Boosting the Performance of Nano-Ni Catalysts by Palladium Doping in Flow Hydrogenation of Sulcatone

 , ,
, ,  , , ,
, , , 
Abstract

1. Introduction
2. Results and Discussion
2.1. Characterization of the Ni Catalysts
2.2. Isotopic Experiments
2.3. DFT Calculations
2.4. Characterization of Pd-Modified Catalysts
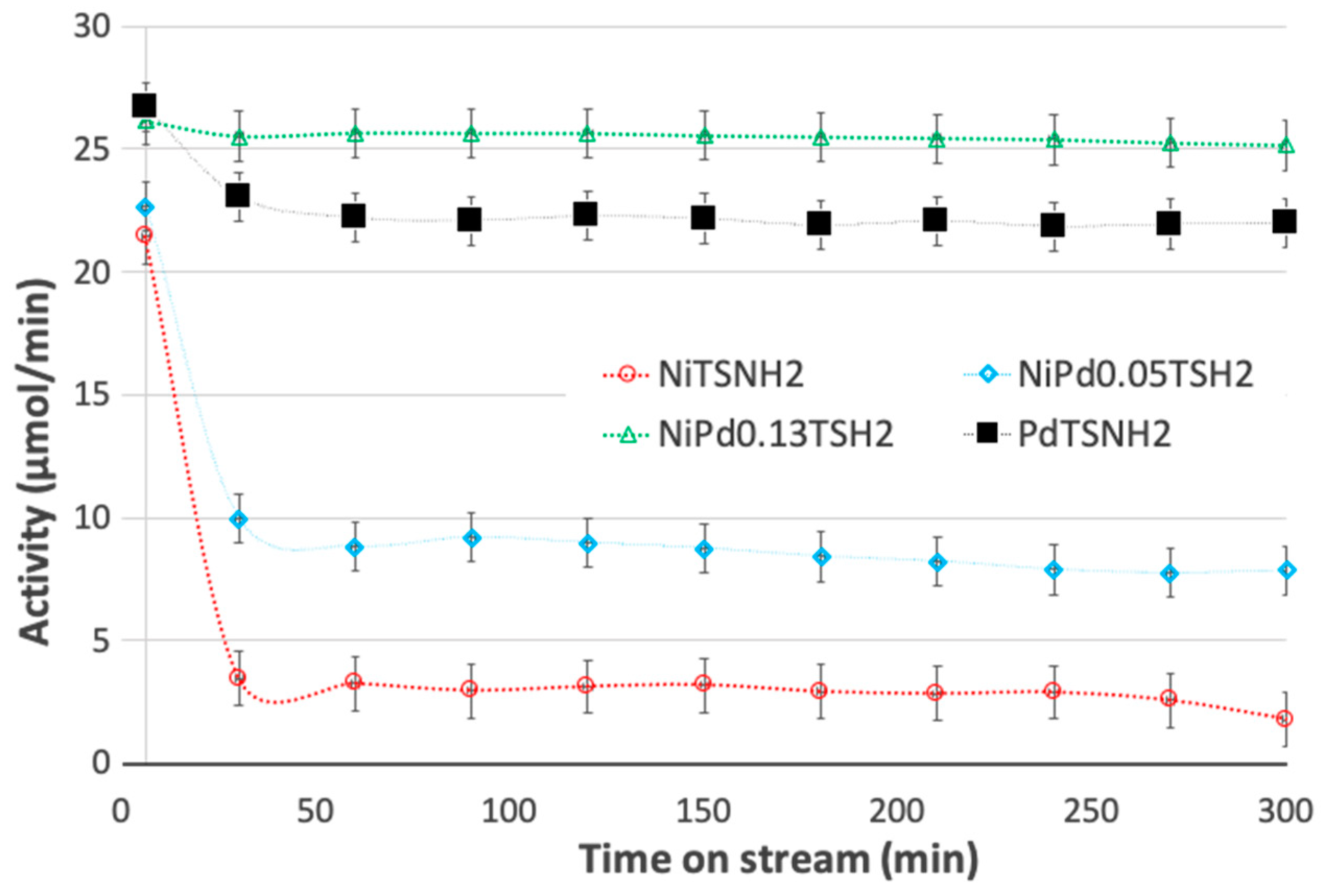
2.5. Catalytic Activity of Ni and Pd-Modified Catalysts in Flow Hydrogenation
3. Experimental Section
3.1. Preparation of the Catalysts
3.1.1. Synthesis of NiTSNH2
3.1.2. Synthesis of PdTSNH2
3.1.3. Synthesis of Pd Modified NiTSNH2
3.2. Catalysts Characterization Technics
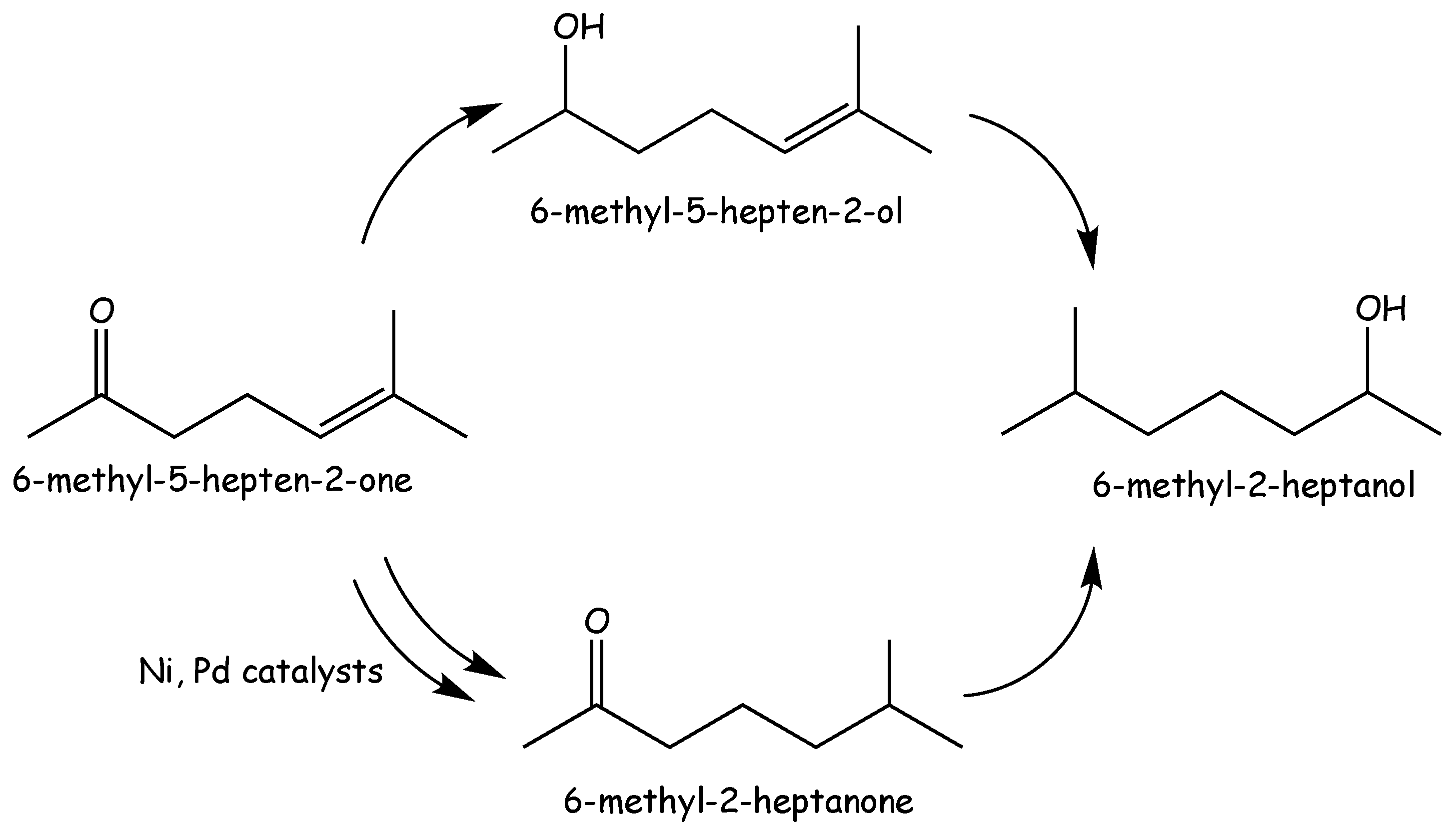
3.3. 6-Methyl-5-Hepten-2-One Hydrogenation
3.4. Quantum Chemical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ranade, V.V.; Joshi, S.S. Industrial Catalytic Processes for Fine and Specialty Chemicals; Elsevier: Amsterdam, The Netherlands, 2016; pp. 1–14. [Google Scholar]
- Weissermel, K.; Arpe, H.J. Industrial Organic Chemistry; Verlag Chemie: Weinheim, Germany, 1978. [Google Scholar]
- Bonrath, W.; Medlock, J.; Schütz, J.; Wüstenberg, B.; Netscher, T. Hydrogenation in the Vitamins and Fine Chemicals Industry—An Overview; Intechopen: London, UK, 2012. [Google Scholar]
- Royo, B. Sustainable Synthesis of Pharmaceuticals: Using Transition Metal Complexes as Catalysts; RSC: London, UK, 2018. [Google Scholar]
- Bauer, K.; Garbe, D. Ullman Encyclopedia; VCH: New York, NY, USA, 1988; p. 141. [Google Scholar]
- Gallezot, P.; Richard, D. Selective hydrogenation of α,β-unsaturated aldehydes. Catal. Rev. Sci. Eng. 1998, 40, 81–126. [Google Scholar] [CrossRef]
- Righi, G.; Rossi, L. Mild regioselective catalytic hydrogenation of α,β-unsaturated carbonyl compounds with lindlar catalyst. Syth. Commun. 1996, 26, 1321–1327. [Google Scholar] [CrossRef]
- Bauer, K.; Garbe, D.; Surburg, H. Common Fragrance and Flavour Materials: Preparation, Properties and Uses, 3rd ed.; Wiley-VCH: Weinheim, Germany, 1977. [Google Scholar]
