Rhodium Nanoparticles Stabilized by PEG-Tagged Imidazolium Salts as Recyclable Catalysts for the Hydrosilylation of Internal Alkynes and the Reduction of Nitroarenes


Abstract
:
1. Introduction
2. Results and Discussion
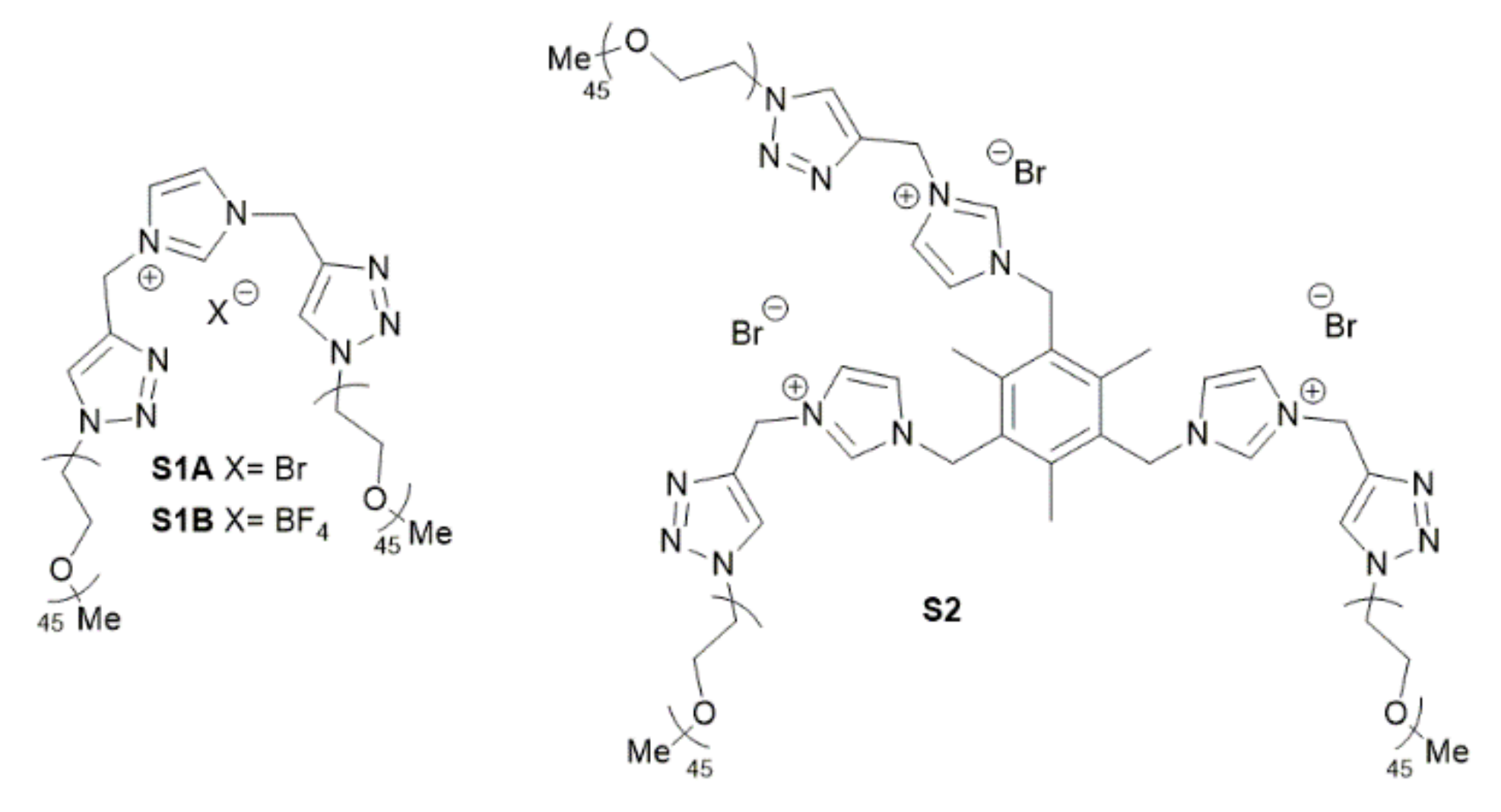
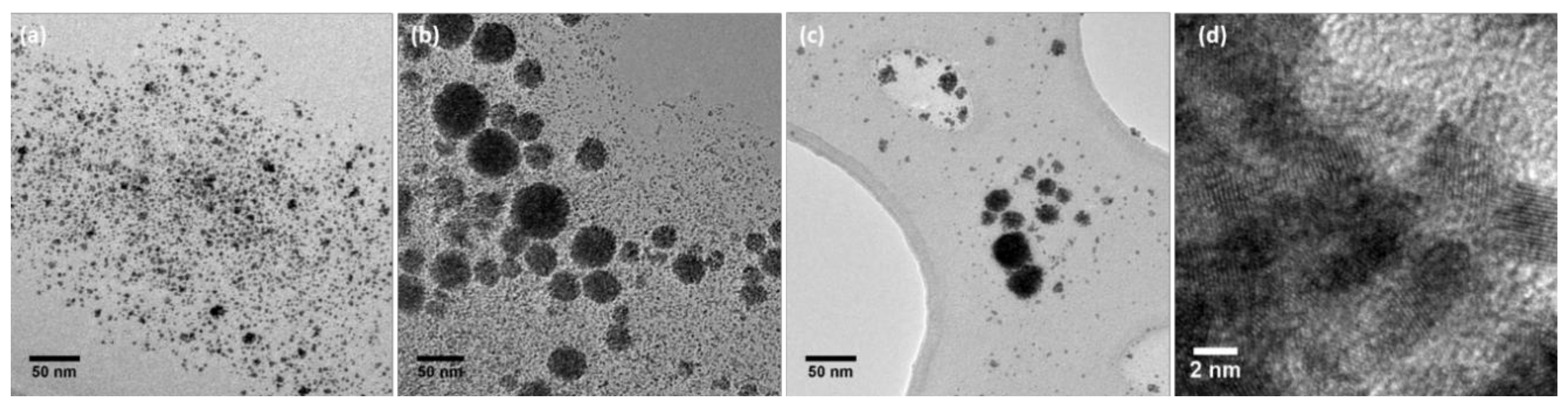
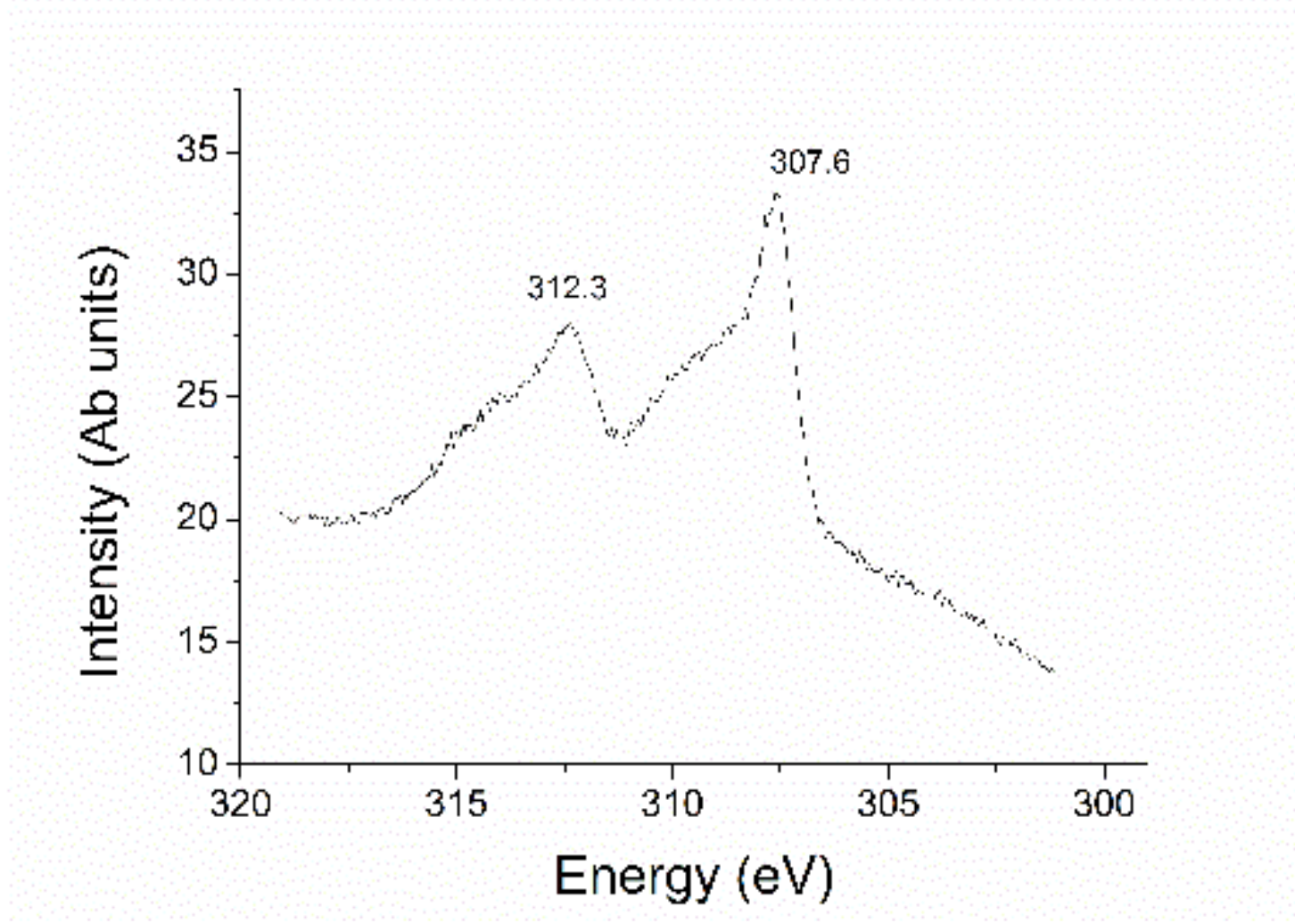
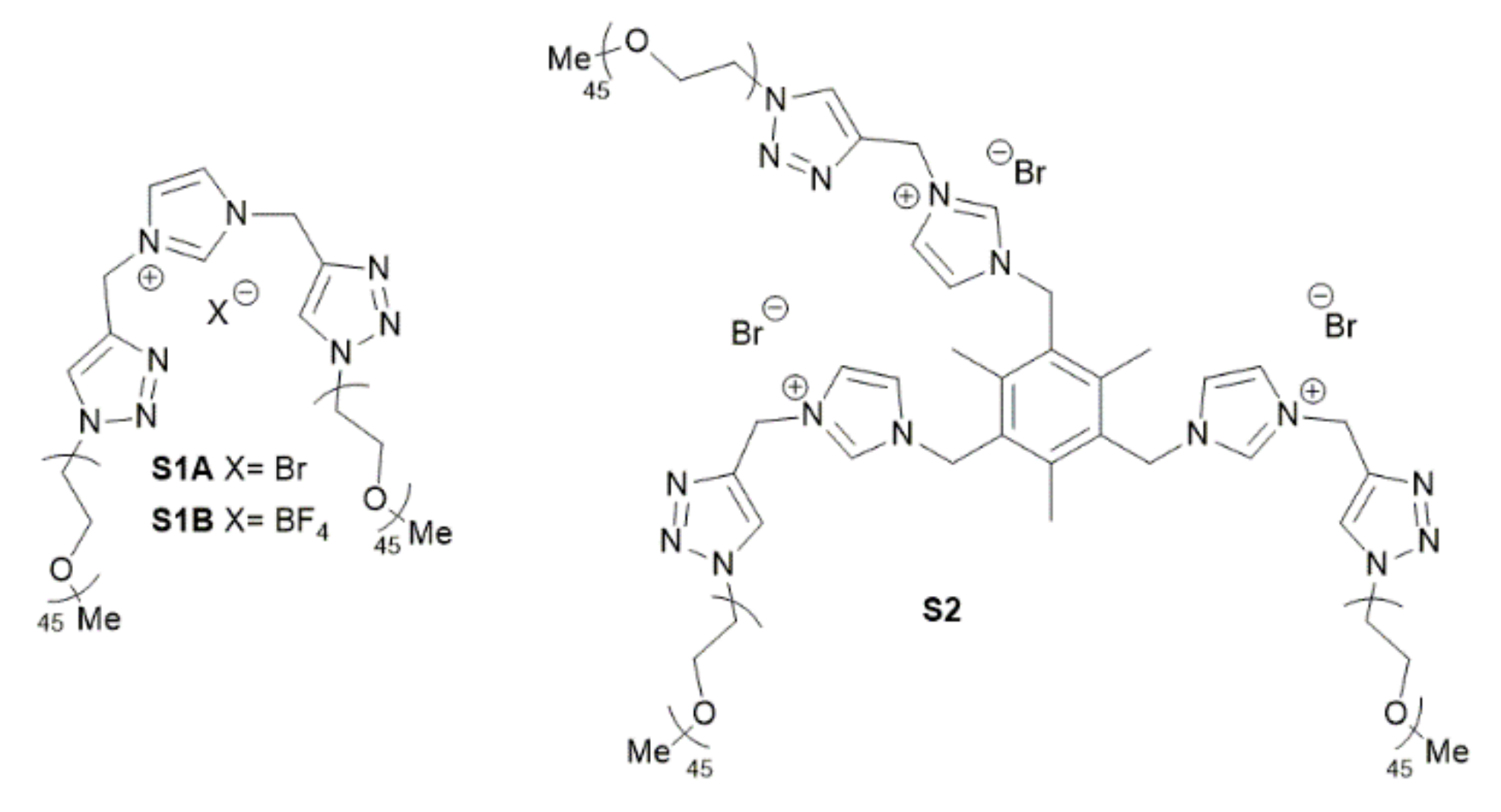
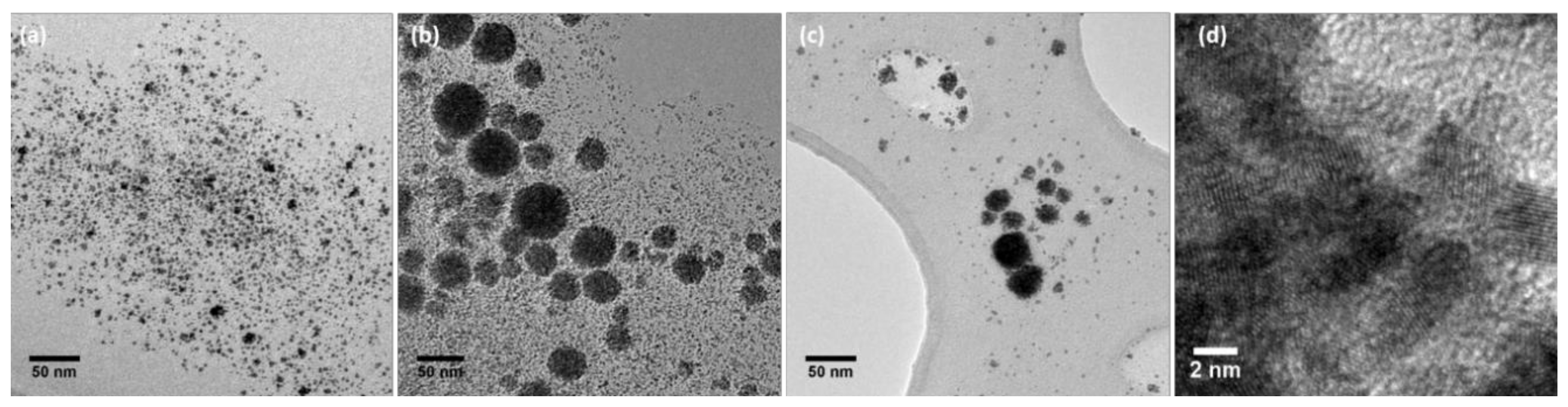
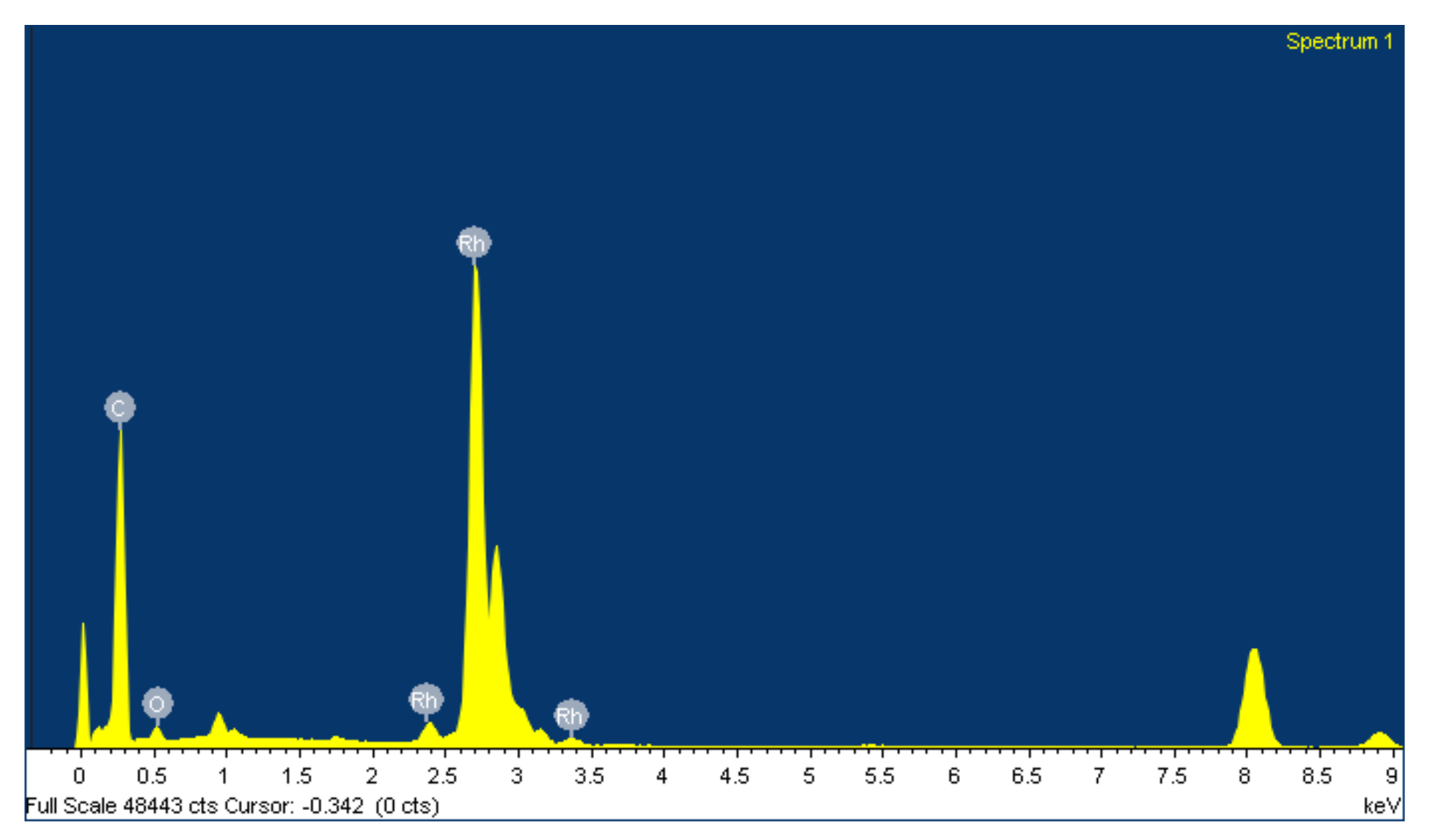
2.1. Preparation and Characterization of Rhodium Nanoparticles

2.2. Hydrosilylation of Alkynes under Catalysis by Rhodium Nanoparticles




2.3. Reduction of Nitroarenes under Catalysis by Rhodium Nanoparticles




3. Materials and Methods
3.1. General Remarks
3.2. General Procedure for the Preparation of Rh NPs (M1A, Table 1, Entry 1)
3.3. General Procedure for the Hydrosilylation of Alkynes by Rh NPs under Neat Conditions (Method A)
3.4. General Procedure for the Hydrosilylation of Alkynes by Rh NPs in THF (Method B)
3.5. General Procedure for the Reduction of Nitroarenes by Rh NPs with Ammonia Borane
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Astruc, D. (Ed.) Nanoparticles and Catalysis; Wiley-VCH: Weinheim, Germany, 2008; pp. 1–640. [Google Scholar]
- Roucoux, A.; Schulz, J.; Patin, H. Reduced Transition Metal Colloids: A Novel Family of Reusable Catalysts? Chem. Rev. 2002, 102, 3757–3778. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Mañas, M.; Pleixats, R. Formation of Carbon-Carbon Bonds under Catalysis by Transition-Metal Nanoparticles. Acc. Chem. Res. 2003, 36, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Widegren, J.A.; Finke, R.G. A review of soluble transition-metal nanoclusters as arene hydrogenation catalysts. J. Mol. Catal. A Chem. 2003, 191, 187–207. [Google Scholar] [CrossRef]
- Astruc, D.; Lu, F.; Ruiz Aranzaes, J. Nanoparticles as recyclable catalysts: The frontier between homogeneous and heterogeneous catalysis. Angew. Chem. Int. Ed. 2005, 44, 7852–7872. [Google Scholar] [CrossRef]
- De Vries, J.G. A unifying mechanism for all high-temperature Heck reactions. The role of palladium colloids and anionic species. Dalton Trans. 2006, 421–429. [Google Scholar] [CrossRef]
- Astruc, D. Palladium nanoparticles as efficient green homogeneous and heterogeneous carbon-carbon coupling precatalysts: A unifying view. Inorg. Chem. 2007, 46, 1884–1894. [Google Scholar] [CrossRef]
- Roucoux, A.; Philippot, K. Homogeneous Hydrogenation. Colloids. Hydrogenation with Noble Metal Nanoparticles. In Handbook of Homogeneous Hydrogenation; De Vries, J.G., Elsevier, C.J., Eds.; Wiley-VCH: Weinheim, Germany, 2007; Volume 9, pp. 217–255. [Google Scholar]
- Corma, A.; García, H. Supported gold nanoparticles as catalysts for organic reactions. Chem. Rev. 2008, 37, 2096–2126. [Google Scholar] [CrossRef]
- Roy, S.; Pericàs, M.A. Functionalized nanoparticles as catalysts for enantioselective processes. Org. Biomol. Chem. 2009, 7, 2669–2677. [Google Scholar] [CrossRef]
- Starkey Ott, L.; Finke, R.G. Transition-metal nanocluster stabilization for catalysis: A critical review of ranking methods and putative stabilizers. Coord. Chem. Rev. 2007, 251, 1075–1100. [Google Scholar]
