Hydrodechlorination of CHClF2 (HCFC-22) over Pd–Pt Catalysts Supported on Thermally Modified Activated Carbon

 , ,
, ,
Abstract
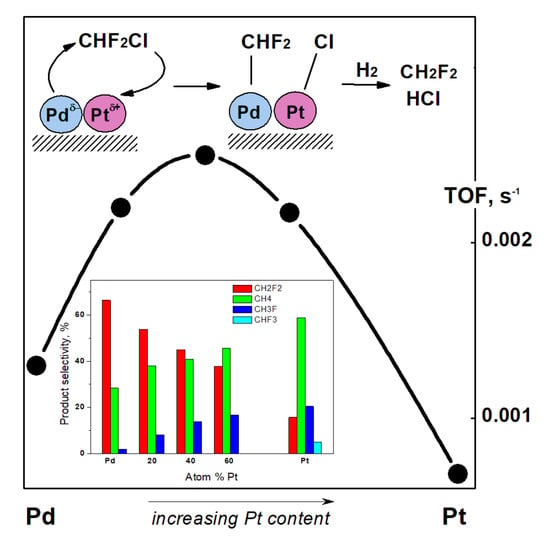
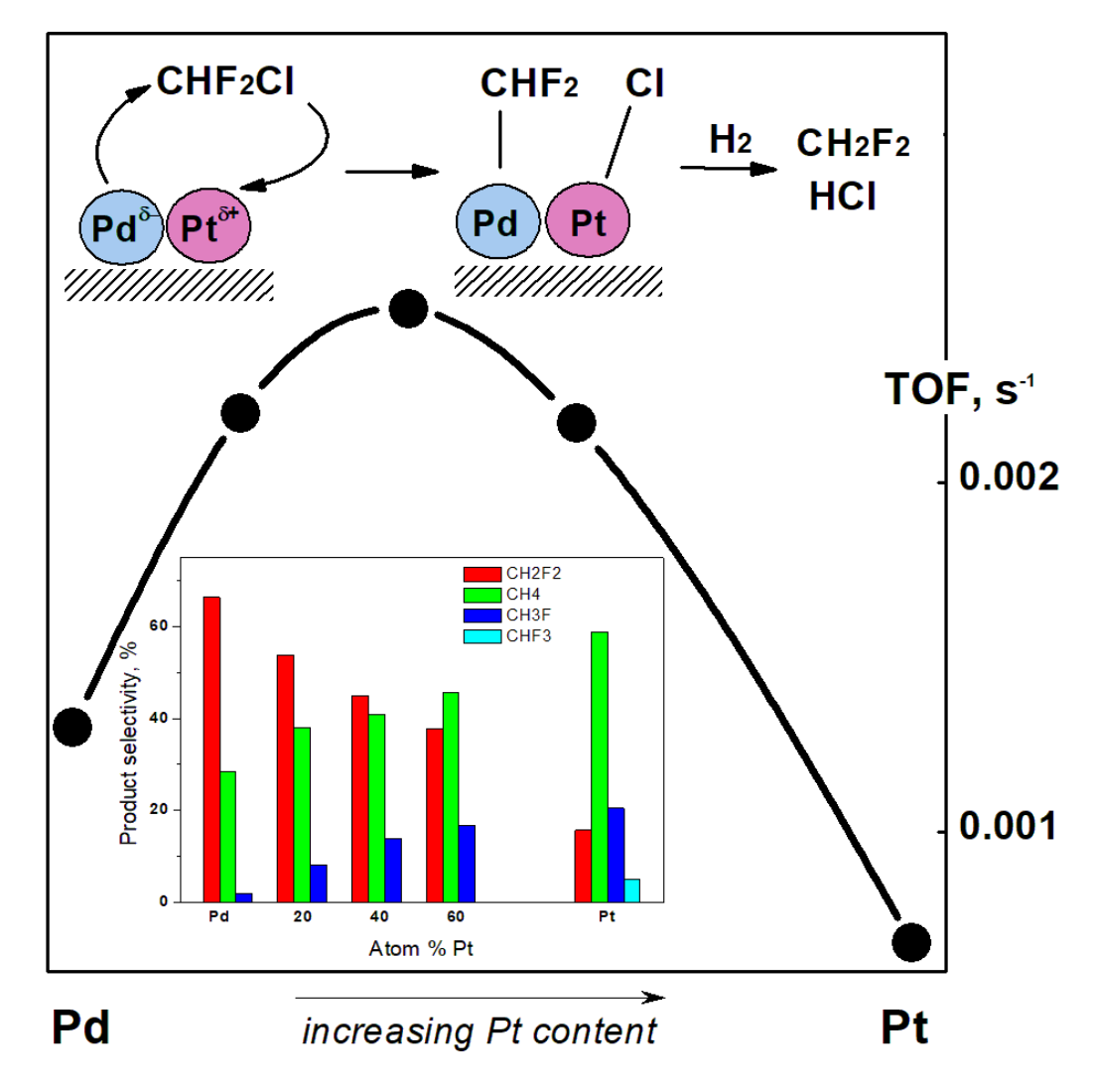
1. Introduction
2. Results
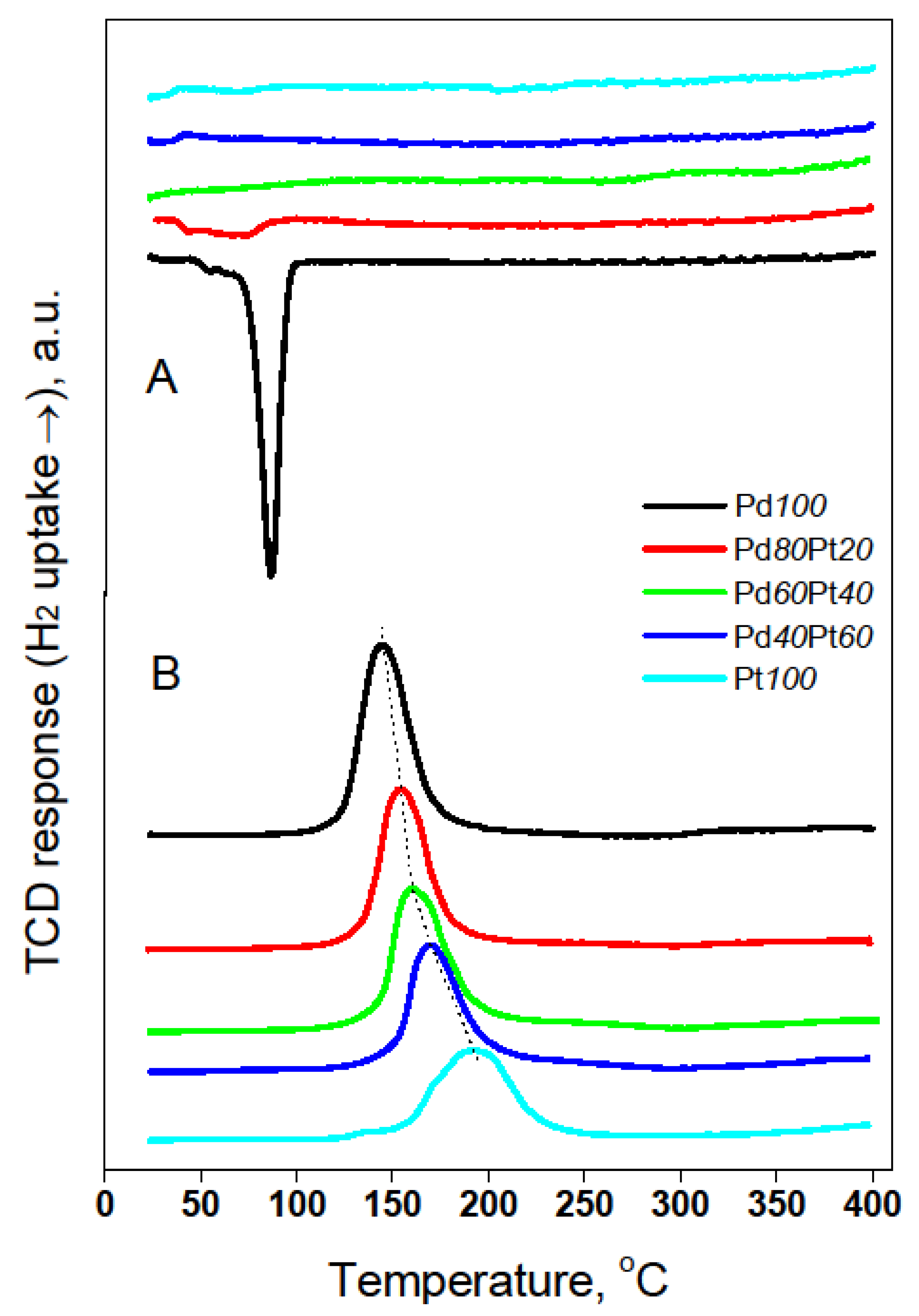
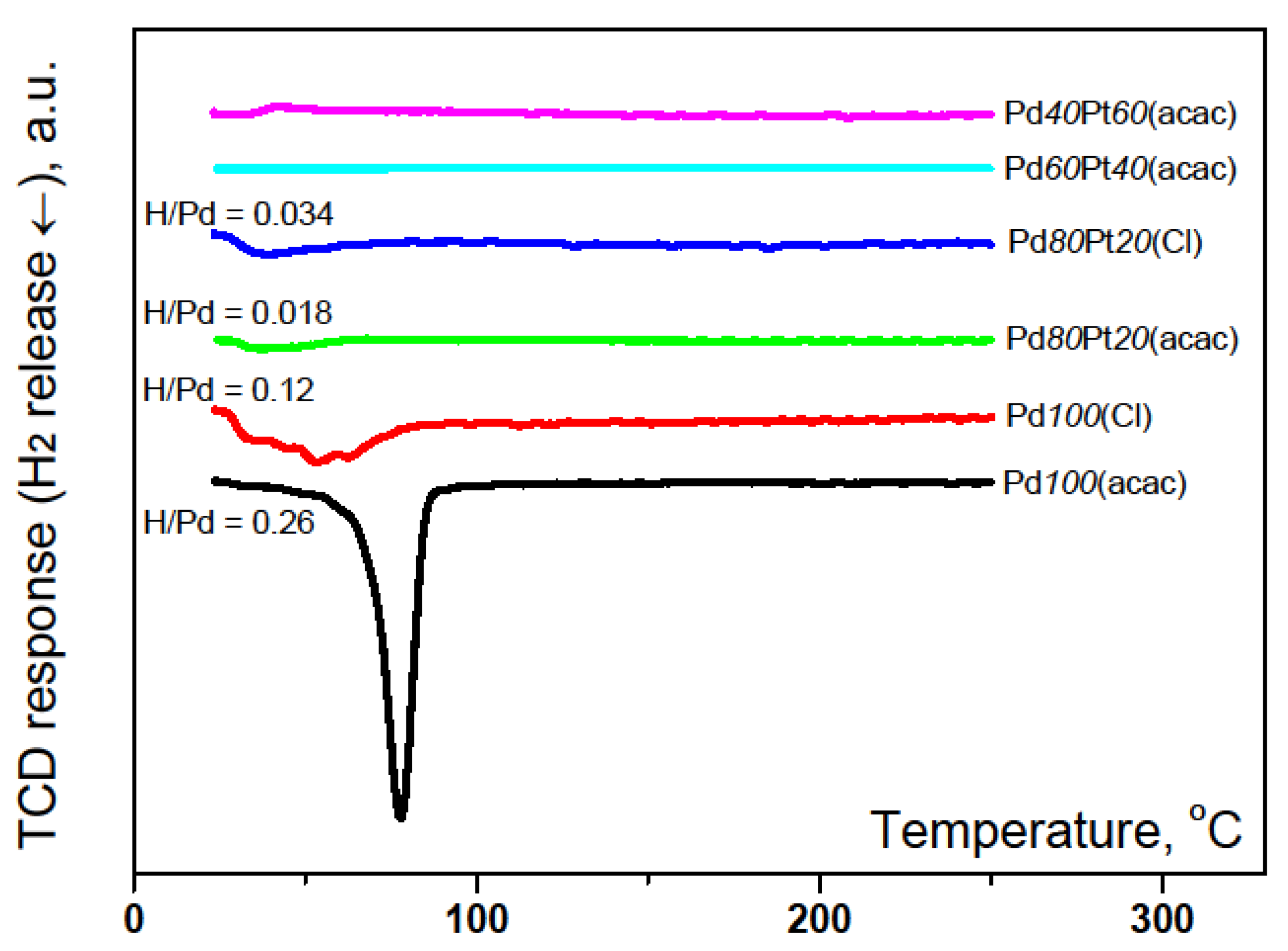
2.1. Catalyst Characterization by Temperature Programmed Methods
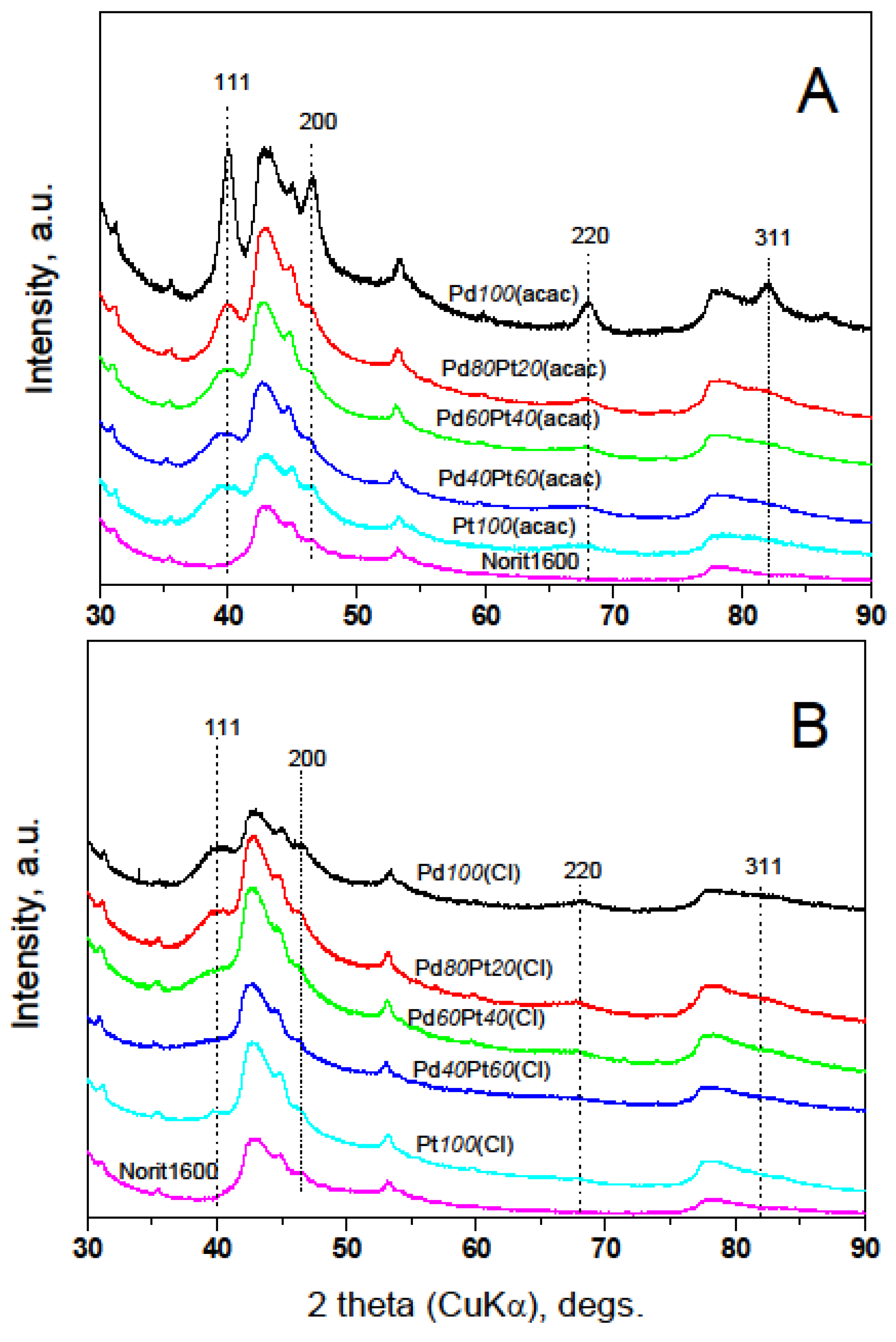
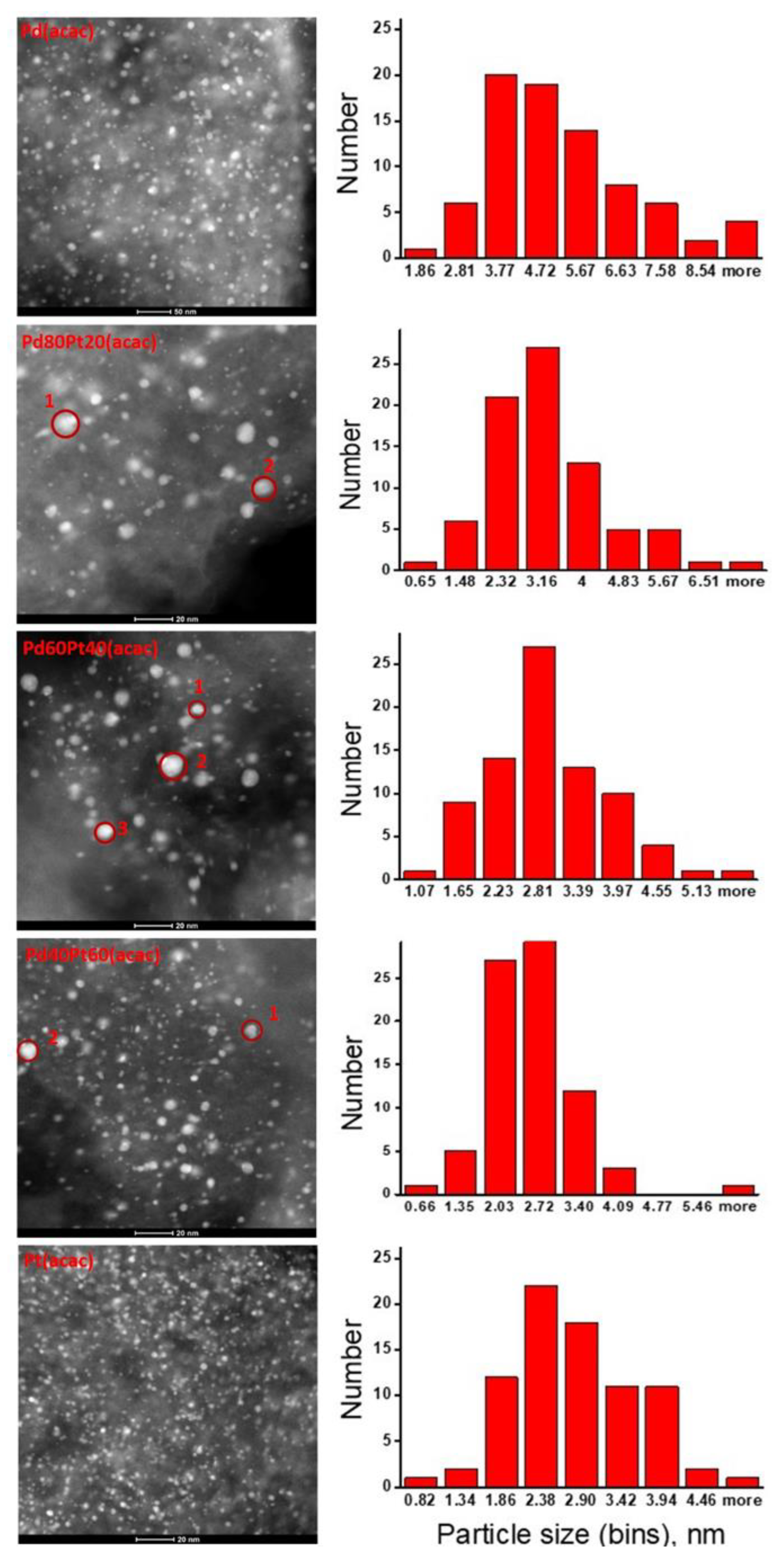
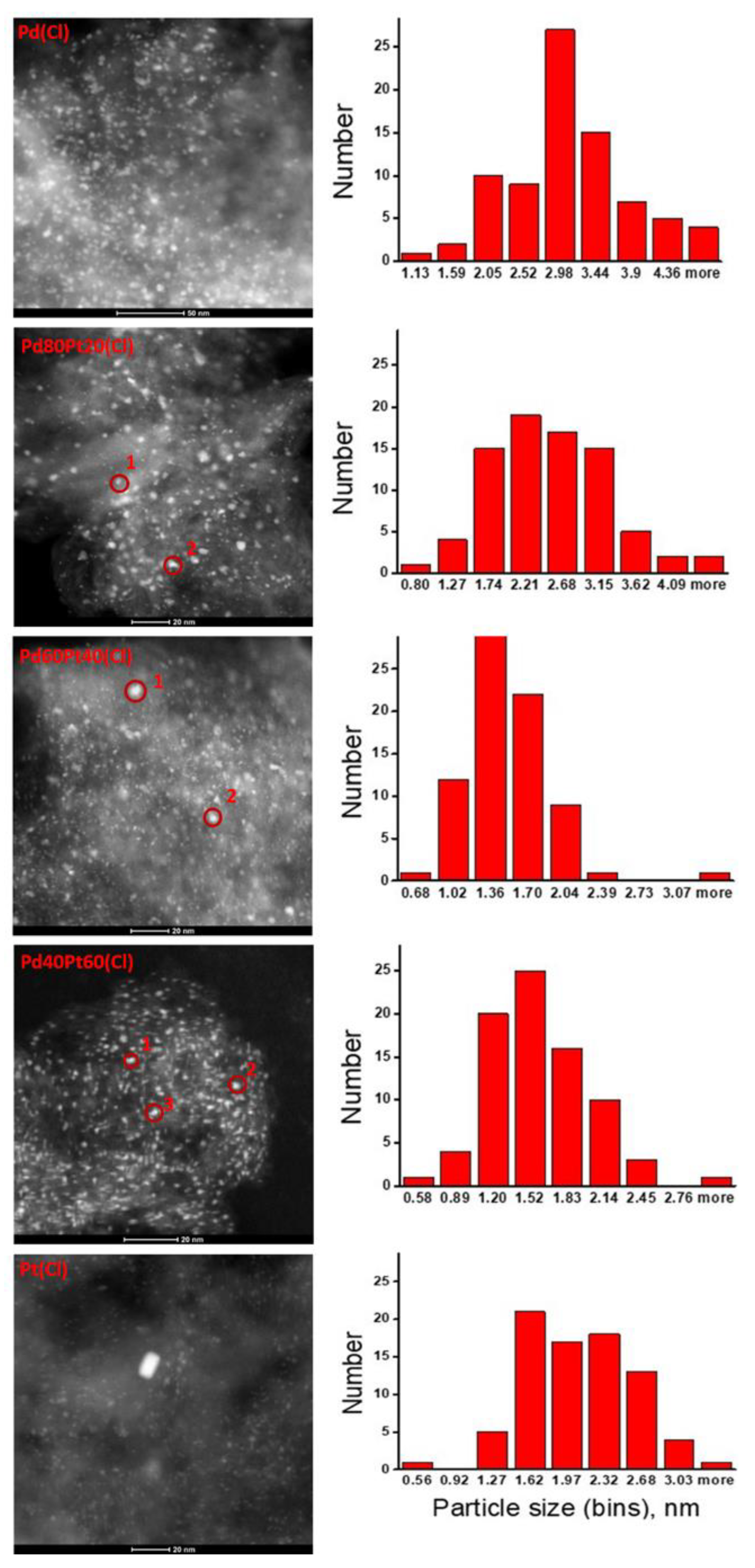
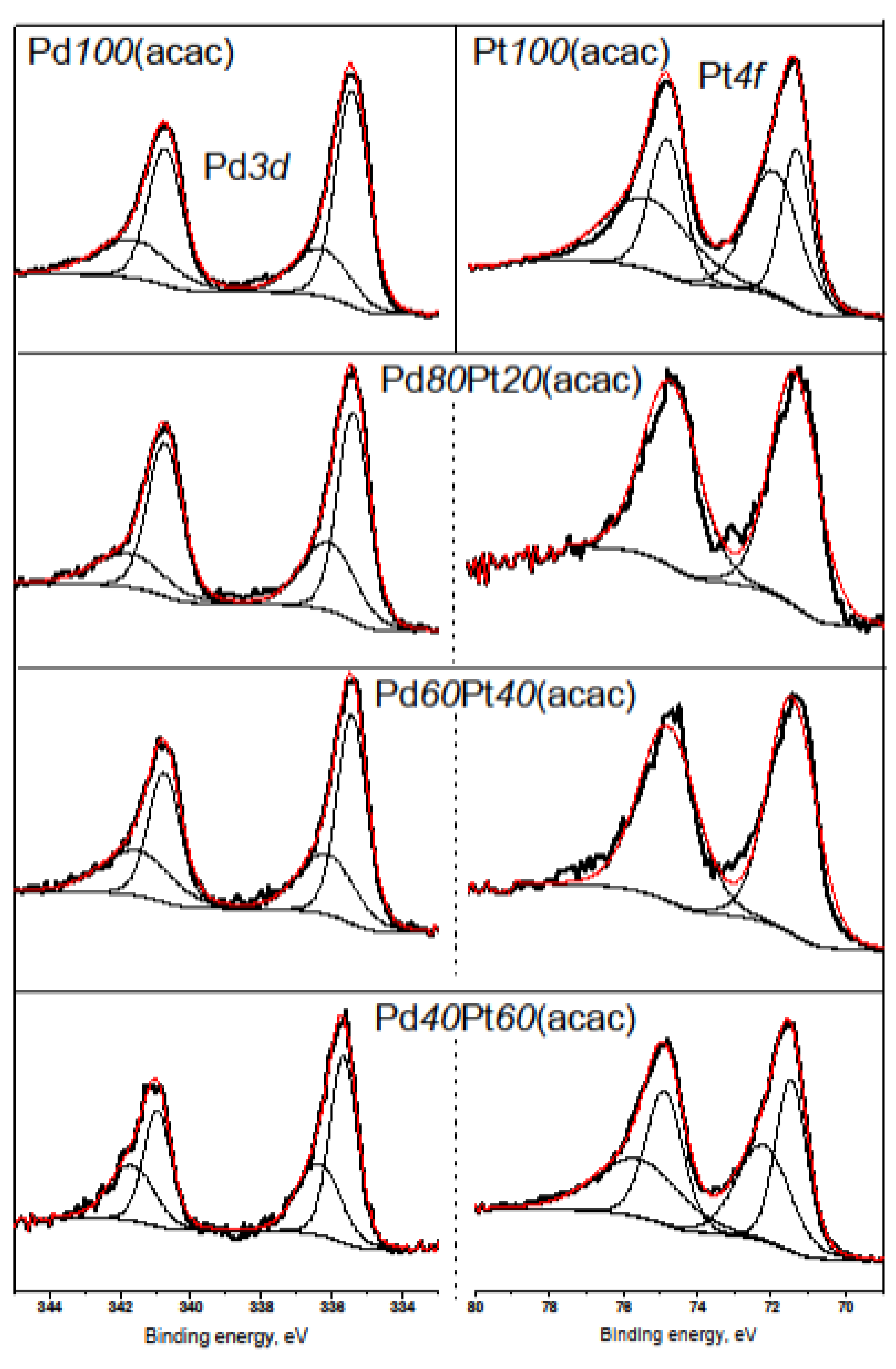
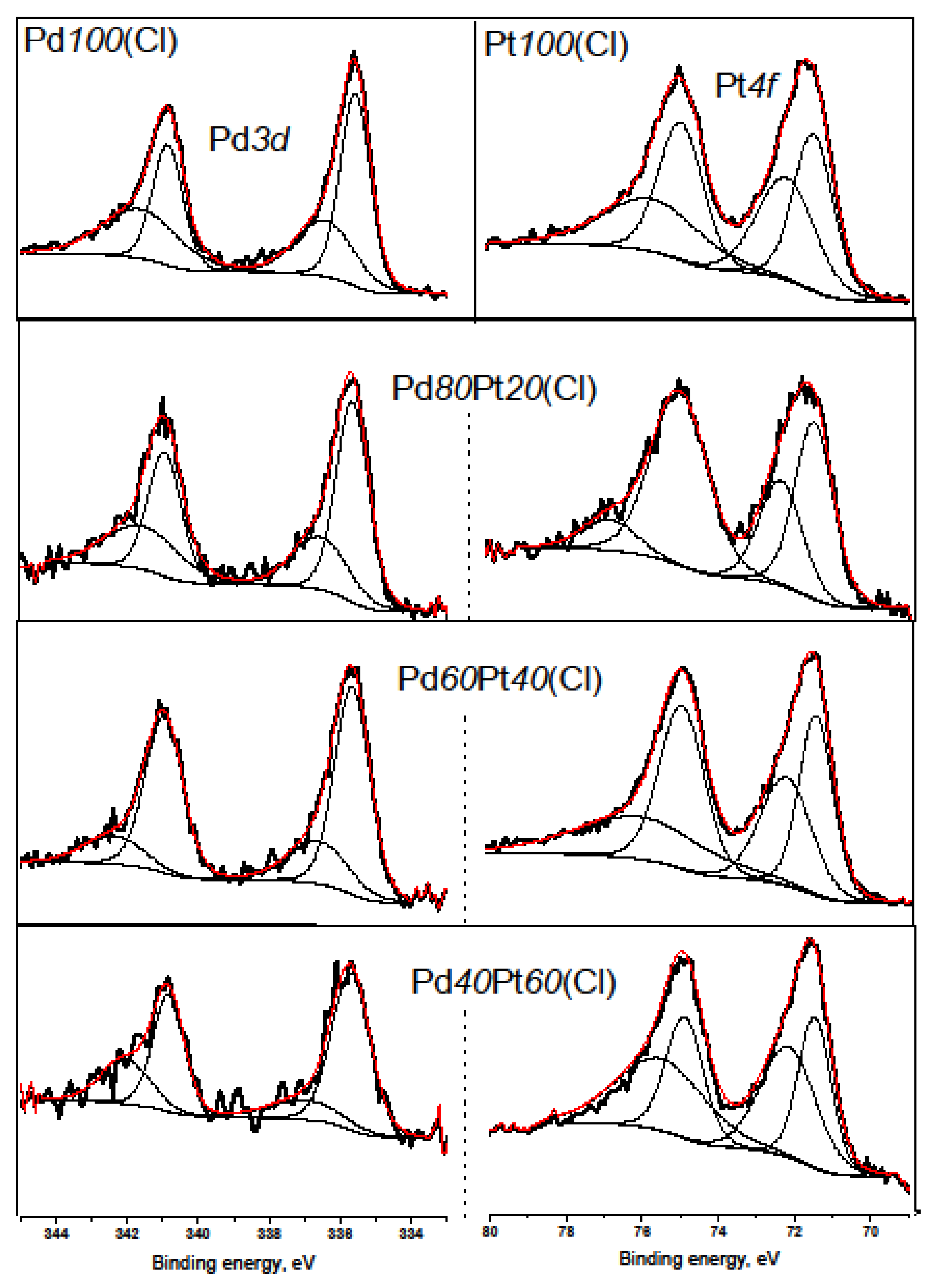
2.2. X-ray Diffraction, Transmission Electron Microscopy, H2 Chemisorption, and X-ray Photoelectron Spectroscopy
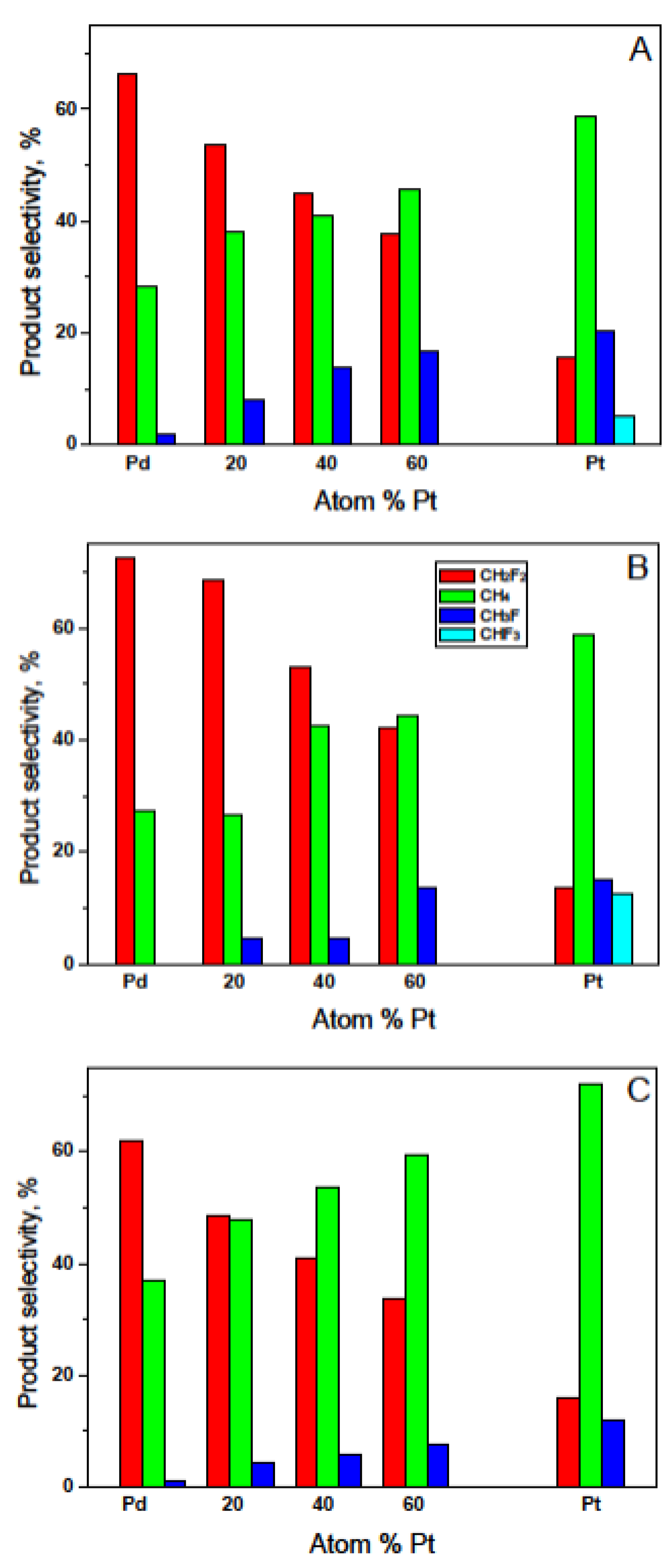
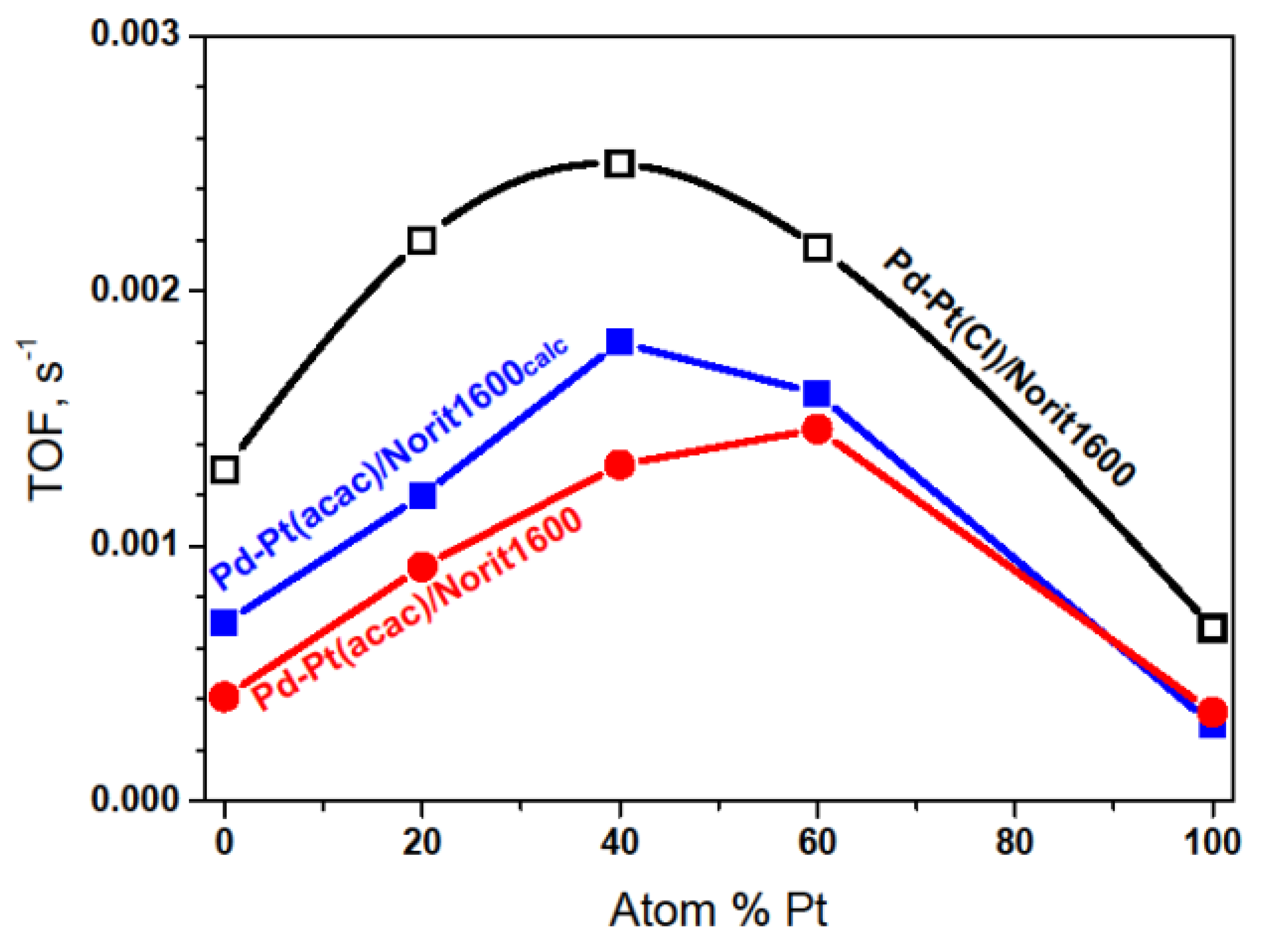
2.3. Hydrodechlorination of CHClF2, Environmentally Relevant Utilization of a Catalytic Probe for the Characterization of Pd–Pt/C Catalysts
3. Methods
3.1. Catalyst Preparation
3.2. Catalytic Tests
3.3. Catalyst Characterization by H2 Chemisorption, Temperature Programmed Techniques, XRD, TEM and XPS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rylander, P.N. Catalytic Hydrogenation over Platinum Metals; Academic Press: London, UK; New York, NY, USA, 1967; pp. 9–11. [Google Scholar]
- Li, H.; Sun, G.; Li, N.; Sun, S.; Su, D.; Xin, Q. Design and preparation of highly active Pt–Pd/C catalyst for the oxygen reduction reaction. J. Phys. Chem. 2007, 111, 5605–5617. [Google Scholar] [CrossRef]
- Martin-Martinez, M.; Gómez-Sainero, L.M.; Bedia, J.; Arevalo-Bastante, A.; Rodriguez, J.J. Enhanced activity of carbon-supported Pd-Pt catalysts in the hydrodechlorination of dichloromethane. Appl. Catal. B 2016, 184, 55–63. [Google Scholar] [CrossRef]
- Baldovino-Medrano, V.; Eloy, P.; Gaigneaux, E.M.; Giraldo, S.A.; Centeno, A. Development of the HYD route of hydrodesulfurization of dibenzothiophenes over Pd-Pt/gamma-Al2O3 catalysts. J. Catal. 2009, 267, 129–139. [Google Scholar] [CrossRef]
- Sterchele, S.; Biasi, P.; Centomo, P.; Canton, P.; Campestrini, S.; Salmi, T.; Zecca, M. Pd-Au and Pd-Pt catalysts for the direct synthesis of hydrogen peroxide in absence of selectivity enhancers. Appl. Catal. A 2013, 468, 160–174. [Google Scholar] [CrossRef]
- Du, M.; Qin, Z.; Ge, H.; Li, X.; Lü, Z.; Wang, J. Enhancement of Pd–Pt/Al2O3 catalyst performance in naphthalene hydrogenation by mixing different molecular sieves in the support. Fuel Process. Technol. 2010, 91, 1655–1661. [Google Scholar] [CrossRef]
- Koutsopoulos, S.; Johannessen, T.; Eriksen, K.M.; Fehrmann, R. Titania-supported Pt and Pt–Pd nanoparticle catalysts for the oxidation of sulfur dioxide. J. Catal. 2006, 238, 206–213. [Google Scholar] [CrossRef]
- Zhang, H.; Jin, M.; Wang, J.; Kim, M.J.; Yang, D.; Xia, Y. Nanocrystals composed of alternating shells of Pd and Pt can be obtained by sequentially adding different precursors. J. Am. Chem. Soc. 2011, 133, 10422–10425. [Google Scholar] [CrossRef]
- Choi, I.; Ahn, S.H.; Kim, J.J.; Kwon, O.J. Preparation of Ptshell–Pdcore nanoparticle with electroless deposition of copper for polymer electrolyte membrane fuel cell. Appl. Catal. B 1996, 102, 608–613. [Google Scholar] [CrossRef]