- Grolig, J. Ullmann’s Encyclopedia of Industrial Chemistry, 7th ed.; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Siddqui, N.; Sarkar, B.; Pendem, C.; Khatum, R.; Konthala, L.N.S.; Sasaki, T.; Bordoloi, A.; Bal, R. Highly selective transfer hydrogenation of α,β-unsaturated carbonyl compounds using Cu-based nanocatalysts. Catal. Sci. Technol. 2017, 7, 2828–2837. [Google Scholar] [CrossRef]
- Farrar-Tobar, R.A.; Tin, S.; de Vries, J.G. Organometallics for Green Chemistry; Springer: Berlin/Heidelberg, Germany, 2018; pp. 193–224. [Google Scholar]
- Mendes-Burak, J.; Ghaffari, B.; Copéret, C. Selective hydrogenation of α,β-unsaturated carbonyl compounds on silica-supported copper nanoparticles. Chem. Commun. 2019, 55, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Bonrath, W.; Netscher, T. Catalytic processes in vitamins synthesis and production. Appl. Catal. A Gen. 2005, 280, 55–73. [Google Scholar] [CrossRef]
- Wang, X.; He, Y.; Liu, Y.; Park, J.; Liang, X. Atomic layer deposited Pt-Co bimetallic catalysts for selective hydrogenation of α,β-unsaturated aldehydes to unsaturated alcohols. J. Catal. 2018, 366, 61–69. [Google Scholar] [CrossRef]
- Du, W.Q.; Rong, Z.M.; Liang, Y.; Wang, Y.; Lu, X.Y.; Wang, Y.F.; Lu, L.H. Chemoselective hydrogenation of α,β-unsaturated aldehydes with modified Pd/C catalyst. Chin. Chem. Lett. 2012, 23, 773–776. [Google Scholar] [CrossRef]
- Goszewska, I.; Giziński, D.; Zienkiewicz-Machnik, M.; Lisovytskiy, D.; Nikiforov, K.; Masternak, J.; Śrębowata, A.; Sá, J. A novel nano-palladium catalyst for continuous-flow chemoselective hydrogenation reactions. Catal. Commun. 2017, 94, 65–68. [Google Scholar] [CrossRef]
- Tessonnier, J.-P.; Pesanat, L.; Ehret, G.; Ledoux, M.J.; Pham-Huu, C. Pd nanoparticles introduced inside multi-walled carbon nanotubes for selective hydrogenation of cinnamaldehyde into hydrocinnamaldehyde. Appl. Catal. A Gen. 2005, 288, 203–210. [Google Scholar] [CrossRef]
- Banerjee, S.; Balasanthiran, V.; Koodali, R.T.; Sereda, G.A. Pd-MCM-48: A novel recyclable heterogeneous catalyst for chemo- and regioselective hydrogenation of olefins and coupling reactions. Org. Biomol. Chem. 2010, 8, 4316–4321. [Google Scholar] [CrossRef]