- Guerrero, M.; Than Chau, N.T.; Noël, S.; Denicourt-Nowicki, A.; Hapiot, F.; Roucoux, A.; Monflier, E.; Philippot, K. About the Use of Rhodium Nanoparticles in Hydrogenation and Hydroformylation Reactions. Curr. Org. Chem. 2013, 17, 364–399. [Google Scholar] [CrossRef]
- Roucoux, A.; Nowicki, A.; Philippot, K. Rhodium and Ruthenium Nanoparticles in Catalysis. In Nanoparticles and Catalysis; Astruc, D., Ed.; Wiley-VCH: Weinheim, Germany, 2008; pp. 349–388, Chapter 11. [Google Scholar]
- Yuan, Y.; Yan, N.; Dyson, P.J. Advances in the Rational Design of Rhodium Nanoparticle Catalysts: Control via Manipulation of the Nanoparticle Core and Stabilizer. ACS Catal. 2012, 2, 1057–1069. [Google Scholar] [CrossRef]
- Kadam, H.K.; Tilve, S.G. Advancements in methodologies for reduction of nitroarenes. RSC Adv. 2015, 5, 83391–83407. [Google Scholar] [CrossRef]
- Zhao, P.; Feng, X.; Huang, D.; Yang, G.; Astruc, D. Basic concepts and recent advances in nitrophenol reduction by gold- and other transition metal nanoparticles. Coord. Chem. Rev. 2015, 287, 114–136. [Google Scholar] [CrossRef]
- Aditya, T.; Pal, A.; Pal, T. Nitroarene reduction: A trusted model reaction to test nanoparticle catalysts. Chem. Commun. 2015, 51, 9410–9431. [Google Scholar] [CrossRef] [PubMed]
- Serna, P.; Corma, A. Transforming Nano Metal Nonselective Particulates into Chemoselective Catalysts for Hydrogenation of Substituted Nitrobenzenes. ACS Catal. 2015, 5, 7114–7121. [Google Scholar] [CrossRef]
- Nakamula, I.; Yamanoi, Y.; Yonezawa, T.; Imaoka, T.; Yamamoto, K.; Nishihara, H. Nanocage catalysts-rhodium nanoclusters encapsulated with dendrimers as accessible and stable catalysts for olefin and nitroarene hydrogenations. Chem. Commun. 2008, 5716–5718. [Google Scholar] [CrossRef]
- Nakamula, I.; Yamanoi, Y.; Imaoka, T.; Yamamoto, K.; Nishihara, H. A Uniform Bimetallic Rhodium/Iron Nanoparticle Catalyst for the Hydrogenation of Olefins and Nitroarenes. Angew. Chem. Int. Ed. 2011, 50, 5830–5833. [Google Scholar] [CrossRef]
- Corma, A.; Serna, P. Chemoselective Hydrogenation of Nitro Compounds with Supported Gold Catalysts. Science 2006, 313, 332–334. [Google Scholar] [CrossRef]
- Chen, Y.; Qiu, J.; Wang, X.; Xiu, J. Preparation and application of highly dispersed gold nanoparticles supported on silica for catalytic hydrogenation of aromatic nitro compounds. J. Catal. 2006, 242, 227–230. [Google Scholar] [CrossRef]
- Lara, P.; Suárez, A.; Collière, V.; Philippot, K.; Chaudret, B. Platinum N-Heterocyclic Carbene Nanoparticles as New and Effective Catalysts for the Selective Hydrogenation of Nitroaromatics. ChemCatChem 2014, 6, 87–90. [Google Scholar] [CrossRef]
- Lara, P.; Philippot, K. The hydrogenation of nitroarenes mediated by platinum nanoparticles: An overview. Catal. Sci. Technol. 2014, 4, 2445–2465. [Google Scholar] [CrossRef]
- Wu, H.; Zhuo, L.; He, Q.; Liao, X.; Shi, B. Heterogeneous hydrogenation of nitrobenzenes over recyclable Pd(0) nanoparticles catalysts stabilized by polyphenol-grafted collagen fibers. Appl. Catal. A Gen. 2009, 366, 44–56. [Google Scholar] [CrossRef]