- Escudero, M.J.; Hontanon, E.; Schwartz, S.; Boutonnet, M.; Daza, L. Development and performance characterisation of new electrocatalysts for PEMFC. J. Power Sources 2002, 106, 206–214. [Google Scholar] [CrossRef]
- Zheng, H.; Matseke, M.S.; Munonde, T.S. The unique Pd@Pt/C core-shell nanoparticles as methanol-tolerant catalysts using sonochemical synthesis. Ultrason. Sonochem. 2019, 57, 166–171. [Google Scholar] [CrossRef]
- Wongkaew, A.; Zhang, Y.; Tengco, J.M.M.; Blom, D.A.; Sivasubramanian, P.K.; Fanson, P.T.; Regalbuto, J.R.; Monnier, J.R. Characterization and evaluation of Pt-Pd electrocatalysts prepared by electroless deposition. Appl. Catal. B 2016, 188, 367–375. [Google Scholar] [CrossRef]
- Rousset, J.L.; Cadrot, A.M.; Lianos, L.; Renouprez, A.J. Structure and reactivity of Pd-Pt clusters produced by laser vaporization of bulk alloys. Investigations on supported bimetallic PdPt nanostructures. Eur. Phys. J. D 1999, 9, 425–428. [Google Scholar] [CrossRef]
- Rousset, J.L.; Khanra, B.C.; Cadrot, A.M.; Cadete Santos Aires, F.J.; Renouprez, A.J.; Pellarin, M. Investigations on supported bimetallic PdPt nanostructures. Surf. Sci. 1996, 352–354, 583–587. [Google Scholar] [CrossRef]
- Wells, P.P.; Crabb, E.M.; King, C.R.; Wiltshire, R.; Bilsborrow, B.; Thompsett, D.; Russell, A.E. Preparation, structure and stability of Pt and Pd monolayer modified Pd and Pt electrocatalysts. Phys. Chem. Chem. Phys. 2009, 11, 5773–5781. [Google Scholar] [CrossRef]
- Cangul, B.; Zhang, L.C.; Aindow, M.; Erkey, C. Preparation of carbon black supported Pd, Pt and Pd–Pt nanoparticles using supercritical CO2 deposition. J. Supercrit. Fluids 2009, 50, 82–90. [Google Scholar] [CrossRef]
- Cho, H.-R.; Regalbuto, J.R. The rational synthesis of Pt-Pd bimetallic catalysts by electrostatic adsorption. Catal. Today 2015, 246, 143–153. [Google Scholar] [CrossRef]
- Huang, L. IR studies on the preservation of RhCo3 clusters on the surface of SiO2. J. Mol. Catal. A Chem. 1996, 112, 69–83. [Google Scholar] [CrossRef]
- Renouprez, A.; Lebas, K.; Bergeret, G. A new method of direct synthesis of bimetallic phases: Silica supported Pd-Cu catalysts from mixed acetylacetonates. J. Mol. Catal. A 1997, 120, 217–225. [Google Scholar] [CrossRef]
- Dossi, C.; Pozzi, A.; Recchia, A.; Fusi, S.; Psaro, R.; Dal Santo, V. An organometallic route to mono and bimetallic Pt and Pt-Pd catalysts supported on magnesium oxide: Thermoanalytical investigation and catalytic behavior in MCP conversion. J. Mol. Catal. A 2003, 204–205, 465–472. [Google Scholar] [CrossRef]
- Renouprez, A.J.; Malhomme, A.; Massardier, J.; Cattenot, M.; Bergeret, G. Sulphur resistant palladium-platinum catalysts prepared from mixed acetylacetonates. Stud. Surf. Sci. Catal. 2000, 130C, 2579–2584. [Google Scholar]
- Bazin, D.; Guillaume, D.; Pichon, C.; Uzio, D.; Lopez, S. Structure and size of bimetallic palladium–platinum clusters in an hydrotreatment catalyst. Oil Gas Sci. Technol. 2005, 60, 801–813. [Google Scholar] [CrossRef]
- Radlik, M.; Śrębowata, A.; Juszczyk, W.; Matus, K.; Małolepszy, A.; Karpiński, Z. n‑Hexane conversion on γ‑alumina supported palladium–platinum catalysts. Adsorption 2019, 25, 843–853. [Google Scholar] [CrossRef]
