1. Introduction
Glycerol (
1) (1,2,3-propanetriol) is a non-toxic, edible, biosustainable and biodegradable compound. It is easily obtained from the fermentation of glucose, hydrogenolysis of sorbitol, and as by-product of biodiesel production from vegetable oils [
1]. Recently, it has gained great attention as an attractive building block of organic synthesis [
2,
3] and as a starting material for the preparation of polyglycerols [
4,
5], which are a class of oligomeric materials that are gaining growing interest in cosmetics, polymers, foods, lubricants, plasticizers, stabilizers, and pharmaceutical industries, as well as in the drug delivery field [
6]. While considerable attention has been focused on hyperbranched polyglycerols synthesis [
7], less success has been attained in the direct selective synthesis of linear polyglycerols [
6,
8], which are innovative oligomers displaying potential applications in biomedical and coating materials fields [
8]. In fact, linear polyglycerols are essentially used as surfactants after conversion to fatty acid esters, and they can replace polyethylene glycol (PEG), which is produced from ethylene oxide of petroleum origin. In the last few years, linear polyglycerols are gaining interest as a promising biomedical material because they may have high hydroxyl content functionality, allowing for a wide range of further modifications and applications [
8].
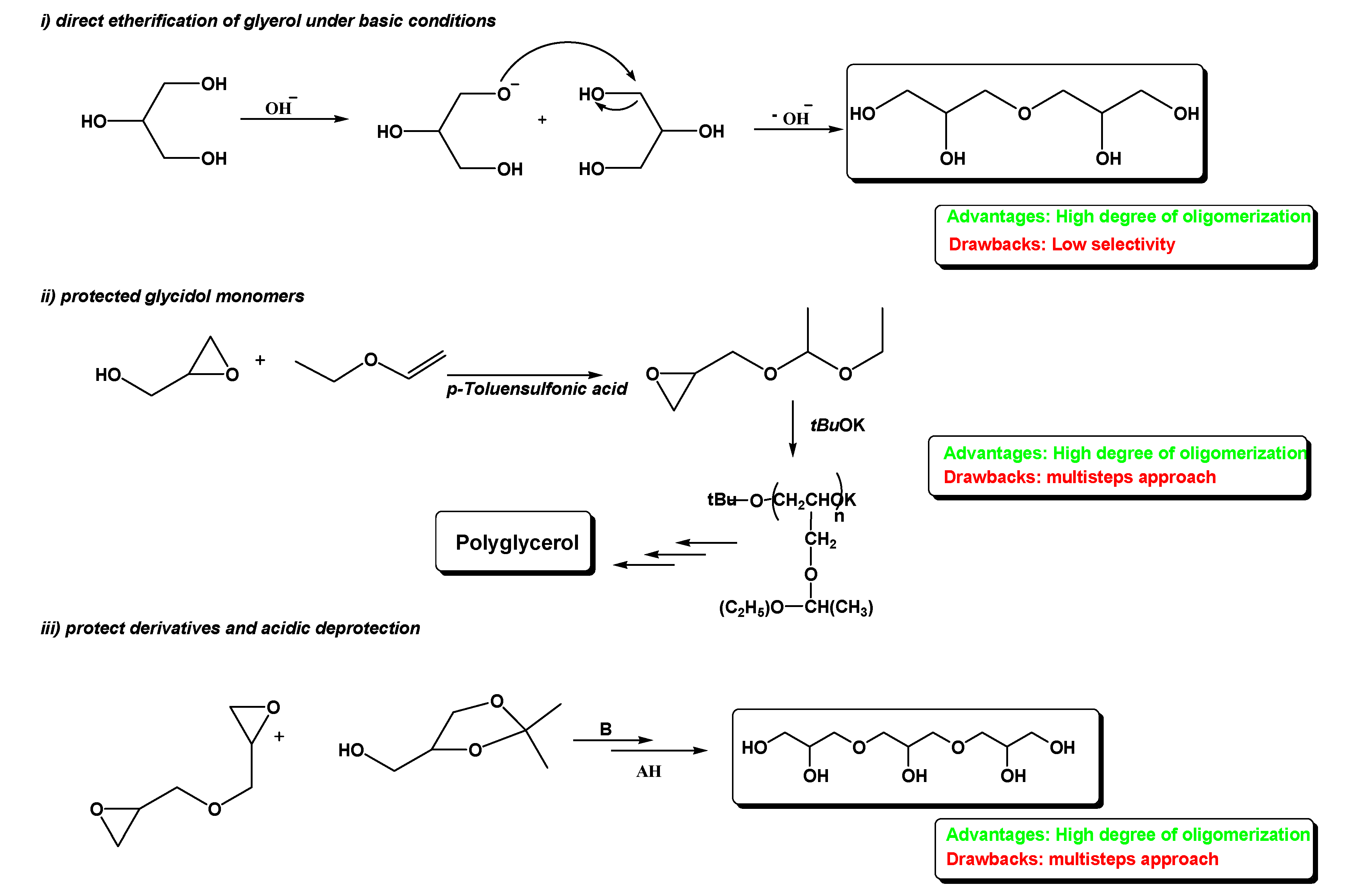
To date, the synthesis of linear polyglycerols can be achieved through three different approaches: (i) direct etherification of unprotected glycerol under basic conditions [
4], (ii) starting from protected glycidol monomers [
4], and (iii) using protected derivatives and acidic deprotection [
9] (
Chart 1).
The base-catalyzed direct etherification of glycerol (way 1) is recognized to be a fast but low selective reaction, yielding PGs (polyglycerols) with a high degree of oligomerization but with many secondary products. Intriguing alternatives make use of glycerol derivatives containing orthogonal protecting groups and oxirane functionalities that could allow an efficient and convergent synthesis (ways 2, 3). However, despite the advantages of high oligomerization degrees, these approaches are complicated by multistep procedures that decrease the synthetic value, especially from the industrial standpoint.
However, the control of the linear selectivity in the polyetherification of glycerol remains an important challenge, which mainly depends on the catalyst properties.
Homogeneous catalysis processes are accomplished with strong acids such as sulfuric or benzenesulfonic acid under harsh conditions (150–280 °C temperature range) that enable very fast but low selective polymerizations, leading to mixtures of cyclic and unsaturated acyclic compounds (alkenes, aldehydes, and ketones) [
10]. Strong bases are also used as homogeneous catalysts, such as alkali hydroxides and carbonates, the latters being more active because of their higher solubility in glycerol [
4,
11]. For instance, high selectivity in linear diglycerol is obtained with CsHCO
3 as a homogeneous catalyst, but at a low conversion of glycerol [
12]. A significant increase of selectivity is observed with heterogeneous catalysts, especially using porous solid materials, which aims at exerting selective effects by reducing the abundant formation of cyclic isomers [
1,
13]. In this context, several studies reported the use of micro- and meso-porous acid zeolites with variable Si/Al ratios, mesoporous aluminosilicates of the MCM-41 type, and acid-modified ion-exchange resins with a macroporous structure (Amberlist 16 and Amberlist 31) [
13]. However, despite their efficiency, these materials still show unsatisfactory linear selectivities, because the reaction takes place with an SN
1 mechanism leading to several undesired secondary and cyclic by-products [
13].