- Li, J.; Shi, X.-Y.; Bi, Y.-Y.; Wei, J.-F.; Chen, Z.-G. Pd Nanoparticles in Ionic Liquid Brush: A Highly Active and Reusable Heterogeneous Catalytic Assembly for Solvent-Free or On-Water Hydrogenation of Nitroarene under Mild Conditions. ACS Catal. 2011, 1, 657–664. [Google Scholar] [CrossRef]
- Liu, W.-J.; Tian, K.; Jiang, H. One-pot synthesis of Ni-NiFe2O4/carbon nanofiber composites from biomass for selective hydrogenation of aromatic nitro compounds. Green Chem. 2015, 17, 821–826. [Google Scholar] [CrossRef]
- Pisiewicz, S.; Formenti, D.; Surkus, A.-E.; Pohl, M.-M.; Radnik, J.; Junge, K.; Topf, C.; Bachmann, S.; Scalone, M.; Beller, M. Synthesis of Nickel Nanoparticles with N-Doped Graphene Shells for Catalytic Reduction Reactions. ChemCatChem 2016, 8, 129–134. [Google Scholar] [CrossRef]
- Huang, L.; Luo, P.; Pei, X.; Liu, X.; Wang, Y.; Wang, J.; Xing, W.; Huang, J. Selective Hydrogenation of Nitroarenes and Olefins over Rhodium Nanoparticles on Hydroxyapatite. Adv. Synth. Catal. 2012, 354, 2689–2694. [Google Scholar] [CrossRef]
- Luo, P.; Xu, K.; Zhang, R.; Huang, L.; Wang, J.; Xing, W.; Huang, J. Highy efficient and selective reduction of nitroarenes with hydrazine over supported rhodium nanoparticles. Catal. Sci. Technol. 2012, 2, 301–304. [Google Scholar] [CrossRef]
- Ganji, S.; Sankar Enumula, S.; Kumar Marella, R.; Seetha Rama Rao, K.; Raju Burri, D. RhNPs/SBA-NH2: A high-performance catalyst for aqueous phase reduction of nitroarenes to aminoarenes at room temperature. Catal. Sci. Technol. 2014, 4, 1813–1819. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Y.; Sun, H.-B.; Tang, Z.; Qi, L.; Liu, L.; Ai, Y.; Li, S.; Shao, Z.; Liang, Q. Porous silica-encapsulated and magnetically recoverable Rh NPs: A highly efficient, stable and green catalyst for catalytic transfer hydrogenation with “slow-release” of stoichiometric hydrazine in water. Green Chem. 2017, 19, 3400–3407. [Google Scholar] [CrossRef]
- Guo, W.; Pleixats, R.; Shafir, A. Water-Soluble Gold Nanoparticles: From Catalytic Selective Nitroarene Reduction in Water to Refractive Index Sensing. Chem. Asian J. 2015, 10, 2437–2443. [Google Scholar] [CrossRef] [Green Version]
- Fernández, G.; Sort, J.; Pleixats, R. Nickel Nanoparticles Stabilized by Tris-imidazolium Salts: Synthesis, Characterization and Application as Recyclable Catalysts for the Reduction of Nitroarenes. ChemistrySelect 2018, 3, 8597–8603. [Google Scholar] [CrossRef]
- Trost, B.M.; Ball, Z.T.; Töge, T. A Chemoselective Reduction of Alkynes to (E)-Alkenes. J. Am. Chem. Soc. 2002, 124, 7922–7923. [Google Scholar] [CrossRef] [PubMed]
- May, T.L.; Dabrowski, J.A.; Hoveyda, A.H. Formation of vinyl-, vinylhalide- or acyl-substituted quaternary carbon stereogenic centers through NHC-Cu-catalyzed enantioselective conjugate additions of Si-containing vinylaluminums to β-substituted cyclic enones. J. Am. Chem. Soc. 2011, 133, 736–739. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.R.; Landais, Y. The oxidation of the carbon-silicon bond. Tetrahedron 1996, 52, 7599–7662. [Google Scholar] [CrossRef]