- Montzka, S.A.; McFarland, M.; Andersen, S.O.; Miller, B.R.; Fahey, D.W.; Hall, B.D.; Hu, L.; Siso, C.; Elkins, J.W. Recent trends in global emissions of hydrochlorofluorocarbons and hydrofluorocarbons: Reflecting on the 2007 Adjustments to the Montreal Protocol. J. Phys. Chem. A 2015, 119, 4439–4449. [Google Scholar] [CrossRef]
- Montzka, S.A.; Dutton, G.S.; Yu, P.; Ray, E.; Portmann, R.W.; Daniel, J.S.; Kuijpers, L.; Hall, B.D.; Mondeel, D.; Siso, C.; et al. An unexpected and persistent increase in global emissions of ozone-depleting CFC-11. Nature 2018, 557, 413–417. [Google Scholar] [CrossRef]
- Prignon, M.; Chabrillat, S.; Minganti, D.; O’Doherty, S.; Servais, C.; Stiller, G.; Toon, G.C.; Vollmer, M.K.; Mahieu, E. Improved FTIR retrieval strategy for HCFC-22 (CHClF2), comparisons with in situ and satellite datasets with the support of models, and determination of its long-term trend above Jungfraujoch. Atmos. Chem. Phys. 2019, 19, 12309–12324. [Google Scholar] [CrossRef]
- Oram, D.E.; Ashfold, M.J.; Laube, J.C.; Gooch, J.L.; Humphrey, S.; Sturges, W.T.; Leedham-Elvidge, E.; Forster, G.L.; Harris, N.R.P.; Iqbal Mead, M.; et al. A growing threat to the ozone layer from short-lived anthropogenic chlorocarbons. Atmos. Chem. Phys. 2017, 17, 11929–11941. [Google Scholar] [CrossRef]
- Coq, B.; Cognion, J.M.; Figuéras, F.; Tournigant, D. Conversion under hydrogen of dichlorodifluoromethane over supported palladium catalysts. J. Catal. 1993, 141, 21–33. [Google Scholar] [CrossRef]
- van de Sandt, E.J.A.X.; Wiersma, A.; Makkee, M.; van Bekkum, H.; Moulijn, J.A. Mechanistic study of the selective hydrogenolysis of CCl2F2 (CFC-12) into CH2F2 (HFC-32) over palladium on activated carbon. Recl. Trav. Pays-Bas 1996, 115, 505–510. [Google Scholar] [CrossRef]
- Moore, G.J.; O’Kell, J. Chemical Process. US Patent 5,877,360, 2 March 1999. [Google Scholar]
- Ohnishi, R.; Wang, W.L.; Ichikawa, M. Selective Hydrogenation of CFC-12 to HFC-32 on Zr-Pd/C Catalyst. Stud. Surf. Sci. Catal. 1994, 90, 101–104. [Google Scholar]
- Schoebrechts, J.-P.; Doiceau, G.; Wilmet, V. Catalytic System Comprising a Hydrogenation Catalyst on a Support and Process for the Hydrodechlorination of Chlorofluorinated Hydrocarbons. US Patent 5,561,069, 1 October 1996. [Google Scholar]
- Morato, A.; Alonso, C.; Medina, F.; Cesteros, Y.; Salagre, P.; Sueiras, J.E.; Tichit, D.; Coq, B. Palladium hydrotalcites as precursors for the catalytic hydroconversion of CCl2F2 (CFC-12) and CHClF2 (HCFC-22). Appl. Catal. B 2001, 32, 167–179. [Google Scholar] [CrossRef]
- Morato, A.; Medina, F.; Sueiras, J.E.; Cesteros, Y.; Salagre, P.; de Menorval, L.C.; Tichit, D.; Coq, B. Characterization and catalytic properties of several KMg1-xPdxF3 with perovskite-like structures for the hydroconversion of CHClF2. Appl. Catal. B 2003, 42, 251–264. [Google Scholar] [CrossRef]
- Yu, H.; Kennedy, E.M.; Uddin, M.A.; Sakata, Y.; Dlugogorski, B.Z. Gas-phase and Pd-catalyzed hydrodehalogenation of CBrClF2, CCl2F2, CHClF2, and CH2F2. Ind. Eng. Chem. Res. 2005, 44, 3442–3452. [Google Scholar] [CrossRef]
- Patil, P.T.; Dimitrov, A.; Kirmse, H.; Neumann, W.; Kemnitz, E. Non-aqueous sol–gel synthesis, characterization and catalytic properties of metal fluoride supported palladium nanoparticles. Appl. Catal. B 2008, 78, 80–91. [Google Scholar] [CrossRef]