- Coloma, F.; Sepúlveda-Escribano, A.; Fierro, J.L.G.; Rodriguez-Reinoso, F. Crotonaldehyde hydrogenation over bimetallic Pt-Sn catalysts supported on pregraphitized carbon black. Effect of the Sn/Pt atomic ratio. Appl. Catal. A Gen. 1996, 136, 231–248. [Google Scholar] [CrossRef]
- Mahata, N.; Goncalves, F.; Fernando, M.; Pereira, R.; Figueiredo, J.L. Selective hydrogenation of cinnamaldehyde to cinnamyl alcohol over mesoporous carbon supported Fe and Zn promoted Pt catalyst. Appl. Catal. A Gen. 2008, 339, 159–168. [Google Scholar] [CrossRef]
- Hu, C.; Creaser, D.; Siahrostami, S.; Grönbeck, H.; Ojagh, H.; Skoglundh, M. Catalytic hydrogenation of C = C and C = O in unsaturated fatty acid methyl esters. Catal. Sci. Technol. 2014, 4, 2427–2444. [Google Scholar] [CrossRef]
- Murillo, L.E.; Menning, C.A.; Chen, J.G. Trend in the C = C and C = O bond hydrogenation of acrolein on Pt–M (M = Ni, Co, Cu) bimetallic surfaces. J. Catal. 2009, 268, 335–342. [Google Scholar] [CrossRef]
- Hudson, R.; Hamasaka, G.; Osako, T.; Yamada, Y.M.A.; Li, C.-J.; Uozumi, Y.; Moores, A. Highly efficient iron(0) nanoparticle-catalyzed hydrogenation in water in flow. Green Chem. 2013, 15, 2141–2148. [Google Scholar] [CrossRef]
- Wang, Y.; Qin, R.; Wang, Y.; Ren, J.; Zhou, W.; Li, L.; Ming, J.; Zhang, W.; Fu, G.; Zheng, N. Chemoselective hydrogenation of nitroaromatics at the nanoscale iron(III)–OH–platinum interface. Angew. Chem. Int. Ed. 2020, 59, 12736–12740. [Google Scholar] [CrossRef]
- Gong, X.; Wang, M.; Fang, H.; Qian, X.; Ye, L.; Duan, X.; Yuan, Y. Copper nanoparticles socketed in situ into copper phyllosilicate nanotubes with enhanced performance for chemoselective hydrogenation of esters. Chem. Commun. 2017, 53, 6933–6936. [Google Scholar] [CrossRef]
- Xavier, K.O.; Sreeekala, R.; Rashid, K.K.A.; Yusuff, K.K.M.; Sen, D. Doping effects of cerium oxide on Ni/Al2O3 catalysts for methanation. Catal. Today 1999, 49, 17–21. [Google Scholar] [CrossRef]
- Hauptmann, H.; Walter, W.F. The action of raney nickel on organic sulfur compounds. Chem. Rev. 1962, 62, 347–404. [Google Scholar] [CrossRef]