- Nakao, Y.; Hiyama, T. Silicon-based cross-coupling reaction: An environmentally benign version. Chem. Soc. Rev. 2011, 40, 4893–4901. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, B. Catalysis of hydrosilylation of carbon-carbon multiple bonds: Recent progress. Silicon Chem. 2002, 1, 155–174. [Google Scholar] [CrossRef]
- Marciniec, B.; Maciejewski, H.; Pietraszuk, C.; Pawluc, P. Hydrosilylation of Alkynes and Their Derivatives. In Hydrosilylation: A Comprehensive Review on Recent Advances (Advances in Silicon Science Series); Marciniec, B., Ed.; Springer: Berlin, Germany, 2009; Volume 1, Chapter 2. [Google Scholar]
- Bali, Z.T. C-E Bond Formation through Hydrosilylation of Alkynes and Related Reactions. In Comprehensive Organometallic Chemistry III; Mingos, D.M.P., Crabtree, R.H., Eds.; Elsevier Ltd.: Oxford, UK, 2007; Volume 10, pp. 789–813. [Google Scholar]
- Dierick, S.; Markó, I.E. NHC Platinum(0) Complexes: Unique Catalysts for the Hydrosilylation of Alkenes and Alkynes. In N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis; Nolan, S.P., Ed.; Wiley-VCH: Weinheim, Germany, 2014; pp. 111–149, Chapter 5. [Google Scholar]
- Sun, J.; Deng, L. Cobalt Complex-Catalyzed Hydrosilylation of Alkenes and Alkynes. ACS Catal. 2016, 6, 290–300. [Google Scholar] [CrossRef]
- Zaranek, M.; Marciniec, B.; Pawluc, P. Ruthenium-catalyzed hydrosilylation of carbon-carbon multiple bonds. Org. Chem. Front. 2016, 3, 1337–1344. [Google Scholar] [CrossRef]
- Sanada, T.; Kato, T.; Mitani, M.; Mori, A. Rhodium-Catalyzed Hydrosilylation of Internal Alkynes with Silane Reagents bearing Heteroatom Substituents. Studies on the Regio-/Stereochemistry and Transformation of the Produced Alkenylsilanes by Rhodium-Catalyzed Conjugate Addition. Adv. Synth. Catal. 2006, 348, 51–54. [Google Scholar] [CrossRef]
- Field, L.D.; Ward, A.J. Catalytic hydrosilylation of acetylenes mediated by phosphine complexes of cobalt(I), rhodium(I), and iridium(I). J. Organomet. Chem. 2003, 681, 91–97. [Google Scholar] [CrossRef]
- Sato, A.; Kinoshita, H.; Shinokubo, H.; Oshima, K. Hydrosilylation of Alkynes with a Cationic Rhodium Species Formed in an Anionic Micellar System. Org. Lett. 2004, 6, 2217–2220. [Google Scholar] [CrossRef]
- Nagashima, H.; Tatebe, K.; Ishibashi, T.; Sakakibara, J.; Itoh, K. (PPh3)3RhCl-catalyzed hydrosilylation of unsaturated molecules by 1,2-bis(dimethylsilyl)ethane: Unprecedented rate difference between two silicon-hydrogen bonds. Organometallics 1989, 8, 2495–2496. [Google Scholar] [CrossRef]
- Cassani, M.C.; Brucka, M.A.; Femoni, C.; Mancinelli, M.; Mazzanti, A.; Mazzoni, R.; Solinas, G. N-Heterocyclic carbene rhodium(I) complexes containing an axis of chirality: Dynamics and catalysis. N. J. Chem. 2014, 38, 1768–1779. [Google Scholar] [CrossRef]
- Rivera, G.; Elizalde, O.; Roa, G.; Montiel, I.; Bernès, S. Fluorinated N-heterocyclic carbenes rhodium(I) complexes and their activity in hydrosilylation of propargylic alcohols. J. Organomet. Chem. 2012, 699, 82–86. [Google Scholar] [CrossRef]
