Addition to Nitroalkenes via Masked Acetaldehyde. Catalysts 2020, 10, 1296. https://doi.org/10.3390/catal10111296
Giorgianni G, Nori V, Baschieri A, Palombi L, Carlone A. Organocatalyzed Michael Addition to Nitroalkenes via Masked Acetaldehyde. Catalysts. 2020; 10(11):1296. https://doi.org/10.3390/catal10111296
Chicago/Turabian StyleGiorgianni, Giuliana, Valeria Nori, Andrea Baschieri, Laura Palombi, and Armando Carlone. 2020. "Organocatalyzed Michael Addition to Nitroalkenes via Masked Acetaldehyde" Catalysts 10, no. 11: 1296. https://doi.org/10.3390/catal10111296
APA StyleGiorgianni, G., Nori, V., Baschieri, A., Palombi, L., & Carlone, A. (2020). Organocatalyzed Michael Addition to Nitroalkenes via Masked Acetaldehyde. Catalysts, 10(11), 1296. https://doi.org/10.3390/catal10111296