- Alonso, F.; Riente, P.; Yus, M. Nickel nanoparticles in hydrogen transfer reactions. Acc. Chem. Res. 2011, 44, 379–391. [Google Scholar] [CrossRef]
- Qi, S.-C.; Zhang, L.; Einaga, H.; Kudo, S.; Norinaga, K.; Hayashi, J.-I. Nano-sized nickel catalyst for deep hydrogenation of lignin monomers and first-principles insight into the catalyst preparation. J. Mater. Chem. A 2017, 5, 3948–3965. [Google Scholar] [CrossRef]
- Jiang, Z.; Xe, J.; Jiang, D.; Wei, X.; Chen, M. Modifiers-assisted formation of nickel nanoparticles and their catalytic application to p-nitrophenol reduction. CrystEngComm 2013, 15, 560–569. [Google Scholar] [CrossRef]
- Kishida, S.; Murakami, Y.; Imanaka, T.; Teranishi, S. Hydrogenation of various ketones on nickel boride catalyst. J. Catal. 1968, 12, 97–101. [Google Scholar] [CrossRef]
- Giziński, D.; Błachucki, W.; Śrębowata, A.; Zienkiewcz-Machnik, M.; Goszewska, I.; Matus, K.; Lisovytskiy, D.; Pisarek, M.; Szlachetko, J.; Sá, J. On-the-fly catalyst accretion and screening in chemoselective flow hydrogenation. ChemCatChem 2018, 10, 3641–3646. [Google Scholar] [CrossRef]
- Budroni, G.; Kondrat, S.A.; Taylor, S.H.; Morgan, D.J.; Carley, A.F.; Williams, P.B.; Hutchings, G.J. Selective deposition of palladium onto supported nickel-bimetallic catalysts for the hydrogenation of crotonaldehyde. Catal. Sci. Technol. 2013, 3, 2746–2754. [Google Scholar] [CrossRef]
- Hammoudeh, A.; Mahmoud, S. Selective hydrogenation of cinnamaldehyde over Pd/SiO2 catalysts: Selectivity promotion by alloyed Sn. J. Mol. Catal. A Chem. 2003, 203, 231–239. [Google Scholar] [CrossRef]
- Fulajtárova, K.; Sotác, T.; Hronec, M.; Vávra, I.; Dobročka, E.; Omastová, M. Aqueous phase hydrogenation of furfural to furfuryl alcohol over Pd–Cu catalysts. Appl. Catal. A Gen. 2015, 502, 78–85. [Google Scholar] [CrossRef]
- Mo, M.; Xie, M.; Guo, X.; Ding, W.; Guo, X. The promoted catalytic hydrogenation performance of bimetallic Ni–Co–B noncrystalline alloy nanotubes. RSC Adv. 2019, 9, 26456–26463. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Tomishige, K. Total hydrogenation of furan derivatives over silica-supported Ni–Pd alloy catalyst. Catal. Commun. 2010, 12, 154–156. [Google Scholar] [CrossRef]