Conversely, alkaline-earth metal oxides, such as CaO, SrO, and BaO, display a superior aptitude in catalyzing the linear etherification of glycerol, affording di- and triglycerols with 80% of conversion after 20 h reaction at 220 °C [
14,
15]. Similar results are obtained with Mg–Al mixed oxides but with lower conversions (50%) [
16]. However, due to the harsh conditions employed (240–280 °C), these catalysts suffer from leaching problems that release alkaline cations from the solid surface, with the consequent inactivation and contamination of glycerol during the reaction [
13]. For example, oxides of alkaline earth metals are partially leached during reaction, forming colloidal and/or transforming into a glyceroxide phase, which makes catalyst separation from reaction medium difficult [
17].
To overcome these drawbacks and maintain the good catalytic performances of metal oxides, we envisioned to exploit rare earth elements. Amongst the most available of these compounds, the clean and cheap lanthanum oxide [
4,
17] has found a wide range of applications [
18,
19,
20], comprising that to function as a catalyst for glycerol polymerization. Previous papers showed that a lanthanum-impregnated catalyst is the most active but the least selective, with glycerol dehydration to acrolein as the main side reaction [
21].
Exploiting our experience in developing green synthetic methods [
22,
23], we envisioned that the catalytic performances of lanthanum oxide could be improved by anchoring on a robust and widely available support such as mesoporous silica gel KIT-6 [
24]. We report here the synthesis, characterization, and catalytic performances of the hybrid material La(III)@KIT-6, which conjugates the amphoteric properties of the lanthanum oxide [
25,
26], enabling a bifunctional catalysis, with the possibility of governing the selectivity of polymerization by means of mesoporous silica gel pores sizes [
17,
21].
2. Results and Discussion
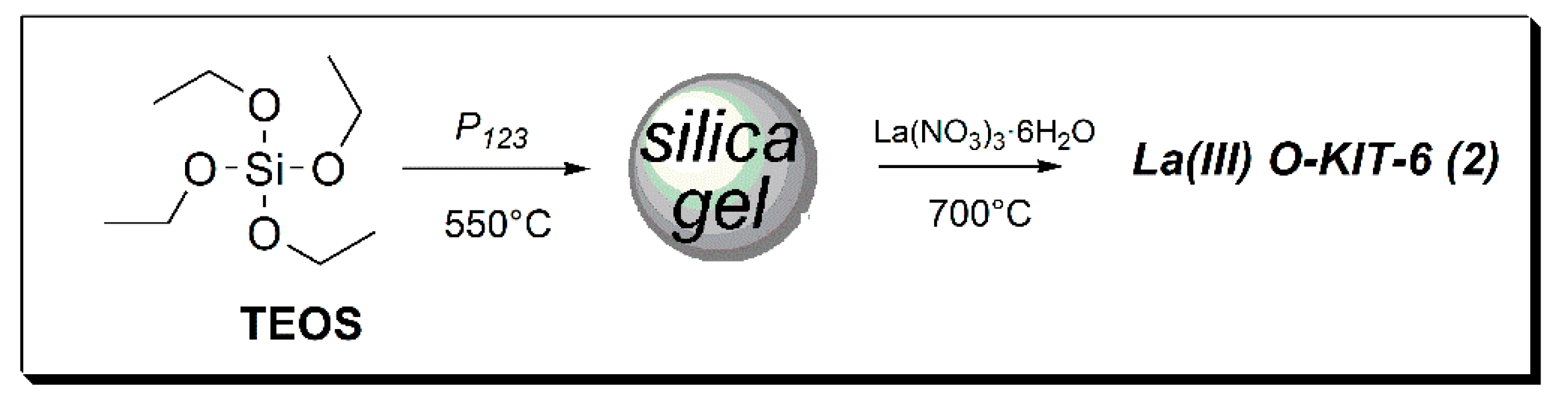
The catalyst was prepared by modifying a literature procedure, [
24] by the impregnation of lanthanum nitrate onto mesoporous silica followed by calcination at 700 °C (
Chart 2). The mesoporous structure was assured by means of a template (P
123), while a high ratio LaNO
3/mesSiO
2 (7 mmol/500 mg) was used to maximize the impregnation of rare earth on the support.
2.1. Characterization of Catalyst La(III)O-KIT-6 (2)
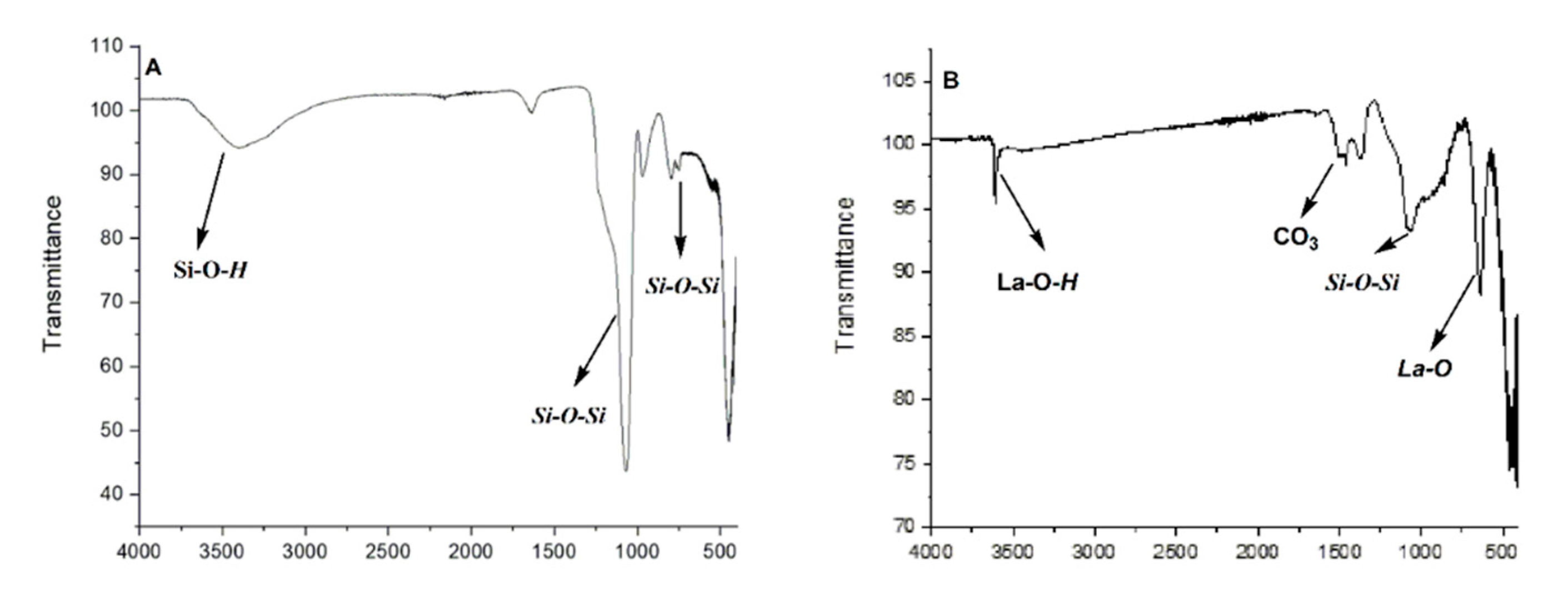
The absorption of metal on silica gel was assured by several techniques. ATR-FTIR analyses of mesoporous silica gel KIT-6 [
24] showed a broad band at 3400–3500 cm
−1 that can be attributed to the stretching of OH bonds, while signals at 1223 to 1092 cm
−1 are ascribed to the asymmetrical and symmetrical stretching of Si–O–Si. In addition, bands at 803 cm
−1 denote the bending of an Si–O framework in the structure (
Figure 1A) [
27]. The FTIR (Fourier transform infrared spectroscopy) spectrum of catalyst La(III)O-KIT-6 (
2) calcined at 700 °C displayed new signals at 644 cm
−1, which can be attributed to the La–O bond, together with 1505 and 1466 cm
−1, that could be assigned to the asymmetric stretching mode of CO
3−2 groups in La-CO
3- (
Figure 1B) [
25,
28].