- Ha, J.-M.; Kim, D.; Kim, J.; Ahn, B.S.; Kim, Y.; Kang, J.W. High-temperature hydrodechlorination of ozone-depleting chlorodifluoromethane (HCFC-22) on supported Pd and Ni catalysts. J. Environ. Sci. Health Part A 2011, 46, 989–996. [Google Scholar] [CrossRef]
- Legawiec-Jarzyna, M.; Śrębowata, A.; Juszczyk, W.; Karpiński, Z. Hydrodechlorination over Pd-Pt/Al2O3 catalysts A comparative study of chlorine removal from dichlorodifluoromethane, carbon tetrachloride and 1,2-dichloroethane. Appl. Catal. A 2004, 271, 61–68. [Google Scholar] [CrossRef]
- Martin-Martinez, M.; Gómez-Sainero, L.M.; Palomar, J.; Omar, S.; Rodriguez, J.J. Dechlorination of dichloromethane by hydrotreatment with bimetallic Pd-Pt/C catalyst. Catal. Lett. 2016, 146, 2614–2621. [Google Scholar] [CrossRef]
- Bedia, J.; Gómez-Sainero, L.M.; Grau, J.M.; Busto, M.; Martin-Martinez, M.; Rodriguez, J.J. Hydrodechlorination of dichloromethane with mono- and bimetallic Pd-Pt on sulfated and tungstated zirconia catalysts. J. Catal. 2012, 294, 207–215. [Google Scholar] [CrossRef]
- Garcia, C.M.; Woolfolk, L.G.; Martin, N.; Granados, A.; de los Reyes, J.A. Evaluation of mono and bimetallic catalysts supported on Al2O3-TiO2 in the reaction of hydrodechlorination of 1,2-dichloroethane. Rev. Mex. Ing. Quim. 2012, 11, 463–468. [Google Scholar]
- Han, W.; Li, X.; Liu, B.; Li, L.; Tang, H.; Li, Y.; Lu, C.H.; Li, X. Microwave assisted combustion of phytic acid for the preparation of Ni2P@C as a robust catalyst for hydrodechlorination. Chem. Commun. 2019, 55, 9279–9282. [Google Scholar] [CrossRef]
- Coloma, F.; Sepúlveda-Escribano, A.; Rodríguez-Reinoso, F. Heat-treated carbon blacks as supports for platinum catalysts. J. Catal. 1995, 154, 299–305. [Google Scholar] [CrossRef]
- Zheng, X.; Zhang, S.; Xu, J.; Wei, K. Effect of thermal and oxidative treatments of activated carbon on its surface structure and suitability as a support for barium promoted ruthenium in ammonia synthesis catalysts. Carbon 2002, 40, 2597–2603. [Google Scholar] [CrossRef]
- Okhlopkova, L.B.; Lisitsyn, A.S.; Likholobov, V.A.; Gurrath, M.; Boehm, H.P. Properties of Pt/C and Pd/C catalysts prepared by reduction with hydrogen of adsorbed metal chlorides, Influence of pore structure of the support. Appl. Catal. A 2000, 204, 229–240. [Google Scholar] [CrossRef]
- Wiersma, A.; van de Sandt, E.J.A.X.; Makkee, M.; Luteijn, C.P.; van Bekkum, H.; Moulijn, J.A. Process for the selective hydrogenolysis of CC12F2 (CFC-12) into CH2F2 (HFC-32). Catal. Today 1996, 27, 257–264. [Google Scholar] [CrossRef]
- van de Sandt, E.J.A.X.; Wiersma, A.; Makkee, M.; van Bekkum, H.; Moulijn, J.A. Selection of activated carbon for the selective hydrogenolysis of CCl2F2 (CFC-12) into CH2F2 (HFC-32) over palladium-supported catalysts. Appl. Catal. A 1998, 173, 161–173. [Google Scholar] [CrossRef]
- Łomot, D.; Juszczyk, W.; Karpiński, Z.; Bozon-Verduraz, F. Evolution of Pd/SiO2 catalysts prepared from chlorine-free precursors. J. Chem. Soc. Faraday Trans. 1997, 93, 2015–2021. [Google Scholar]
- Chou, C.-W.; Chu, S.-J.; Chiang, H.-J.; Huang, C.-Y.; Lee, C.-J.; Sheen, S.-R.; Perng, T.P.; Yeh, C.-T. Temperature-programmed reduction study on calcination of nano-palladium. J. Phys. Chem. B 2001, 105, 9113–9117. [Google Scholar] [CrossRef]