- Tan, L.; Li, T.; Zhou, J.; Chen, H.; Jiang, F. Liquid-phase hydrogenation of N-nitrosodimethylamine over Pd-Ni supported on CeO2-TiO2: The role of oxygen vacancies. Coll. Surf. A 2018, 558, 211–218. [Google Scholar] [CrossRef]
- McAllister, M.I.; Boulho, C.; Gilpin, L.F.; McMillan, L.; Brennan, C.; Lenon, D. Hydrogenation of benzonitrile over supported pd catalysts: Kinetic and mechanistic insight. Org. Process Res. Dev. 2019, 23, 977–989. [Google Scholar] [CrossRef]
- Liguori, F.; Barbaro, P.; Sawa, H. Continuous flow hydrogenation reactions by Pd catalysts onto hybrid ZrO2/PVA materials. Appl. Catal. A Gen. 2014, 488, 58–65. [Google Scholar] [CrossRef]
- Sá, J.; Medlin, J.W. On-the-fly catalyst modification: Strategy to improve catalytic processes selectivity and understanding. ChemCatChem 2019, 11, 3355–3365. [Google Scholar] [CrossRef]
- Cossar, P.J.; Hizartzidis, L.; Simone, M.I.; McCluskey, A.; Gordon, C.P. The expanding utility of continuous flow hydrogenation. Org. Biomol. Chem. 2015, 13, 7119–7130. [Google Scholar] [CrossRef]
- Mallia, C.J.; Baxendale, I.R. The use of gases in flow synthesis. Org. Process Res. Dev. 2016, 20, 327–360. [Google Scholar] [CrossRef]
- Jones, R.V.; Godorhazy, L.; Varga, N.; Szalay, D.; Urge, L.; Darvas, F. Continuous-flow high pressure hydrogenation reactor for optimization and high-throughput synthesis. J. Comb. Chem. 2006, 8, 110–116. [Google Scholar] [CrossRef]
- Zienkiewicz-Machnik, M.; Goszewska, I.; Śrębowata, A.; Kubas, A.; Giziński, D.; Słowik, G.; Matus, K.; Lisovytskiy, D.; Pisarek, M.; Sá, J. Tuning nano-nickel selectivity with tin in flow hydrogenation of 6-methyl-5-hepten-2-one by surface organometallic chemistry modification. Catal. Today 2018, 308, 38–44. [Google Scholar] [CrossRef]
- Zienkiewcz-Machnik, M.; Goszewska, I.; Giziński, D.; Śrębowata, A.; Kuzmowicz, K.; Kubas, A.; Matus, K.; Lisovytskiy, D.; Pisarek, M.; Sá, J. Tuning nano-nickel catalyst hydrogenation aptitude by on-the-fly zirconium doping. ChemCatChem 2020, 12, 3132–3138. [Google Scholar] [CrossRef]