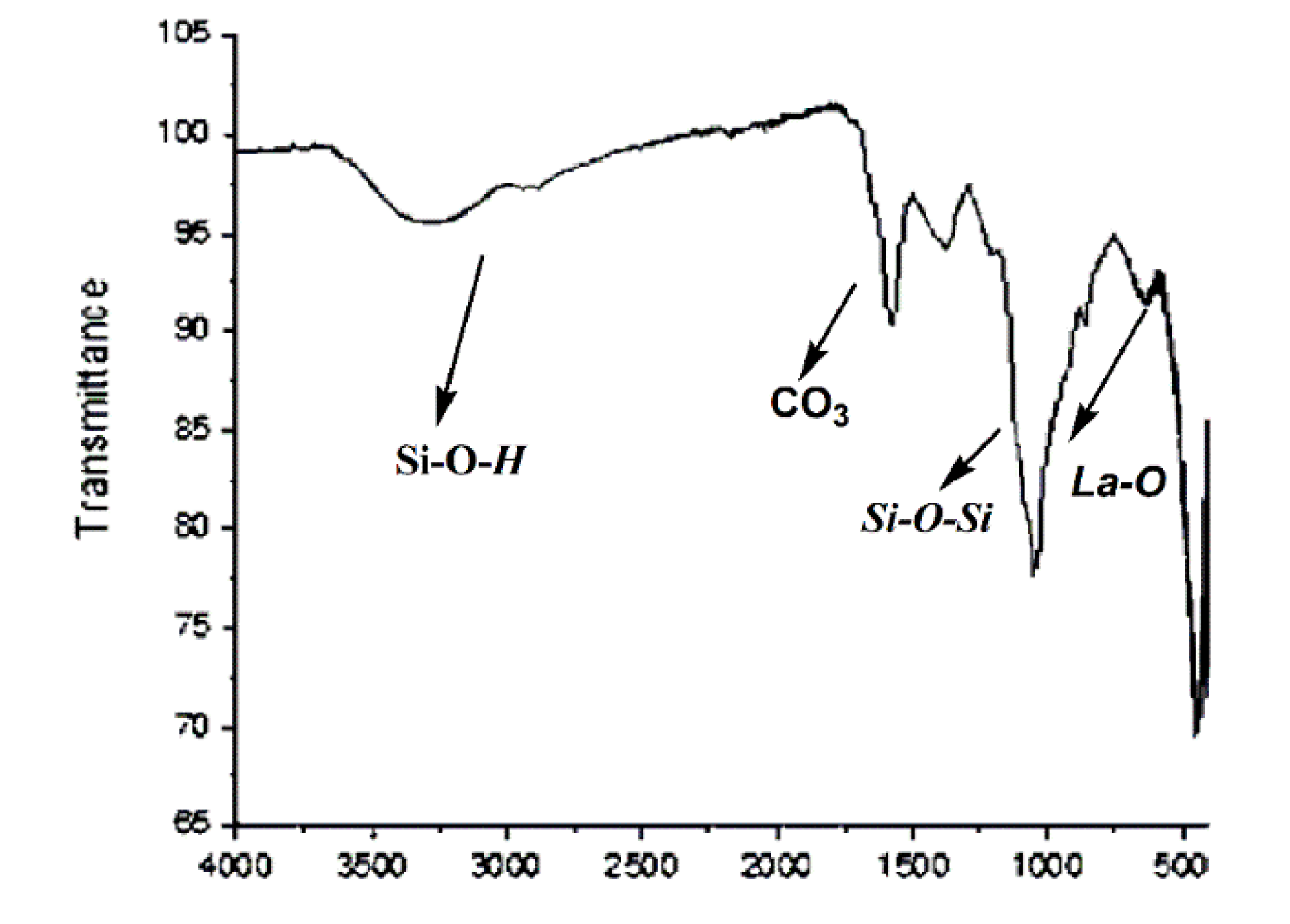
After the reaction, catalyst (
2) was brown; then, it was washed several times with methanol and was again calcined at 700 °C and reused for a new cycle. After three cycles, the catalyst continued to show the signals of CO
3−2, while an intensity of the bands of La-OH and La-O lower than the fresh catalyst (
2) was obtained (
Figure 2).