- Amorim, A.; Yuan, G.; Patterson, P.M.; Keane, M.A. Catalytic hydrodechlorination over Pd supported on amorphous and structured carbon. J. Catal. 2005, 234, 268–281. [Google Scholar] [CrossRef]
- Coloma, F.; Sepúlveda-Escribano, A.; Fierro, J.L.G.; Rodriguez-Reinoso, F. Preparation of platinum supported on pregraphitized carbon blacks. Langmuir 1994, 10, 750–755. [Google Scholar] [CrossRef]
- Gurrath, M.; Kuretzky, T.; Boehm, H.P.; Okhlopkova, L.B.; Lisitsyn, A.S.; Likholobov, V.A. Palladium catalysts on activated carbon supports. Influence of reduction temperature, origin of the support and pretreatments of the carbon surface. Carbon 2000, 38, 1241–1255. [Google Scholar] [CrossRef]
- Bonarowska, M.; Karpiński, Z. Characterization of supported Pd-Pt catalysts by chemical probes. Catal. Today 2008, 137, 498–503. [Google Scholar] [CrossRef]
- Noh, H.; Flanagan, T.B.; Sonoda, T.; Sakamoto, Y. Solubility and thermodynamics of hydrogen in homogeneous f.c.c. Pd-Pt alloys. J. Alloys Compd. 1995, 228, 164–171. [Google Scholar] [CrossRef]
- Rosantha Kumara, L.S.R.; Sakata, O.; Kobayashi, H.; Song, C.; Kohara, S.; Ina, T.; Yoshimoto, T.; Yoshioka, S.; Matsumura, S.; Kitagawa, H. Hydrogen storage and stability properties of Pd–Pt solid-solution nanoparticles revealed via atomic and electronic structure. Sci. Rep. 2017, 7, 14606. [Google Scholar] [CrossRef]
- Vannice, M.A. Kinetics of Catalytic Reactions; Springer Science+Business Media, Inc.: New York, NY, USA, 2005; Ch. 3.3.2.1. Line Broadening of X-Ray Diffraction (XRD) Peaks, p. 20. [Google Scholar]
- Ichikawa, S.; Poppa, H.; Boudart, M. Disproportionation of CO on small particles of silica-supported palladium. J. Catal. 1985, 91, 1–10. [Google Scholar] [CrossRef]
- Rioux, R.M.; Vannice, M.A. Dehydrogenation of isopropyl alcohol on carbon-supported Pt and Cu–Pt catalysts. J. Catal. 2005, 233, 147–165. [Google Scholar] [CrossRef]
- Krishnankutty, N.; Li, J.; Vannice, M.A. The effect of Pd precursor and pretreatment on the adsorption and absorption behavior of supported Pd catalysts. Appl. Catal. A 1998, 173, 137–144. [Google Scholar] [CrossRef]
- Tengco, J.M.M.; Lugo-José, Y.K.; Monnier, J.R.; Regalbuto, J.R. Chemisorption–XRD particle size discrepancy of carbon supported palladium: Carbon decoration of Pd? Catal. Today 2015, 246, 9–14. [Google Scholar] [CrossRef]
- Simonov, P.A.; Romanenko, A.V.; Prosvirin, I.P.; Moroz, E.M.; Boronin, A.I.; Chuvilin, A.L.; Likholobov, V.A. On the nature of the interaction of H2PdCl4 with the surface of graphite-like carbon materials. Carbon 1997, 35, 73–82. [Google Scholar] [CrossRef]
- Wojnicki, M.; Socha, R.P.; Pędzich, Z.; Mech, K.; Tokarski, T.; Fitzner, K. Palladium(II) chloride complex ion recovery from aqueous solutions using adsorption on activated carbon. J. Chem. Eng. Data 2018, 63, 702–711. [Google Scholar] [CrossRef]
- de Pedro, Z.M.; Gómez-Sainero, L.M.; González-Serrano, E.; Rodriguez, J.J. Gas-phase hydrodechlorination of dichloromethane at low concentrations with palladium/carbon catalysts. Ind. Eng. Chem. Res. 2006, 45, 7760–7766. [Google Scholar] [CrossRef]
- Álvarez-Montero, M.A.; Gómez-Sainero, L.M.; Martín-Martínez, M.; Heras, F.; Rodriguez, J.J. Hydrodechlorination of chloromethanes with Pd on activated carbon catalysts for the treatment of residual gas streams. Appl. Catal. B 2010, 96, 148–156. [Google Scholar] [CrossRef]