- Ayats, C.; Henseler, A.H.; Pericas, M.A. A solid-supported organocatalyst for continuous-flow enantioselective aldol reactions. ChemSusChem 2012, 5, 320–325. [Google Scholar] [CrossRef]
- Canellas, S.; Ayats, C.; Henseler, A.H.; Pericas, M.A. A highly active polymer-supported catalyst for asymmetric robinson annulations in continuous Flow. ACS Catal. 2017, 7, 1383–1391. [Google Scholar] [CrossRef]
- Krill, S.; Huthmacher, K. Process for the production of 6-methyl heptanone. Patent No. US6417406B1, 10 September 2001. [Google Scholar]
- Giziński, D.; Goszewska, I.; Zieliński, M.; Lisovytskiy, D.; Nikiforov, K.; Masternak, J.; Zienkiewcz-Machnik, M.; Śrębowata, A.; Sá, J. Chemoselective flow hydrogenation of α,β—Unsaturated aldehyde with nano-nickel. Catal. Commun. 2017, 98, 17–21. [Google Scholar] [CrossRef]
- Quaino, P.; Juarez, F.; Santos, E.; Schmickler, W. Volcano plots in hydrogen electrocatalysis—Uses and abuses. Beilstein J. Nanotechnol. 2014, 5, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Laursen, A.B.; Varela, A.S.; Dionigi, F.; Fanchiu, H.; Miller, C.; Trinhammer, O.L.; Rossmeisl, J.; Dahl, S. Electrochemical hydrogen evolution: Sabatier’s principle and the volcano plot. J. Chem. Educ. 2012, 89, 1595–1599. [Google Scholar] [CrossRef]
- Van Meerten, R.; Morales, A.; Barbier, J.; Maurel, R. Isotope effects in the hydrogenation and exchange of benzene on platinum and nickel. J. Catal. 1979, 58, 43–51. [Google Scholar] [CrossRef]
- Adams, B.D.; Chen, A. The role of palladium in a hydrogen economy. Mater. Today 2011, 14, 282–289. [Google Scholar] [CrossRef]
- Pan, D.; Jian, J.K.; Ablat, A.; Li, J.; Sun, Y.F.; Wu, R. Structure and magnetic properties of Ni-doped AlN films. J. Appl. Phys. 2012, 112, 053911. [Google Scholar] [CrossRef]
- Pulm, H.; Hohlneicher, G.; Freund, H.-J.; Schuster, H.-U.; Drews, J.; Eberv, U. Charge distribution in some ternary vintl phases as studied by v-ray photoelectron spectroscopy. J. Less Common Met. 1986, 115, 127–143. [Google Scholar] [CrossRef]