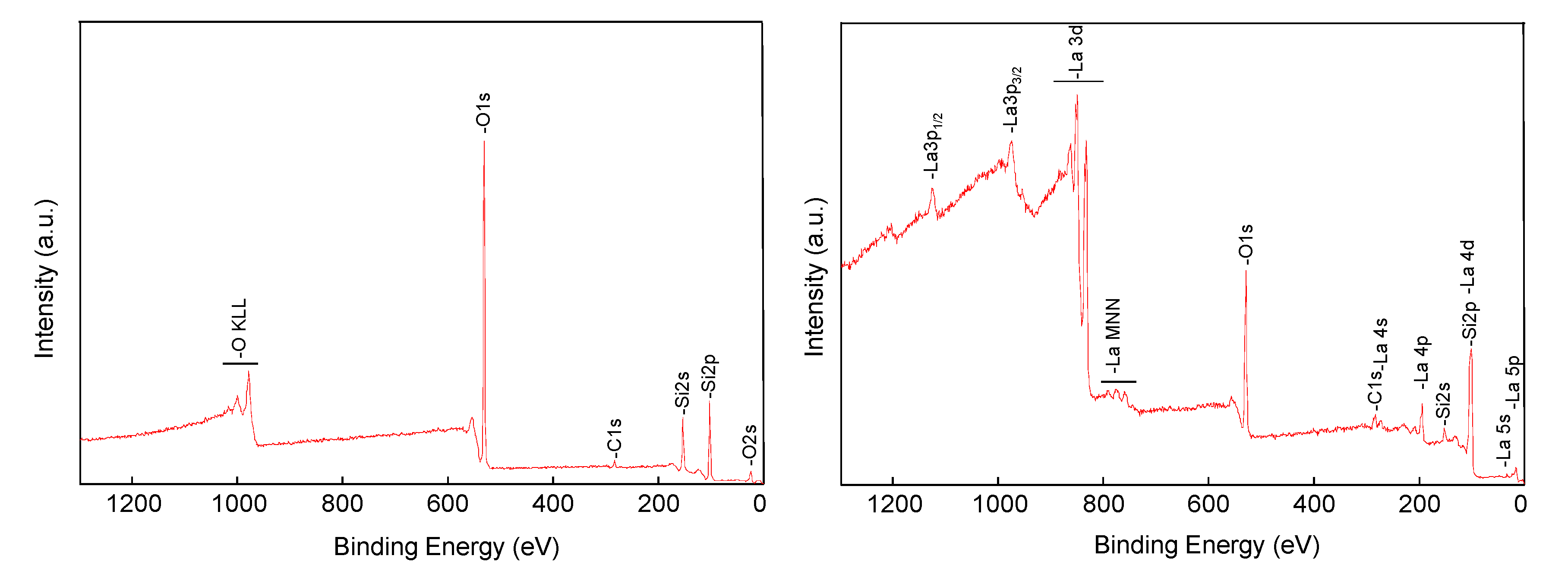
The XPS (X-ray photoelectron spectroscopy) spectrum of support (
Figure 3, left) showed the expected main components, Si and O, with a little C contamination (ca. 5 at. %). Instead, the spectrum of the catalyst (
2) was dominated by the peaks of lanthanum (
Figure 3, right), indicating that the rare earth was successfully supported onto the mesoporous silica. Indeed, peaks ascribed to Si were still present, although with reduced intensity. More specifically, the Si2p signal overlapped with that of La4d, becoming no longer diagnostic, while the Si2s peak was still clearly visible.
The elemental composition of support of catalyst (
2), as determined by XPS peaks integration (
Table 1), was clearly characterized by the presence of lanthanum(III) in accordance with the literature [
28,
29], while the presence of carbon can be attributed to the CO
3−2 group, confirming that lanthanum is present both as La(OH)
3 and as LaO
2CO
3 [
30,
31]. The component of carbon is increased in the reused catalyst after three times, due to the absorption of the oligomer on its surface, as well as the formation of La
2O
2CO
3 (see
Supplementary Materials Figure S1). Meanwhile, the quantity of lanthanum is practically the same, confirming that the catalyst does not undergo leaching (
Supplementary Materials Figure S2).
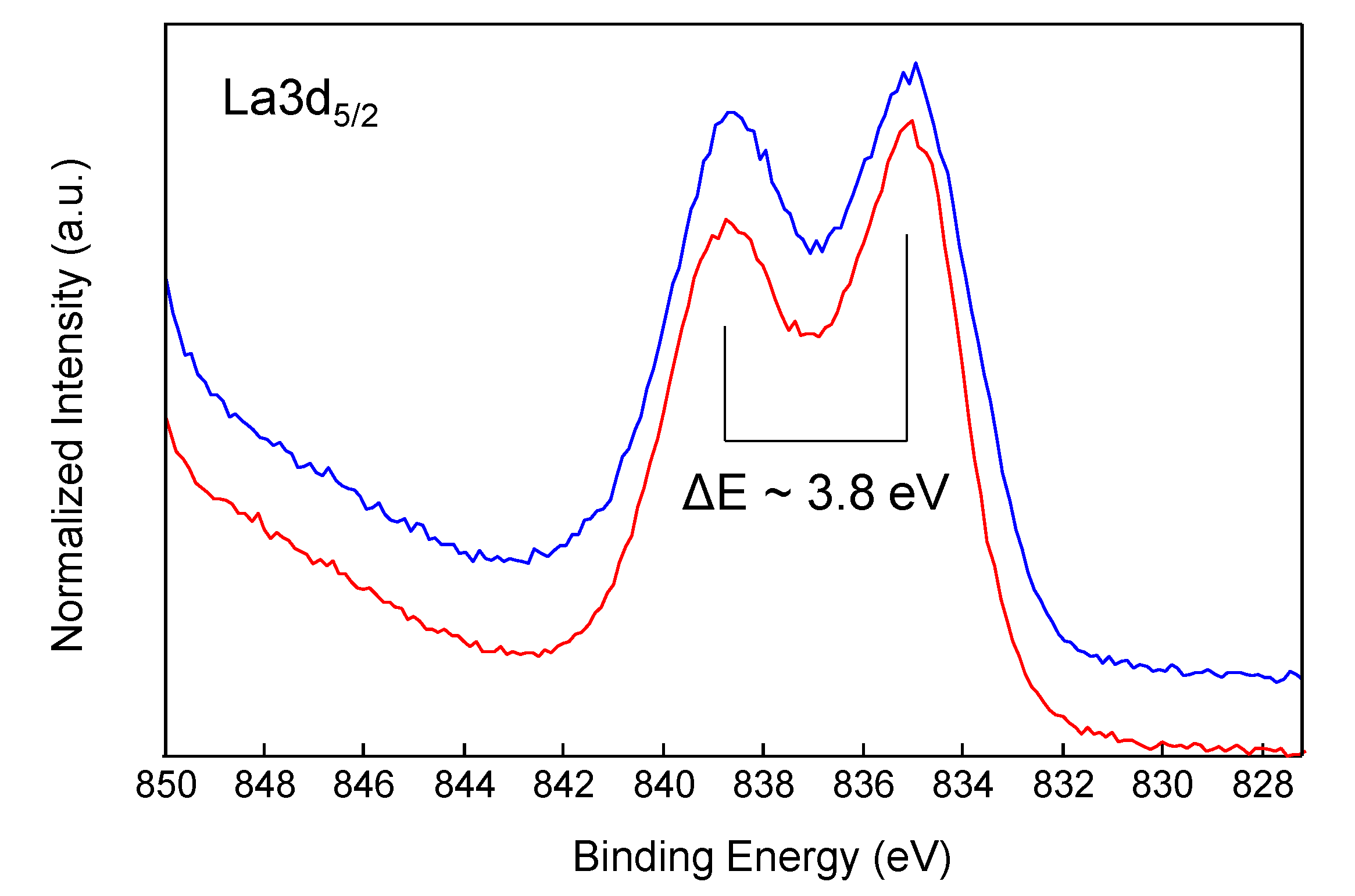
Figure 4 shows a comparison of the high-resolution La3d
5/2 spectra of catalyst before and after three cycles. The La3d region has well separated spin-orbit components, which were each further split by multiplet splitting. The magnitude of the multiplet splitting and intensity ratio of each multiplet-split component are chemically diagnostic [
29]. Both spectra of
Figure 4 show a peak separation of nearly 3.8 eV, which is indicative of the presence of La(OH)
3. However, comparing these two spectra with that of pure La(OH)
3 reported in the literature, a pronounced valley between the multiplet-split components is observed, which could be indicative of the presence of two compounds, one of which is a lanthanum dioxycarbonate La
2O
2CO
3 [
29,
31].