- Solomonsz, W.A.; Rance, G.A.; Khlobystov, A.N. Evaluating the Effects of Carbon Nanoreactor Diameter and Internal Structure on the Pathways of the Catalytic Hydrosilylation Reaction. Small 2014, 10, 1866–1872. [Google Scholar] [CrossRef] [PubMed]
- Alonso, F.; Buitrago, R.; Moglie, Y.; Ruiz-Martínez, J.; Sepúlveda-Escribano, A.; Yus, M. Hydrosilylation of alkynes catalysed by platinum on titania. J. Organomet. Chem. 2011, 696, 368–372. [Google Scholar] [CrossRef] [Green Version]
- Alonso, F.; Buitrago, R.; Moglie, Y.; Sepúlveda-Escribano, A.; Yus, M. Selective Hydrosilylation of 1,3-Diynes Catalyzed by Titania-Supported Platinum. Organometallics 2012, 31, 2336–2342. [Google Scholar] [CrossRef]
- Cano, R.; Yus, M.; Ramón, D.J. Impregnated Platinum on Magnetite as an Efficient, Fast, and Recyclable Catalyst for the Hydrosilylation of Alkynes. ACS Catal. 2012, 2, 1070–1078. [Google Scholar] [CrossRef]
- Solomonsz, W.A.; Rance, G.A.; Harris, B.J.; Khlobystov, A.N. Competitive hydrosilylation in carbon nanoreactors: Probing the effect of nanoscale confinement on selectivity. Nanoscale 2013, 5, 12200–12205. [Google Scholar] [CrossRef]
- Hu, W.; Xie, H.; Yue, H.; Prinsen, P.; Luque, R. Super-microporous silica-supported platinum catalyst for highly regioselective hydrosilylation. Catal. Commun. 2017, 97, 51–55. [Google Scholar] [CrossRef]
- Rivero-Crespo, M.A.; Leyva-Pérez, A.; Corma, A. A Ligand-Free Pt3 Cluster Catalyzes the Markovnikov Hydrosilylation of Alkynes with up to 106 Turnover Frequencies. Chem. Eur. J. 2017, 23, 1702–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chauhan, B.P.S.; Sarkar, A. Functionalized vinylsilanes via highly efficient and recyclable Pt-nanoparticle catalysed hydrosilylation of alkynes. Dalton Trans. 2017, 46, 8709–8715. [Google Scholar] [CrossRef] [PubMed]
- Duke, B.J.; Akeroyd, E.N.; Bhatt, S.V.; Onyeagusi, C.I.; Bhatt, S.V.; Adolph, B.R.; Fotie, J. Nano-dispersed platinum(0) in organically modified silicate matrices as sustainable catalysts for a regioselective hydrosilylation of alkenes and alkynes. N. J. Chem. 2018, 42, 11782–11795. [Google Scholar] [CrossRef]
- Fernández, G.; Pleixats, R. Soluble Pt Nanoparticles Stabilized by a Tris-Imidazolium Tetrafluoroborate as Efficient and Recyclable Catalyst for the Stereoselective Hydrosilylation of Alkynes. ChemistrySelect 2018, 3, 11486–11493. [Google Scholar] [CrossRef]
- Planellas, M.; Guo, W.; Alonso, F.; Yus, M.; Shafir, A.; Pleixats, R.; Parella, T. Hydrosilylation of Internal Alkynes Catalyzed by Tris-Imidazolium Salt-Stabilized Palladium Nanoparticles. Adv. Synth. Catal. 2014, 356, 179–188. [Google Scholar] [CrossRef]
- Duan, Y.; Ji, G.; Zhang, S.; Chen, X.; Yang, Y. Additive-modulated switchable reaction pathway in the addition of alkynes with organosilanes catalyzed by supported Pd nanoparticles: Hydrosilylation versus semihydrogenation. Cat. Sci. Technol. 2018, 8, 1039–1050. [Google Scholar] [CrossRef]
- Solomonsz, W.A.; Rance, G.A.; Suyetin, M.; La Torre, A.; Bichoutskaia, E.; Khlobystov, A.N. Controlling the Regioselectivity of the Hydrosilylation Reaction in Carbon Nanoreactors. Chem. Eur. J. 2012, 18, 13180–13187. [Google Scholar] [CrossRef]