- Kumar, G.; Blackburn, J.R.; Albridge, R.G.; Moddeman, W.E.; Jones, M.M. Photoelectron spectroscopy of coordination compounds. II. Palladium complexes. Inorg. Chem. 1972, 11, 296–300. [Google Scholar] [CrossRef]
- Vogt, C.; Groeneveld, E.; Kamsma, G.; Nachtegaal, M.; Lu, L.; Kiely, C.J.; Berben, P.H.; Meirer, F.; Weckhuysen, B.M. Unravelling structure sensitivity in CO2 hydrogenation over nickel. Nat. Catal. 2018, 1, 127–134. [Google Scholar] [CrossRef]
- Barbier, J. Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Weitkamp, J., Eds.; Wiley: Weinheim, Germany, 1997; p. 257. [Google Scholar]
- Epron, F.; Gauthard, F.; Pinéda, C.; Barbier, J. Catalytic reduction of nitrate and nitrite on Pt–Cu/Al2O3 catalysts in aqueous solution: Role of the interaction between copper and platinum in the reaction. J. Catal. 2001, 198, 309–318. [Google Scholar] [CrossRef]
- Sá, J.; Vinek, H. Catalytic hydrogenation of nitrates in water over a bimetallic catalyst. Appl. Catal. B Environ. 2005, 57, 247–256. [Google Scholar] [CrossRef]
- Sá, J.; Gross, S.; Vinek, H. Effect of the reducing step on the properties of Pd-Cu bimetallic catalysts used for denitration. Appl. Catal. A Gen. 2005, 294, 226–234. [Google Scholar] [CrossRef]
- Błachucki, W.; Czapla-Masztafiak, J.; Sá, J.; Szlachetko, J. A laboratory-based double X-ray spectrometer for simultaneous X-ray emission and X-ray absorption studies. J. Anal. At. Spectrom. 2019, 34, 1409–1415. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Weigend, F. Accurate coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 8, e1327. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef]
- Prins, R. Hydrogen spillover. Facts and fiction. Chem. Rev. 2012, 112, 2714–2738. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
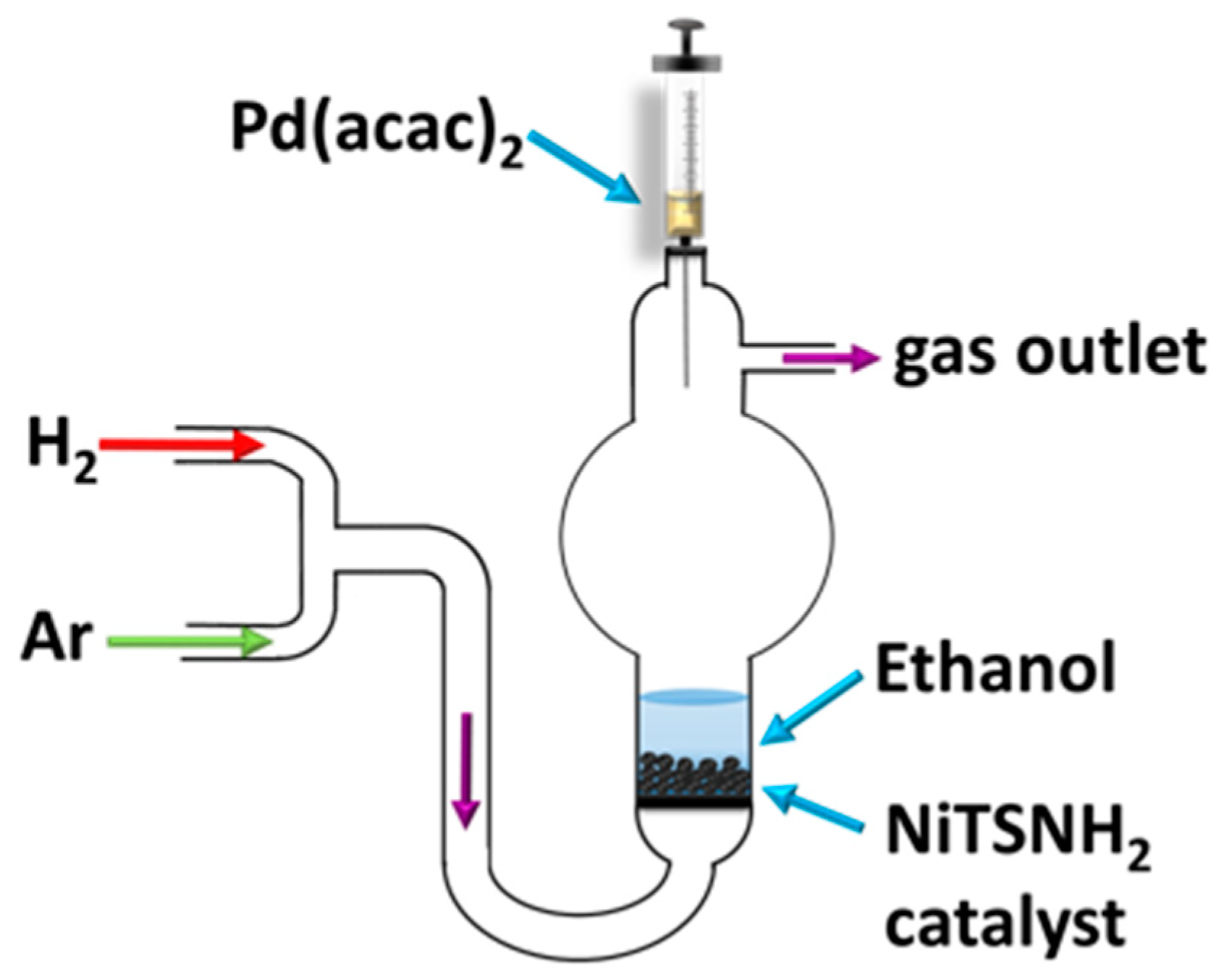{kind=link}
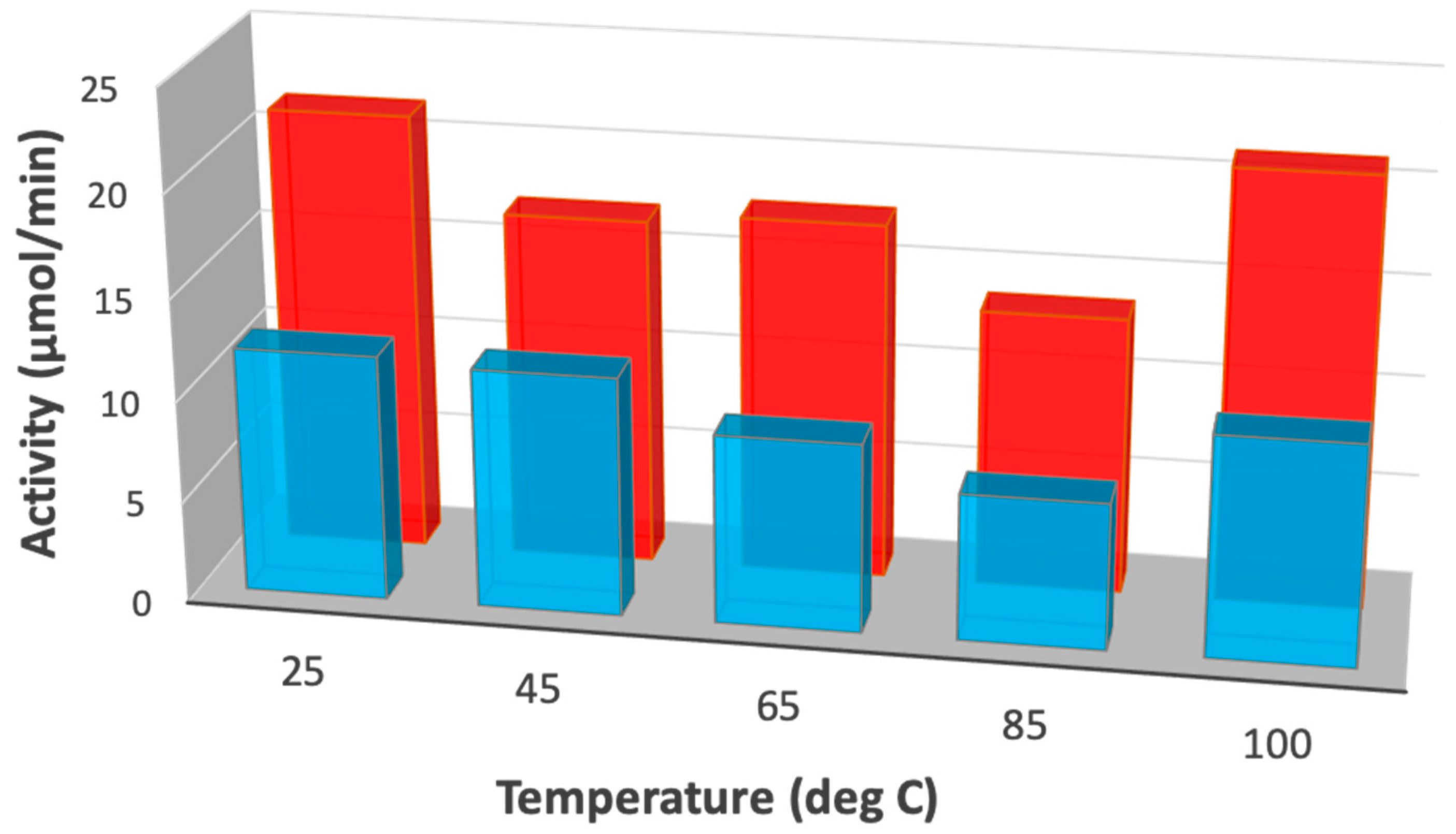
| Temperature (°C) | KIE | Gas Pressure (Bar) | KIE |
|---|---|---|---|
| 25 | 1.81 ± 0.38 | 10 | 1.96 ± 0.78 |
| 45 | 1.29 ± 0.16 | 20 | 1.72 ± 0.18 |
| 85 | 1.96 ± 0.05 | 40 | 1.71 ± 0.39 |
| 100 | 2.11 ± 0.55 | 60 | 1.79 ± 0.20 |
| Ni13 | Pd13 | Ni13Pd | |
|---|---|---|---|
| H2 binding [H2 + M → M(H2)] | −2.0 | −4.7 | −6.7 |