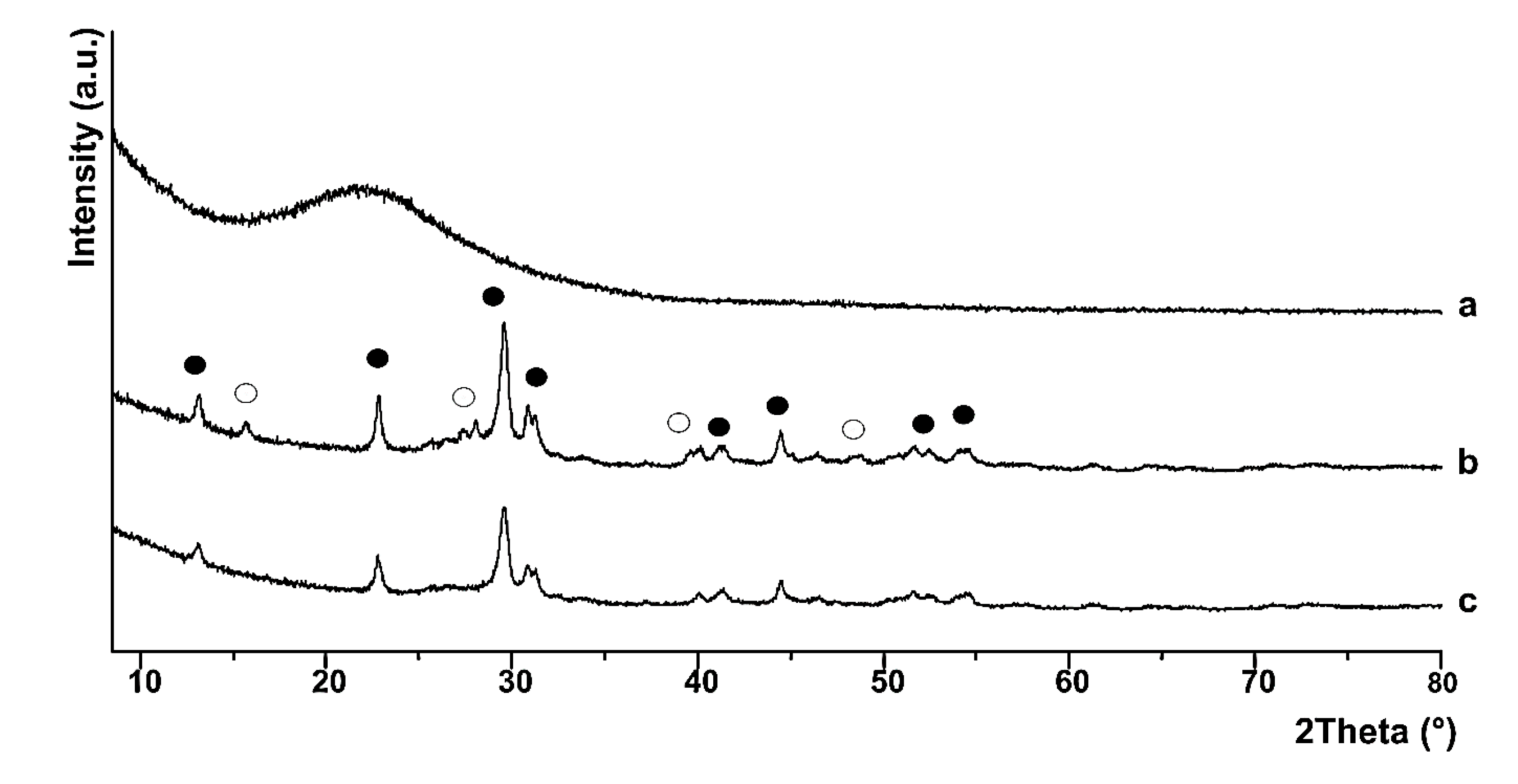
These conclusions were confirmed by powder X-ray diffraction (PXRD) spectra, where the broad amorphous halo in the 2Θ range 20–30° confirms the formation of amorphous silica under the chosen experimental conditions (
Figure 5a). The reaction of La(NO
3)
3·6H
2O with citric acid in the presence of amorphous silica in solution, followed by calcination at 700 °C, brought about the formation of two crystalline La-phases, which are lanthanum oxycarbonate (monoclinic and hexagonal phase) (major compound) [
30,
31] and lanthanum hydroxide La(OH)
3 (hexagonal phase) (
Figure 5, trace b) [
27,
29]. Both crystalline phases cover entirely the amorphous SiO
2 phase, which is confirmed by the absence of the characteristic broad hump for amorphous SiO
2 in trace b [
30]. The PXRD spectrum of the recovered catalyst (
Figure 5, trace c) shows the typical XRD pattern for lanthanium oxycarbonate, which is indeed the only crystalline phase on the SiO
2-based support as in trace b, in accordance with FTIR analysis.
To evaluate the specific surface area and pore sizes distribution, Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods were used, respectively. N
2 adsorption–desorption isotherms (see
Figures S3–S5 in Supplemental Materials) show a hysteresis located in the relative pressure range of 0.5–1.0, which is characteristic of mesoporous materials [
24], and
Table 2 shows the specific surface area of both the support and catalyst. The BET surface area, the BJH pore size, and the pore volume of the mesoporous silica KIT-6 are in accord with the literature [
24]. The presence of lanthanum oxide on pore surfaces is confirmed by the decrease of all these three parameters in catalyst
2. In addition, the decrease of the pore sizes is due to the acid/base interactions between the lanthanum oxide, which is formed during the calcination, and the silanol groups (Si-OH) of the mesoporous silica surface (see the full spectra for BET analysis
Figures S3–S5 in the
Supplemental Materials) [
17].