- Martin-Martinez, M.; Gómez-Sainero, L.M.; Alvarez-Montero, M.A.; Bedia, J.; Rodriguez, J.J. Comparison of different precious metals in activated carbon-supported catalysts for the gas-phase hydrodechlorination of chloromethanes. Appl. Catal. B 2013, 132–133, 256–265. [Google Scholar] [CrossRef]
- Chen, N.; Rioux, R.M.; Barbosa, L.A.M.M.; Ribeiro, F.H. Kinetic and theoretical study of the hydrodechlorination of CH4-xClx (x = 1-4) compounds on palladium. Langmuir 2010, 26, 16615–16624. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Toba, M.; Matsui, T.; Harada, M.; Ichihashi, Y.; Bando, K.K.; Yasuda, H.; Ishihara, H.; Morita, Y.; Kameoka, T. Active phases and sulfur tolerance of bimetallic Pd-Pt catalysts used for hydrotreatment. Appl. Catal. A 2007, 322, 152–171. [Google Scholar] [CrossRef]
- Yu, Y.; Fonfé, B.; Jentys, A.; Haller, G.L.; van Veen, J.A.R.; Gutiérrez, O.Y.; Lercher, J.A. Bimetallic Pt-Pd/silica-alumina hydrotreating catalysts-Part I: Physicochemical characterization. J. Catal. 2012, 292, 1–12. [Google Scholar] [CrossRef]
- Matsubayashi, N.; Yasuda, H.; Imamura, M.; Yoshimura, Y. EXAFS study on Pd-Pt catalyst supported on USY zeolite. Catal. Today 1998, 45, 375–380. [Google Scholar] [CrossRef]
- Kappers, M.; Dossi, C.; Psaro, R.; Recchia, S.; Fusi, A. DRIFT study of CO chemisorption on organometallics derived Pd/MgO catalysts: The effect of chlorine. Catal. Lett. 1996, 39, 183–189. [Google Scholar] [CrossRef]
- Dal Santo, V.; Dossi, C.; Recchi, S.; Colavita, P.E.; Vlaic, G.; Psaro, R. Carbon tetrachloride hydrodechlorination with organometallics-based platinum and palladium catalysts on MgO. J. Mol. Catal. A 2002, 182–183, 157–166. [Google Scholar] [CrossRef]
- Dal Santo, V.; Sordelli, L.; Dossi, C.; Recchia, S.; Fonda, E.; Vlaic, G.; Psaro, R. Characterization of Pd/MgO catalysts: Role of organometallic precursor-surface interactions. J. Catal. 2001, 198, 296–308. [Google Scholar] [CrossRef]
- Jujjuri, S.; Ding, E.; Hommel, E.L.; Shore, S.G.; Keane, M.A. Synthesis and characterization of novel silica-supported Pd/Yb bimetallic catalysts: Application in gas-phase hydrodechlorination and hydrogenation. J. Catal. 2006, 239, 486–500. [Google Scholar] [CrossRef]
- Shore, S.G.; Ding, E.; Park, C.; Keane, M.A. Vapor phase hydrogenation of phenol over silica supported Pd and Pd-Yb catalysts. Catal. Commun. 2002, 3, 77–84. [Google Scholar] [CrossRef]
- Jujjuri, S.; Ding, E.; Shore, S.G.; Keane, M.A. Gas-phase hydrodechlorination of chlorobenzenes over silica-supported palladium and palladium-ytterbium. Appl. Organometal. Chem. 2003, 17, 493–498. [Google Scholar] [CrossRef]
- Öcal, M.; Maciejewski, M.; Baiker, A. Conversion of CCl2F2 (CFC-12) in the presence and absence of H2 on sol–gel derived Pd/Al2O3 catalysts. Appl. Catal. B 1999, 21, 279–289. [Google Scholar] [CrossRef]
- Bonarowska, M.; Malinowski, A.; Juszczyk, W.; Karpiński, Z. Hydrodechlorination of CCl2F2 (CFC-12) over silica-supported palladium–gold catalysts. Appl. Catal. B 2001, 30, 187–193. [Google Scholar] [CrossRef]
- Juszczyk, W.; Malinowski, A.; Karpiński, Z. Hydrodechlorination of CCl2F2 (CFC-12) over γ-Al2O3 Supported Palladium Catalysts. Appl. Catal. A 1998, 166, 311–319. [Google Scholar] [CrossRef]