| H2 dissociation TS [M(H2) → M(H2)‡] * | 6.7 (7.0) | 1.7 (2.2) | 2.6 (3.1) |
| H2 dissociation [M(H2) → M(2H)] | −8.5 | −24.6 | −18.2 |
| overall [H2 + M → M(2H)] | −10.4 | −29.2 | −24.9 |
| Sample | Ni wt% | Pd wt% | Pd/Ni Ratio | Pd Deposition Efficacy (%) |
|---|---|---|---|---|
| NiTSNH2 | 0.86 ± 0.03 | - | - | - |
| PdTSNH2 | - | 2.16 ± 0.13 | - | - |
| NiPd0.05TSNH2 | 0.85 ± 0.03 | 0.08 ± 0.03 | 0.05 | 70 |
| NiPd0.13TSNH2 | 0.88 ± 0.04 | 0.20 ± 0.02 | 0.13 | 88 |
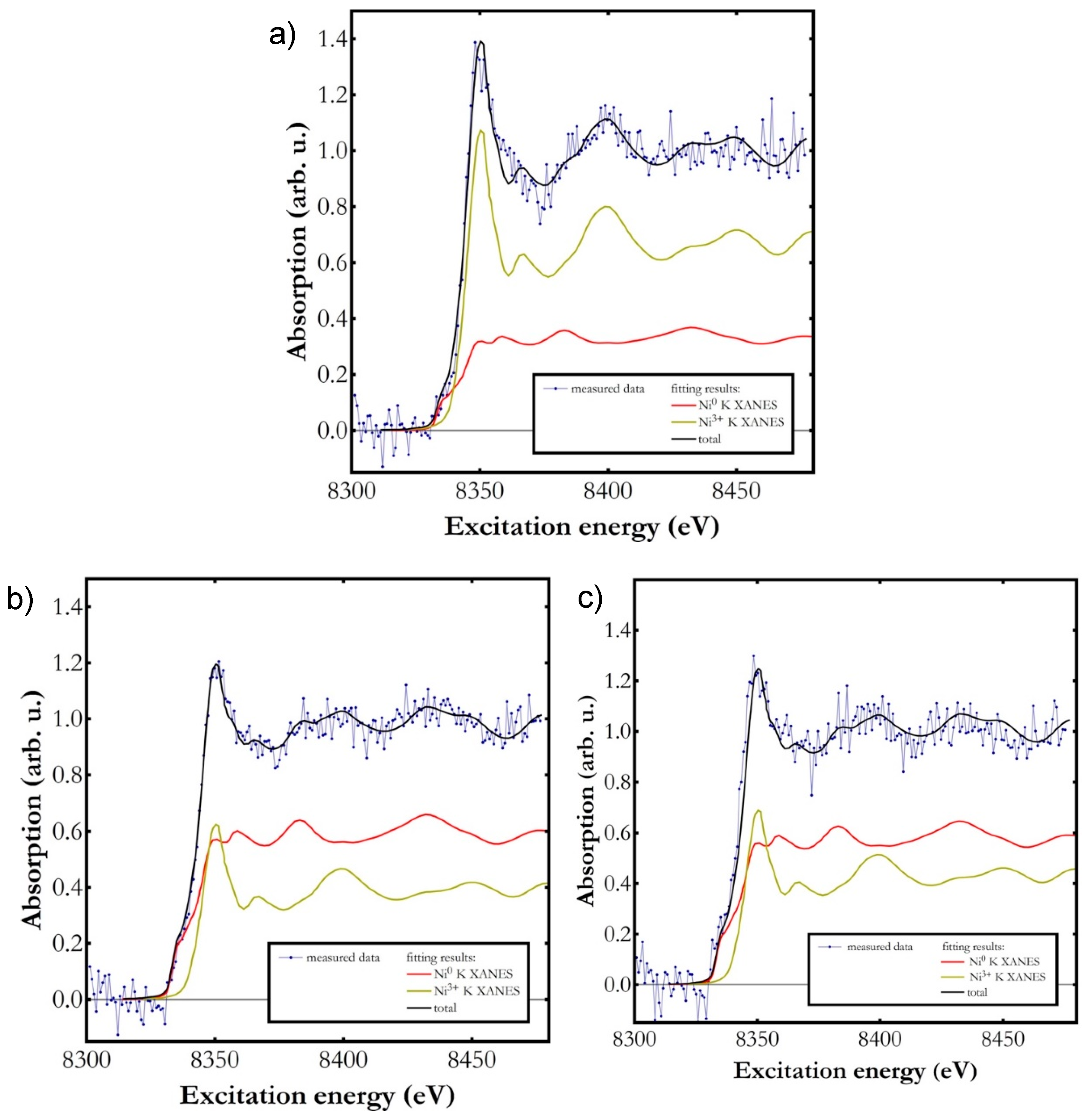
| Sample | Ni0 (%) | Ni3+ (%) |
|---|---|---|
| NiTSNH2 | 34 ± 2.0 | 66 ± 2.0 |
| NiPd0.05TSNH2 | 61 ± 1.5 | 39 ± 1.5 |
| NiPd0.13TSNH2 | 58 ± 2.2 | 42 ± 2.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goszewska, I.; Zienkiewicz-Machnik, M.; Błachucki, W.; Kubas, A.; Giziński, D.; Matus, K.; Nikiforow, K.; Lisovytskiy, D.; Śrębowata, A.; Szlachetko, J.; et al. Boosting the Performance of Nano-Ni Catalysts by Palladium Doping in Flow Hydrogenation of Sulcatone. Catalysts 2020, 10, 1267. https://doi.org/10.3390/catal10111267
Goszewska I, Zienkiewicz-Machnik M, Błachucki W, Kubas A, Giziński D, Matus K, Nikiforow K, Lisovytskiy D, Śrębowata A, Szlachetko J, et al. Boosting the Performance of Nano-Ni Catalysts by Palladium Doping in Flow Hydrogenation of Sulcatone. Catalysts. 2020; 10(11):1267. https://doi.org/10.3390/catal10111267
Chicago/Turabian StyleGoszewska, Ilona, Małgorzata Zienkiewicz-Machnik, Wojciech Błachucki, Adam Kubas, Damian Giziński, Krzysztof Matus, Kostiantyn Nikiforow, Dmytro Lisovytskiy, Anna Śrębowata, Jakub Szlachetko, and et al. 2020. "Boosting the Performance of Nano-Ni Catalysts by Palladium Doping in Flow Hydrogenation of Sulcatone" Catalysts 10, no. 11: 1267. https://doi.org/10.3390/catal10111267
APA StyleGoszewska, I., Zienkiewicz-Machnik, M., Błachucki, W., Kubas, A., Giziński, D., Matus, K., Nikiforow, K., Lisovytskiy, D., Śrębowata, A., Szlachetko, J., & Sá, J. (2020). Boosting the Performance of Nano-Ni Catalysts by Palladium Doping in Flow Hydrogenation of Sulcatone. Catalysts, 10(11), 1267. https://doi.org/10.3390/catal10111267