As expected, after three cycles, a further decrease of the pore sizes is observed, due to the effect of calcinations at 700 °C during the recycling process to eliminate the organic phase, as shown by the XPS analysis, and thanks to the adsorption of organic material in the exhausted catalyst. (See
Figure S1 C1s spectra for the catalyst La(III)OKIT-6 before (A) and after (B) three recycling experiments in
Supplementary Materials)
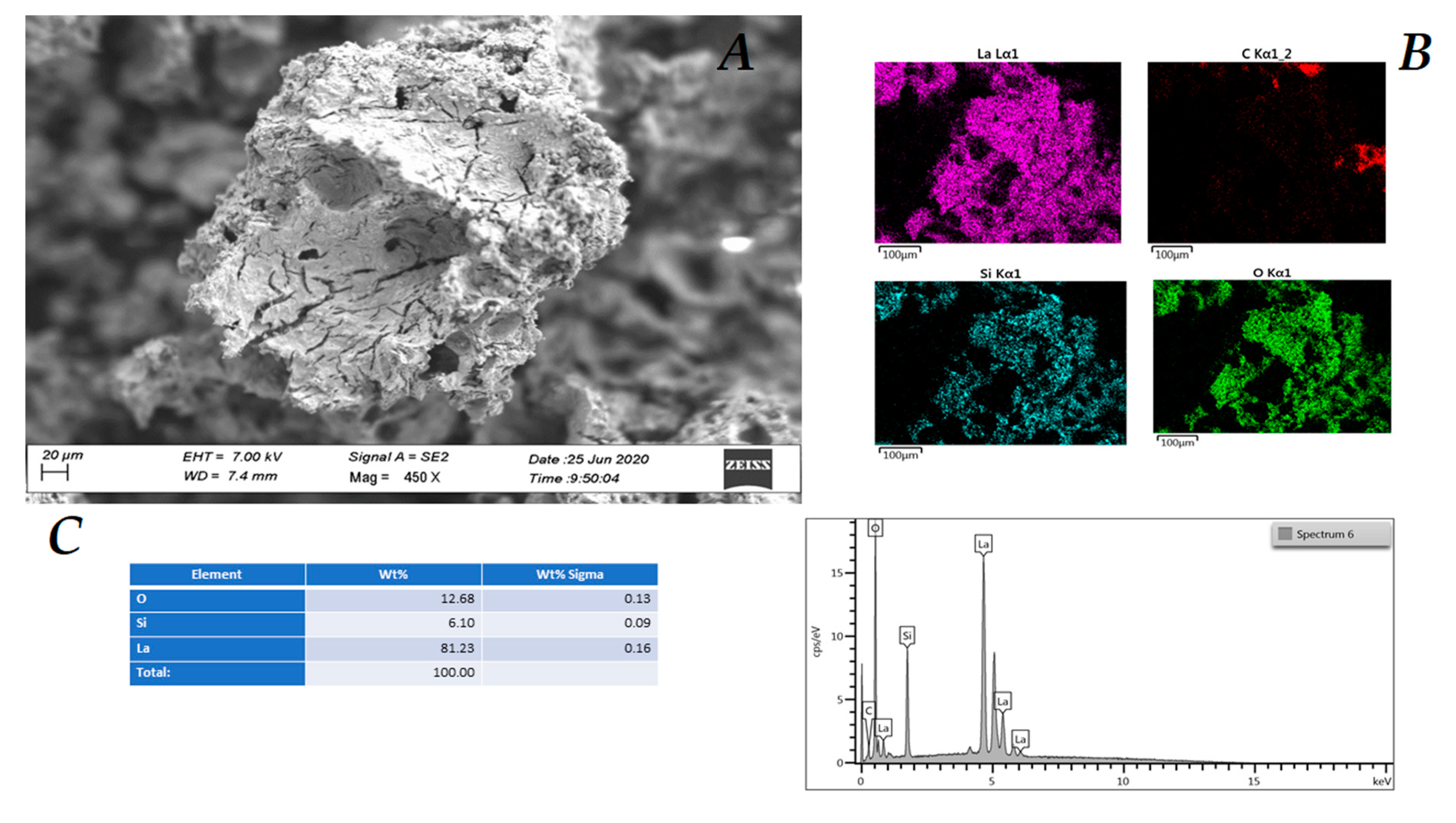
Further confirmations of the change in the morphology of the catalyst were given by the SEM (scanning electron microscope) analyses. The SEM image of catalyst La(III) O-KIT-6 (2) shows the presence of pores, while the distribution of lanthanum on silica is homogeneous, as shown by the purple color in
Figure 6 (see also the
Supplementary Materials Figure S6–S8 for the complete analysis).
After the third cycle, EDS (energy dispersive X-ray analysis) analysis (insert C,
Figure 7) shows that the chemical composition of the catalyst (
2) does not change significantly, which is also in agreement with the XPS analysis, while its morphology undergoes evident changes, in particular its surface roughness (
Figure 7A and
Supplementary Materials Figures S9 and S10).
2.2. Catalytic Tests
According to most of the reported procedures (see
Table 3), catalytic experiments were conducted in a batch, at 240 °C and under solvent-free conditions, in the presence of 2%
w/w of catalyst (2) (see
Table 3). Glycerol conversions were evaluated by GC/MS after derivatization (see the Experimental section for the complete procedure), HPLC analyses, and monitoring of the water amounts produced during reaction by means of a Dean–Stark apparatus (see the experimental section) [
32,
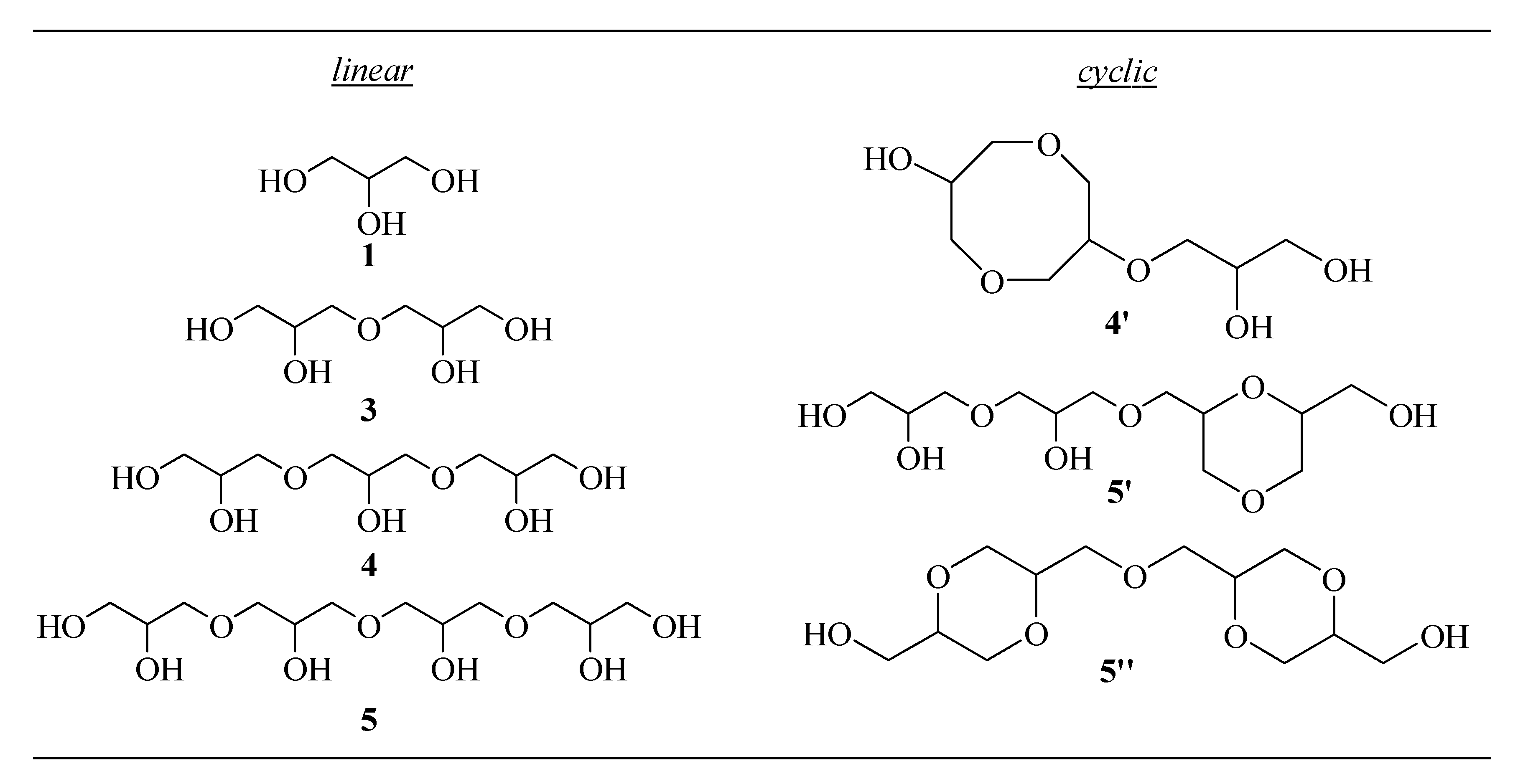
33]. The identification of linear (
3–5) and cyclic (
4′,5′,5′’) glycerol oligomers listed in
Scheme 1 was accomplished by the HPLC-IT-TOF technique, and the spectra obtained are in accordance with the literature [
10,
34,
35,
36] and the NIST DATA BASE for EI mode.