- Psyllaki, A.; Lykakis, I.N.; Stratakis, M. Reaction of hydrosilanes with alkynes catalyzed by gold nanoparticles supported on TiO2. Tetrahedron 2012, 68, 8724–8731. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Yamamoto, Y.; Asao, N. Selective hydrosilylation of alkynes with a nanoporous gold catalyst. Catal. Sci. Technol. 2013, 3, 2902–2905. [Google Scholar] [CrossRef]
- Guo, W.; Pleixats, R.; Shafir, A.; Parella, T. Rhodium Nanoflowers Stabilized by a Nitrogen-Rich PEG-Tagged Substrate as Recyclable Catalyst for the Stereoselective Hydrosilylation of Internal Alkynes. Adv. Synth. Catal. 2015, 357, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Fernández, G.; Bernardo, L.; Villanueva, A.; Pleixats, R. Gold nanoparticles stabilized by PEG-tagged imidazolium salts as recyclable catalysts for the synthesis of propargylamines and the cycloisomerization of γ-alkynoic Acids. N. J. Chem. 2020, 44, 6130–6141. [Google Scholar] [CrossRef]
- Yao, Q.; Lu, Z.-H.; Jia, Y.; Chen, X.; Liu, X. In situ facile synthesis of Rh nanoparticles supported on carbon nanotubes as highly active catalysts for H2 generation from NH3BH3 hydrolysis. Int. J. Hydrogen Energy 2015, 40, 2207–2215. [Google Scholar] [CrossRef]
- Gopiraman, M.; Saravanamooorthy, S.; Ullah, S.; Ilangovan, A.; Kim, I.S.; Chung, I.M. Reducing-agent-free facile preparation of Rh nanoparticles uniformly anchored on onion-like fullerene for catalytic applications. RSC Adv. 2020, 10, 2545–2559. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhang, J.; Liu, H.; Qiu, J.; Zhang, X. Heterogeneous Ligand-Free Rhodium Oxide Catalyst Embedded within Zeolitic Microchannel to Enhance Regioselectivity in Hydroformylation. Ind. Eng. Chem. Res. 2019, 58, 21285–21295. [Google Scholar] [CrossRef]
- Zahmakiran, M.; Özkar, S. Transition Metal Nanoparticles in Catalysis for the Hydrogen Generation from the Hydrolysis of Ammonia-Borane. Top. Catal. 2013, 56, 1171–1183. [Google Scholar] [CrossRef]
- Zhao, T.-J.; Zhang, Y.-N.; Wang, K.-X.; Su, J.; Wei, X.; Li, X.-H. General transfer hydrogenation by activating ammonia-borane over cobalt nanoparticles. RSC Adv. 2015, 5, 102736–102740. [Google Scholar] [CrossRef]
- Wang, W.; Ciganda, R.; Wang, C.; Escobar, A.; Martínez-Villacorta, A.M.; Ramírez, M.A.; Hernández, R.; Moya, S.E.; Ruiz, J.; Hamon, J.-R.; et al. High catalytic activity of Rh nanoparticles generated from cobaltocene and RhCl3 in aqueous solution. Inorg. Chem. Front. 2019, 6, 2704–2708. [Google Scholar] [CrossRef]
- Akbayrak, S.; Tonbul, Y.; Özkar, S. Magnetically Separable Rh0/Co3O4 Nanocatalyst Provides over a Million Turnovers in Hydrogen Release from Ammonia Borane. ACS Sustain. Chem. Eng. 2020, 8, 4216–4224. [Google Scholar] [CrossRef]
- Luo, W.; Zhao, X.; Cheng, W.; Zhang, Y.; Wang, Y.; Fan, G. A simple and straightforward strategy for synthesis of N, P co-doped porous carbon: An efficient support for Rh nanoparticles for dehydrogenation of ammonia borane and catalytic application. Nanoscale Adv. 2020, 2, 1685–1693. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
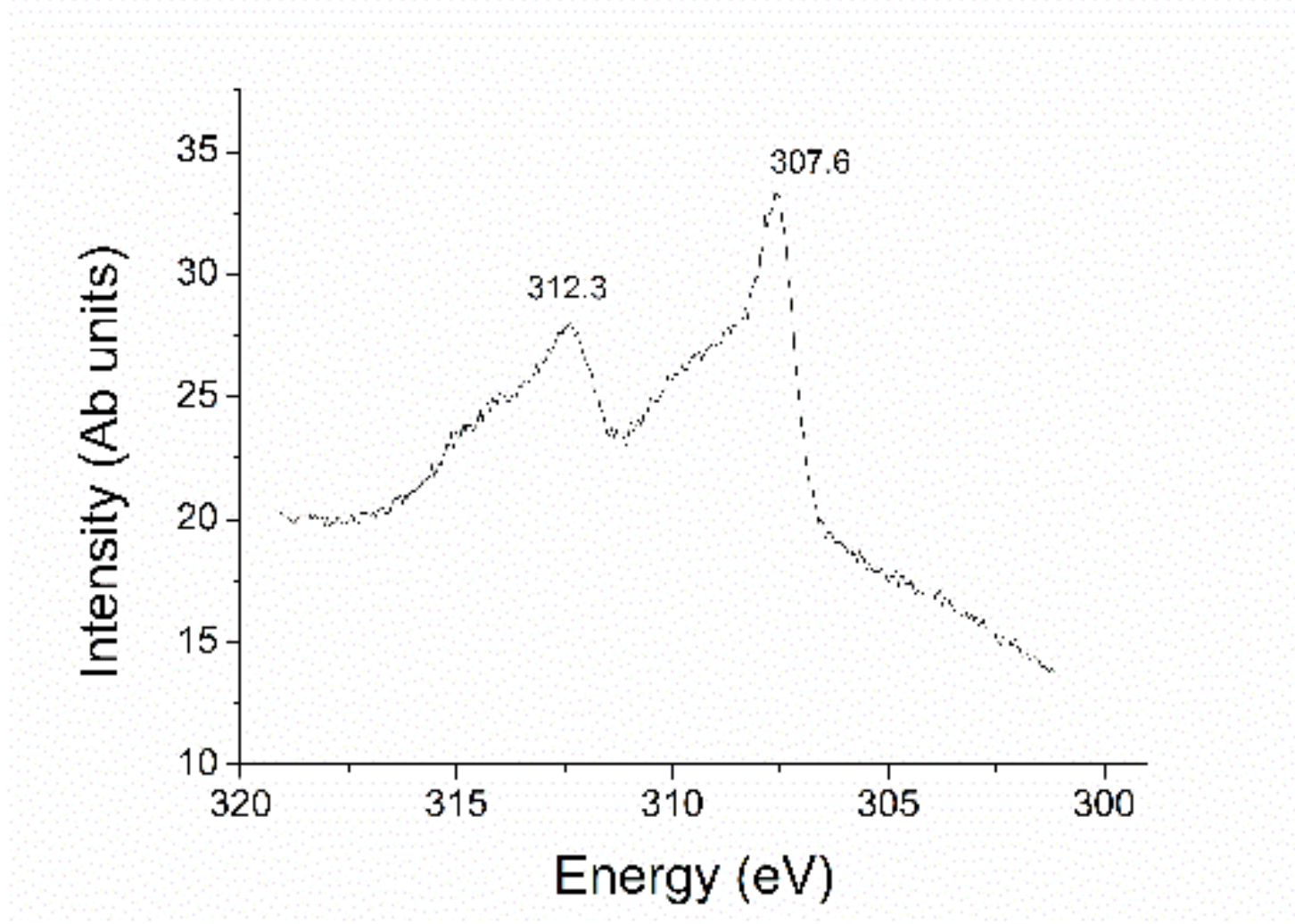{kind=link}