- Malinowski, A.; Łomot, D.; Karpiński, Z. Hydrodechlorination of CH2Cl2 over Pd/γ-Al2O3. Correlation with Hydrodechlorination of CCl2F2 (CFC-12). Appl. Catal. B 1998, 19, L79–L86. [Google Scholar]
- Ben-David, Y.; Gozin, M.; Portnoy, M.; Milstein, D. Reductive dichlorination of aryl chlorides catalyzed by palladium complexes containing basing, chelating phosphines. J. Mol. Catal. 1992, 73, 173–180. [Google Scholar] [CrossRef]
- Bartholomew, C.A. Hydrogen adsorption on supported cobalt, iron and nickel. Catal. Lett. 1990, 7, 27–52. [Google Scholar] [CrossRef]
- Moulder, F.; Stickle, W.F.; Sobol, P.E.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy; Chastain, J., King, R.C., Jr., Eds.; Physical Electronics Inc.: Eden Prairie, MN, USA, 1995. [Google Scholar]
- NIST X-ray Photoelectron Spectroscopy Database 20, Version 4.1. Available online: https://srdata.nist.gov/xps (accessed on 8 November 2020).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst a | Particle Size from TEM (nm) b | Crystallite Size, from XRD c (nm) | MFE from TEM d | H/(Pd + Pt) from Chemisorption | ||||
|---|---|---|---|---|---|---|---|---|
| SD e | SE e | |||||||
| Pd100(acac) | 4.75 | 1.66 | 0.19 | 5.91 | 6.47 | 7.5 | 0.19 | 0.097 |
| Pd80Pt20(acac) | 2.87 | 1.23 | 0.14 | 3.97 | 4.57 | 2.8 | 0.28 | 0.155 |
| Pd60Pt40(acac) | 2.69 | 0.9 | 0.10 | 3.28 | 3.58 | 2.5 | 0.34 | 0.222 |
| Pd40Pt60(acac) | 2.24 | 0.79 | 0.09 | 2.87 | 3.35 | 2.1 | 0.39 | 0.276 |
| Pt100(acac) | 2.59 | 0.80 | 0.09 | 3.06 | 3.28 | 3.0 | 0.37 | 0.416 |
| Pd100(Cl) | 2.84 | 0.73 | 0.09 | 3.24 | 3.43 | 3.9 | 0.35 | 0.229 |
| Pd80Pt20(Cl) | 2.31 | 0.75 | 0.08 | 2.79 | 3.01 | ~2 | 0.40 | 0.214 |
| Pd60Pt40(Cl) | 1.36 | 0.40 | 0.05 | 1.63 | 1.85 | ~2 | 0.70 | 0.235 |
| Pd40Pt60(Cl) | 1.45 | 0.42 | 0.05 | 1.70 | 1.83 | ~2 | 0.66 | 0.339 |
| Pt100(Cl) | 1.93 | 0.52 | 0.06 | 2.19 | 2.31 | <2 | 0.59 | 0.596 |
| Catalyst | Pt0 | Pt2+ | Pd0 | Pd2+ |
|---|---|---|---|---|
| Pd100(acac) | 67.2 | 32.8 | ||
| Pd80Pt20(acac) | 100 | 0 | 66.6 | 33.4 |
| Pd60Pt40(acac) | 100 | 0 | 60.6 | 39.4 |
| Pd40Pt60(acac) | 50.5 | 49.5 | 59.2 | 40.8 |
| Pt100(acac) | 42.2 | 57.8 | ||
| Pd100(Cl) | 55.5 | 44.5 | ||
| Pd80Pt20(Cl) | 74.6 | 25.4 | 61.3 | 38.7 |
| Pd60Pt40(Cl) | 54.9 | 45.1 | 76.0 | 24.0 |
| Pd40Pt60(Cl) | 42.8 | 57.2 | 70.8 | 29.2 |
| Pt100(Cl) | 52.7 | 47.3 |
| Catalyst a | Reaction Temperature °C | TOF b s−1 | Product Selectivity, % | Activation Energy, kJ/mol | |||
|---|---|---|---|---|---|---|---|
| CH4 | CH3F | CHF3 | CH2F2 | ||||
| Pd100(acac) | 272 | 4.1 × 10−4 | 28.5 | 1.9 | - | 66.3 | 100.7 ± 13.4 |
| 262 | 2.85 × 10−4 | 25 | 4 | - | 69.9 | ||
| 252 | 1.7 × 10−4 | 28 | - | - | 72 | ||
| Pd80Pt20(acac) | 272 | 9.2 × 10−4 | 30.3 | 8.2 | 7.7 | 53.8 | 110.1 ± 8.5 |
| 262 | 5.25 × 10−4 | 22.3 | 4.9 | 21.2 | 51.5 | ||
| 251 | 3.6 × 10−3 | 19 | 5 | 25 | 51 | ||
| Pd60Pt40(acac) | 272 | 1.32 × 10−3 | 41 | 14 | - | 45 | 83.1 ± 4.6 |
| 261 | 9.4 × 10−4 | 36.7 | 14.5 | - | 48.8 | ||
| 251 | 6.3 × 10−4 | 33.5 | 14.5 | - | 52.0 | ||
| Pd40Pt60(acac) | 272 | 1.46 × 10−3 | 45.6 | 16.7 | - | 37.7 | 77.1 ± 5.2 |
| 262 | 1.09 × 10−3 | 42.0 | 14.9 | - | 44.6 | ||
| 252 | 7.44 × 10−4 | 37.5 | 17.7 | - | 44.8 | ||