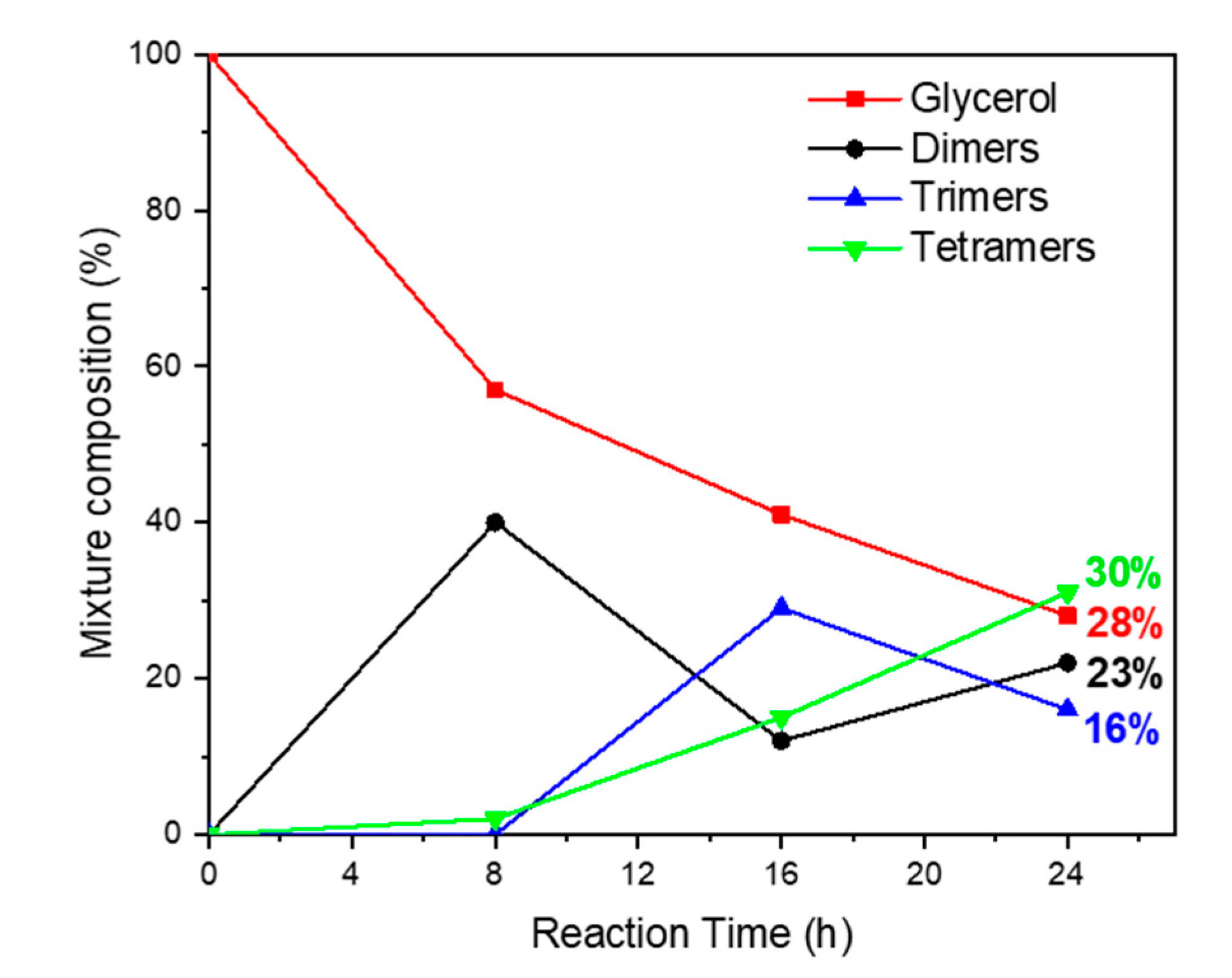
Catalyst efficiency was evaluated monitoring oligomerization products in the range of 24 h (
Figure 8). A survey of this screening clearly indicated that the oligomerization promoted by catalyst
2, within the time range investigated, lead to good yields (about 40%) of linear tetramer
5 and in general of linear oligomers (about 96%,
Table 3, entry 1) without the formation of oxidation and dehydration by-products, which is a result representing a significant improvement if compared with the recent literature (see
Table 3)
.A careful analysis of trend over time shows that after 8 h, a good conversion in diglycerol (3) is accompanied by trace amounts of trimer (4) and a small but interesting percentage (4%) of linear tetramer 5, which is indicative that at the beginning of the process, surely due to the influence of support, dimer (3) undergoes more rapidly a dimerization reaction rather than a chain prolonging the reaction with glycerol.
This result could be rationalized by assuming that the mesoporous structure of support, due to the pore sizes, would allow hosting dimer (3) in the cavity enabling mainly its linear homocoupling. Subsequently, the formation of trimer (4) becomes rapid, reaching the maximum yield in 16 h, after which the formation of tetramer (5) becomes dominant. After 24 h, glycerol conversion reached 72%, with a selectivity toward a linear tetramer (5) of 42%, followed by 32% of dimer (3), 22% of trimer (4), and 4% of cyclic oligomers.