| Entry | S | S/Rh/NaBH4 | [Rh] (mM) | Diameter (nm) 1,2 | % Rh | Yield (%) 3 | Nanomaterial | |
|---|---|---|---|---|---|---|---|---|
| Theor | Expt | |||||||
| 1 | S1A | 0.02/1/14 | 1 | 2.9 ± 3 | 30.3 | 31.7 | 40 | M1A |
| 2 | S1B | 0.02/1/14 | 1 | 3.3 ± 2 | 30.3 | 29.9 | 27 | M1B |
| 3 | S2 | 0.025/1/18 | 0.8 | 5.6 ± 3 | 24.2 | 27.4 | 41 | M2 |
 | hlk | dhlk (nm) | |
| Exper. | Theor. [a] | ||
| (111) | 0.2186 | 0.2196 | |
| (200) | 0.1907 | 0.1902 | |
| (220) | 0.1340 | 0.1345 | |
| (311) | 0.1157 | 0.1147 | |
| Entry | Catalyst | Product | Method A 1 | Method B 2 | ||
|---|---|---|---|---|---|---|
| Time (h) | Yield (%) 3 | Time (h) | Yield (%) 3 | |||
| 1 | M1A | 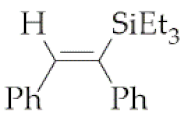 1 1 | 16 | 76 | 24 | 79 |
| 2 | M1B | 16 | 84 | - 4 | - 4 | |
| 3 | M2 | 20 | 68 | - 4 | - 4 | |
| 4 | M1A |  2 2 | 16 | 84 | - 4 | - 4 |
| 5 | M1B | 16 | 66 | - 4 | - 4 | |
| 6 | M2 | 16 | 78 | - 4 | - 4 | |
| 7 | M1A | 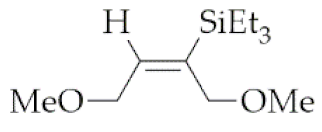 3 3 | 16 | 68 | 24 | 97 |
| 8 | M1B | 16 | 56 | - 4 | - 4 | |
| 9 | M2 | - 4 | - 4 | 24 | 90 | |
| 10 | M1A | 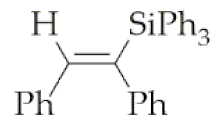 4 4 | - 4 | - 4 | 24 | 86 |
| Entry 1 | Catalyst | Products | Time (h) | Yield (%) 2 | 5:6 3 |
|---|---|---|---|---|---|
| 1 | M1A | 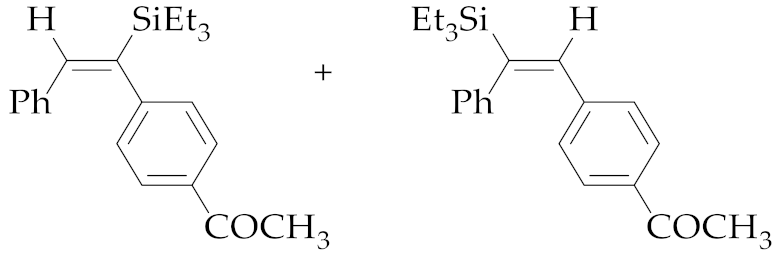 (E)-5a + (E)-6a | 16 | 98 | 65:35 |
| 2 | M1B | 16 | 73 | 68:32 | |
| 3 | M2 | 16 | 85 | 64:36 | |
| 4 | M1A | 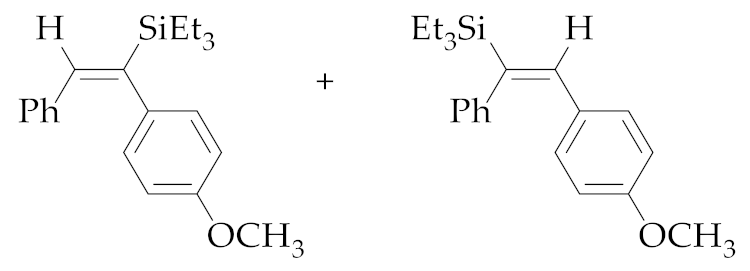 (E)-5b + (E)-6b | 48 | 85 | 43:57 |
| 5 | M1B | 24 | 97 | 44:56 | |
| 6 | M2 | 24 | 92 | 36:64 |
| Entry | Cat | Products | Method A 1 | Method B 2 | ||||
|---|---|---|---|---|---|---|---|---|
| t (h) | Yield (%) 3 | 7:8 4 | t (h) | Yield (%) 3 | 7:8 4 | |||
| 1 | M1A | 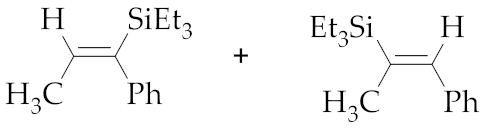 (E)-7a + (E)-8a | 15 | 69 | 76:24 | 24 | 98 | 76:24 |
| 2 | M1B | 16 | 75 | 77:23 | 24 | 94 | 77:23 | |
| 3 | M2 | - 5 | - 5 | - | 24 | 99 | 78:22 | |
| 4 | M1A | 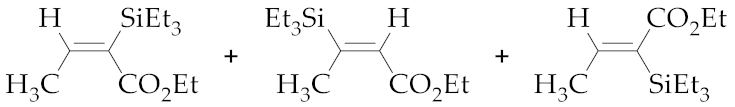 (E)-7b + (E)-8b + (Z)-7b | - 5 | - 5 | - | 24 | 80 | 11:15:74 |
| 5 | M1B | - 5 | - 5 | - | 24 | 85 | 10:19:71 | |
| 6 | M2 | - 5 | - 5 | - | 24 | 79 | 14:15:71 | |
| 7 | M1A |  (E)-7c + 8c + (Z)-7c | 20 | 85 | 65:25:10 | - 5 | - 5 | - |
| 8 | M1B | 20 | 79 | 59:30:11 | - 5 | - 5 | - | |
| 9 | M1A | 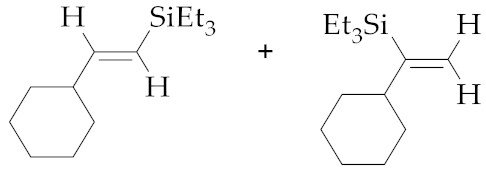 (E)-7d + 8d | 23 | 36 | 80:20 | - 5 | - 5 | - |
| 10 | M1B | 23 | 47 | 80:20 | - 5 | - 5 | - | |
| 11 | M1A | 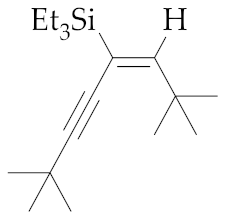 (E)-9 | 16 | 99 | - | - 5 | - 5 | - |
| 12 | M1B | 16 | 64 | - | - 5 | - 5 | - | |
| Catalyst | Cycle 1 | (E)-7a:(E)-8a 2 | Yield (%) 3 |
|---|---|---|---|
| M1A | 1 | 76:24 | 98 |
| 2 | 75:25 | 91 | |
| 3 | 76:24 | 99 | |
| 4 | 75:25 | 84 | |
| 5 | 78:22 | 80 | |
| M1B | 1 | 77:23 | 94 |
| 2 | 77:23 | 93 | |
| 3 | 78:22 | 97 | |
| 4 | 79:21 | 94 | |
| 5 | 78:22 | 85 | |
| M2 | 1 | 76:24 | 80 |
| 2 | 78:22 | 99 | |
| 2 | 79:21 | 99 | |
| 4 | 78:22 | 92 | |
| 5 | 77:23 | 86 |
| Entry 1 | Cat. | Product | ArNH2/ArNO2 2 | Time (h) | Yield (%) 3 |
|---|---|---|---|---|---|
| 1 4 | M2 | 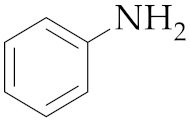 10 10 | >99:1 | 0.25 | 92 |