| Pt100(acac) | 271 | 3.5 × 10−4 | 58.8 | 20.5 | 5.0 | 15.7 | 87.8 ± 6.6 |
| 262 | 2.5 × 10−4 | 56.5 | 20.6 | - | 22.9 | ||
| 251 | 1.65 × 10−4 | 53.5 | 18.6 | 11.0 | 16.9 | ||
| Pd100(Cl) | 271 | 1.3 × 10−3 | 36.8 | 1.2 | - | 62.0 | 88.8 ± 2.9 |
| 261 | 9.2 × 10−4 | 33.6 | 1.2 | - | 65.2 | ||
| 251 | 6.15 × 10−4 | 30.1 | - | - | 69.9 | ||
| Pd80Pt20(Cl) | 271 | 2.2 × 10−3 | 47.6 | 4.0 | - | 48.4 | 84.7 ± 4.2 |
| 262 | 1.6 × 10−3 | 44.1 | 4.1 | - | 51.8 | ||
| 252 | 1.08 × 10−3 | 39.8 | 4.2 | - | 56.0 | ||
| Pd60Pt40(Cl) | 271 | 2.5 × 10−3 | 53.4 | 5.6 | - | 41.0 | 76.1 ± 1.1 |
| 262 | 1.87 × 10−3 | 50.9 | 5.9 | - | 43.2 | ||
| 252 | 1.35 × 10−3 | 47.4 | 6.3 | - | 46.2 | ||
| Pd40Pt60(Cl) | 272 | 2.17 × 10−3 | 59.4 | 7.0 | - | 33.5 | 92.2 ± 1.5 |
| 262 | 1.5 × 10−3 | 55.6 | 7.6 | - | 36.8 | ||
| 252 | 1 × 10−3 | 44.5 | 7.6 | 6.4 | 41.4 | ||
| Pt100(Cl) | 272 | 6.8 × 10−4 | 71.9 | 11.9 | 0.3 | 15.8 | 75.7 ± 2.1 |
| 262 | 4.9 × 10−4 | 68.8 | 12.6 | - | 18.5 | ||
| 252 | 3.6 × 10−4 | 66.5 | 13.7 | - | 19.8 | ||
| Catalyst a,b | Reaction Temperature °C | TOF s−1 | Product Selectivity, % | Activation Energy, kJ/mol | |||
|---|---|---|---|---|---|---|---|
| CH4 | CH3F | CHF3 | CH2F2 | ||||
| Pd100(acac) H/Pd = 0.110 | 272 | 7.0 × 10−4 | 27.4 | - | - | 72.6 | 89.4 ± 10.0 |
| 262 | 4.5 × 10−4 | 24.4 | 4 | - | 75.6 | ||
| 252 | 3.3 × 10−4 | 21.5 | - | - | 78.5 | ||
| Pd80Pt20(acac) H/(Pd + Pt) = 0.182 | 272 | 1.20 × 10−3 | 26.8 | 4.6 | - | 68.6 | 88.1 ± 5.4 |
| 262 | 8.0 × 10−4 | 24.7 | 5.2 | - | 70.1 | ||
| 251 | 5.5 × 10−4 | 24.9 | 6.0 | - | 69.1 | ||
| Pd60Pt40(acac) H/(Pd + Pt) = 0.210 | 272 | 1.80 × 10−3 | 42.4 | 4.5 | - | 53.0 | 85.1 ± 4.6 |
| 261 | 1.2 × 10−3 | 35.9 | 9.1 | - | 55.0 | ||
| 251 | 8.9 × 10−4 | 32.6 | 9.8 | - | 57.6 | ||
| Pd40Pt60(acac) H/(Pd + Pt) = 0.282 | 272 | 1.6 × 10−3 | 44.4 | 13.5 | - | 42.1 | 77.1 ± 5.2 |
| 262 | 1.05 × 10−3 | 14.0 | 14.9 | - | 45.7 | ||
| 252 | 7.2 × 10−4 | 14.9 | 17.7 | - | 49.1 | ||
| Pt100(acac) H/Pt = 0.351 | 271 | 3.0 × 10−4 | 58.9 | 14.9 | 12.6 | 13.6 | 86.2 ± 12.0 |
| 262 | 2.0 × 10−4 | 59.2 | 16.8 | 9.0 | 15.0 | ||
| 251 | 1.5 × 10−4 | 80.3 | 19.7 | - | - | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radlik, M.; Juszczyk, W.; Matus, K.; Raróg-Pilecka, W.; Karpiński, Z. Hydrodechlorination of CHClF2 (HCFC-22) over Pd–Pt Catalysts Supported on Thermally Modified Activated Carbon. Catalysts 2020, 10, 1291. https://doi.org/10.3390/catal10111291
Radlik M, Juszczyk W, Matus K, Raróg-Pilecka W, Karpiński Z. Hydrodechlorination of CHClF2 (HCFC-22) over Pd–Pt Catalysts Supported on Thermally Modified Activated Carbon. Catalysts. 2020; 10(11):1291. https://doi.org/10.3390/catal10111291
Chicago/Turabian StyleRadlik, Monika, Wojciech Juszczyk, Krzysztof Matus, Wioletta Raróg-Pilecka, and Zbigniew Karpiński. 2020. "Hydrodechlorination of CHClF2 (HCFC-22) over Pd–Pt Catalysts Supported on Thermally Modified Activated Carbon" Catalysts 10, no. 11: 1291. https://doi.org/10.3390/catal10111291
APA StyleRadlik, M., Juszczyk, W., Matus, K., Raróg-Pilecka, W., & Karpiński, Z. (2020). Hydrodechlorination of CHClF2 (HCFC-22) over Pd–Pt Catalysts Supported on Thermally Modified Activated Carbon. Catalysts, 10(11), 1291. https://doi.org/10.3390/catal10111291