| 2 | M2 | 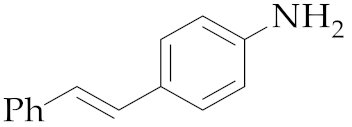 11 11 | >99:1 | 20 | 91 5 |
| 3 | M1A | >99:1 | 18 | 85 | |
| 4 | M1B | >99:1 | 18 | 77 6 | |
| 5 | M2 |  12 12 | >99:1 | 0.83 | 81 |
| 6 | M2 | 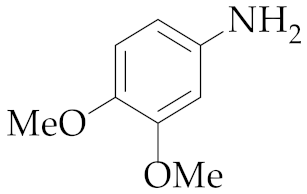 13 13 | >99:1 | 0.83 | 89 |
| 7 | M1A | >99:1 | 0.83 | 92 | |
| 8 | M1B | >99:1 | 0.83 | 48 | |
| 9 | M2 | 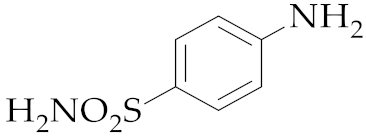 14 14 | >99:1 | 0.83 | 58 |
| 10 | M2 | 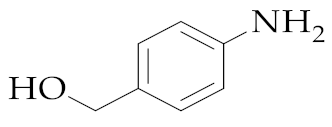 157 157 | >99:1 | 1 | 64 |
| 11 | M1A | >99:1 | 1 | 72 | |
| 12 | M1B | >99:1 | 1 | 55 | |
| 13 | M2 | 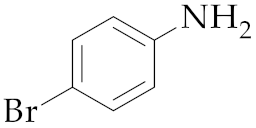 16 16 | >99:1 | 1 | 76 |
| 14 | M2 | 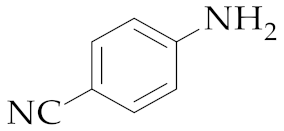 17 17 | >99:1 | 18 | 85 |
| 15 | M2 | 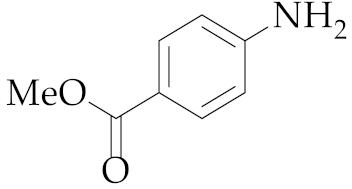 18 18 | >99:1 | 1 | 66 |
| 16 | M2 | 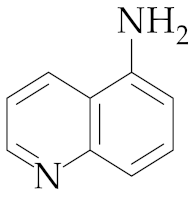 19 19 | >99:1 | 1 | 83 |
| 17 | M2 | 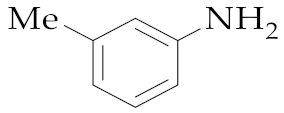 20 20 | >99:1 | 18 | 53 |
| 18 | M2 |  218 218 | >99:1 | 2 | 78 |
| 19 | M2 | 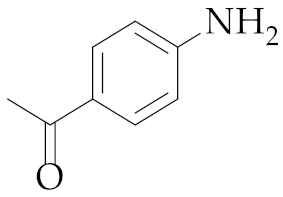 22 22 | >99:1 | 1 | 64 9 |
| 20 | M1A | 69:31 | 1 | 56 9 | |
| 21 | M1B | 82:18 | 1 | 71 9 |
| Cycle 1 | Time (h) | Yield (%) 2 |
|---|---|---|
| 1 | 0.83 | 92 |
| 2 | 1.25 | 88 |
| 3 | 18 | 90 |
| 4 | 18 | 85 |
| 5 | 24 | 87 |
| Entry 1 | Cat. | Product | ArNH2/ArNO2 2 | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | M2 | 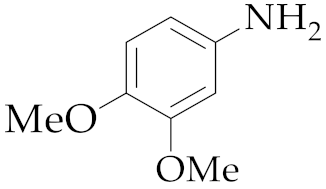 13 13 | >99:1 | 0.83 | 66 3 |
| 2 | M2 | 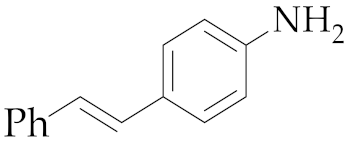 11 11 | >99:1 | 24 | 77 4 |
| 3 | M1A | >99:1 | 18 | 92 5 | |
| 4 | M1B | >99:1 | 18 | 70 6 | |
| 5 | M2 | 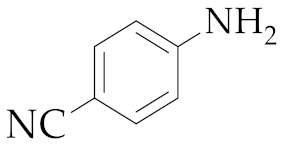 17 17 | >99:1 | 18 | 83 3 |
| 6 | M2 | 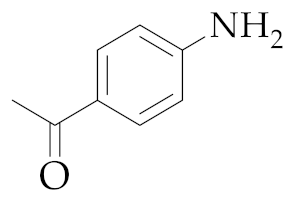 22 22 | >99:1 | 0.5 | - 7 |
| 7 | M1A | 69:31 | 1 | 58 8 | |
| 8 | M1B | >99:1 | 1 | 10 9 |
| Entry 1 | Cat. | Product | ArNH2/ArNO2 2 | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | M2 | 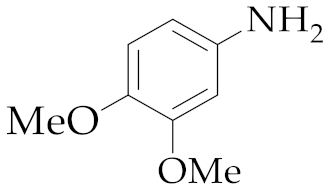 13 13 | >99:1 | 16 | 70 3 |
| 2 | M1A | 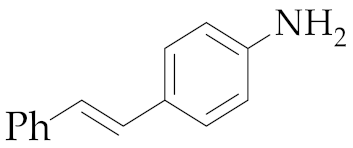 11 11 | 62:38 | 48 | 48 4 |
| 3 | M1B | 29:71 | 48 | 23 4 | |
| 4 | M2 | 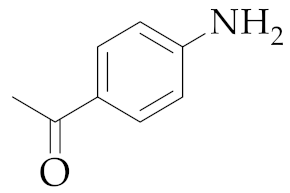 22 22 | >99:1 | 2 | - 5 |
| 5 | M1A | >99:1 | 24 | - 5 | |
| 6 | M1B | 87:13 | 24 | - 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernández, G.; Pleixats, R. Rhodium Nanoparticles Stabilized by PEG-Tagged Imidazolium Salts as Recyclable Catalysts for the Hydrosilylation of Internal Alkynes and the Reduction of Nitroarenes. Catalysts 2020, 10, 1195. https://doi.org/10.3390/catal10101195
Fernández G, Pleixats R. Rhodium Nanoparticles Stabilized by PEG-Tagged Imidazolium Salts as Recyclable Catalysts for the Hydrosilylation of Internal Alkynes and the Reduction of Nitroarenes. Catalysts. 2020; 10(10):1195. https://doi.org/10.3390/catal10101195
Chicago/Turabian StyleFernández, Guillem, and Roser Pleixats. 2020. "Rhodium Nanoparticles Stabilized by PEG-Tagged Imidazolium Salts as Recyclable Catalysts for the Hydrosilylation of Internal Alkynes and the Reduction of Nitroarenes" Catalysts 10, no. 10: 1195. https://doi.org/10.3390/catal10101195