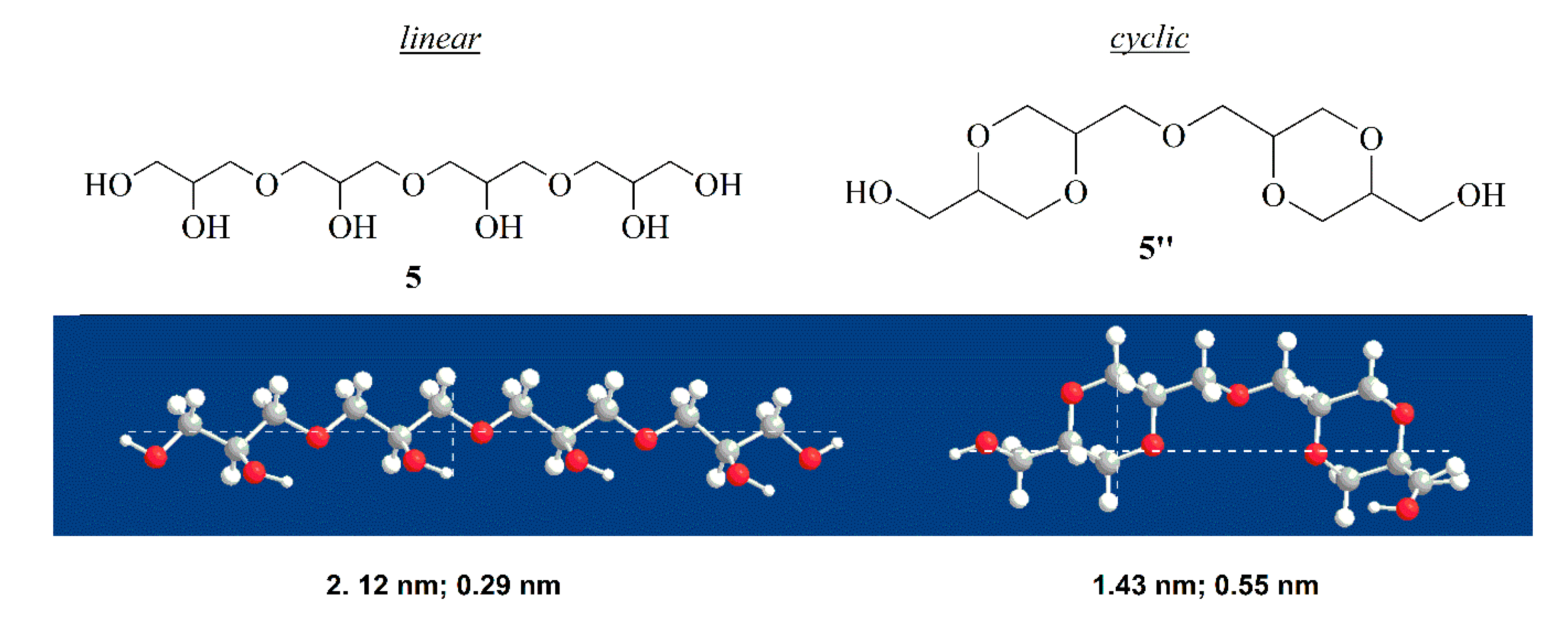
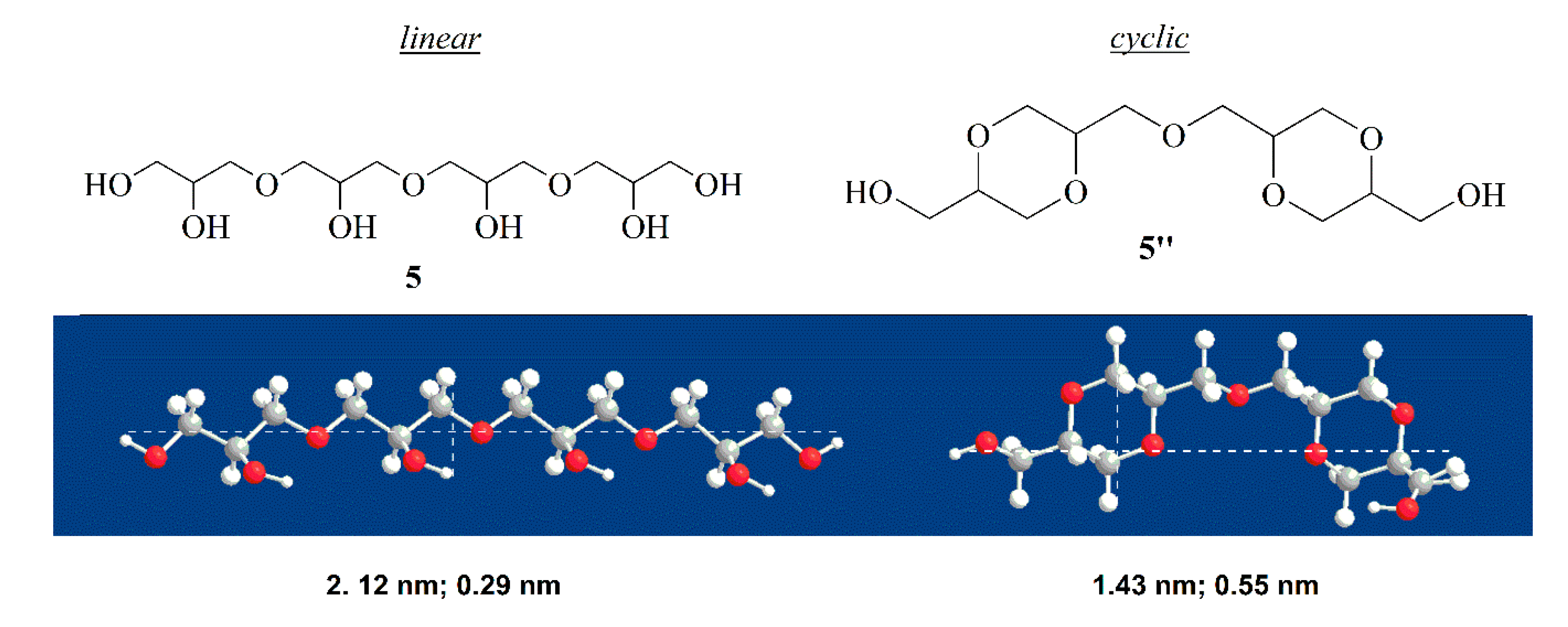
To support our hypothesis, we determined the dimension of linear tetramer
5 and the corresponding cyclic compound
5”.
Figure 9 shows that cyclic tetramer possess a diameter roughly twice that of linear isomer, which is a condition that can cause difficulty accommodating into the pores of the catalyst.
Neither alkenes nor oxidation by-products were observed during the range time investigated.
All these data show that catalyst
2 is efficient in giving essentially the formation of linear polyglycerol.
Table 3 compares catalytic performances of our catalyst La(III)OKIT-6 (
2) (run 1) with those of some analogous heterogeneous catalysts reported in the literature. Data in the table clearly highlight the better activity of 2 in giving higher yields and longer linear oligomers with respect to the basic oxides CaO and MgO (runs 2–3). Likewise, better performances of support silica gel KIT-6 are observed compared with the most used mesoporous material MCM-41 (runs 4–5). In this latter case, dehydration or oxidation by-products such as acrolein are formed during the reaction (run 5). A further advantage of using rare earth metal oxides as catalysts originates from an easy separation of the catalyst achieved by centrifugation of the suspension and washing of the catalytic material with methanol.
Finally, a blank reaction with silica gel support KIT-6 affording very low glycerol conversion to dimer (3) and trimer (4) indicates that lanthanum oxide is a true catalyst for this process (run 6).
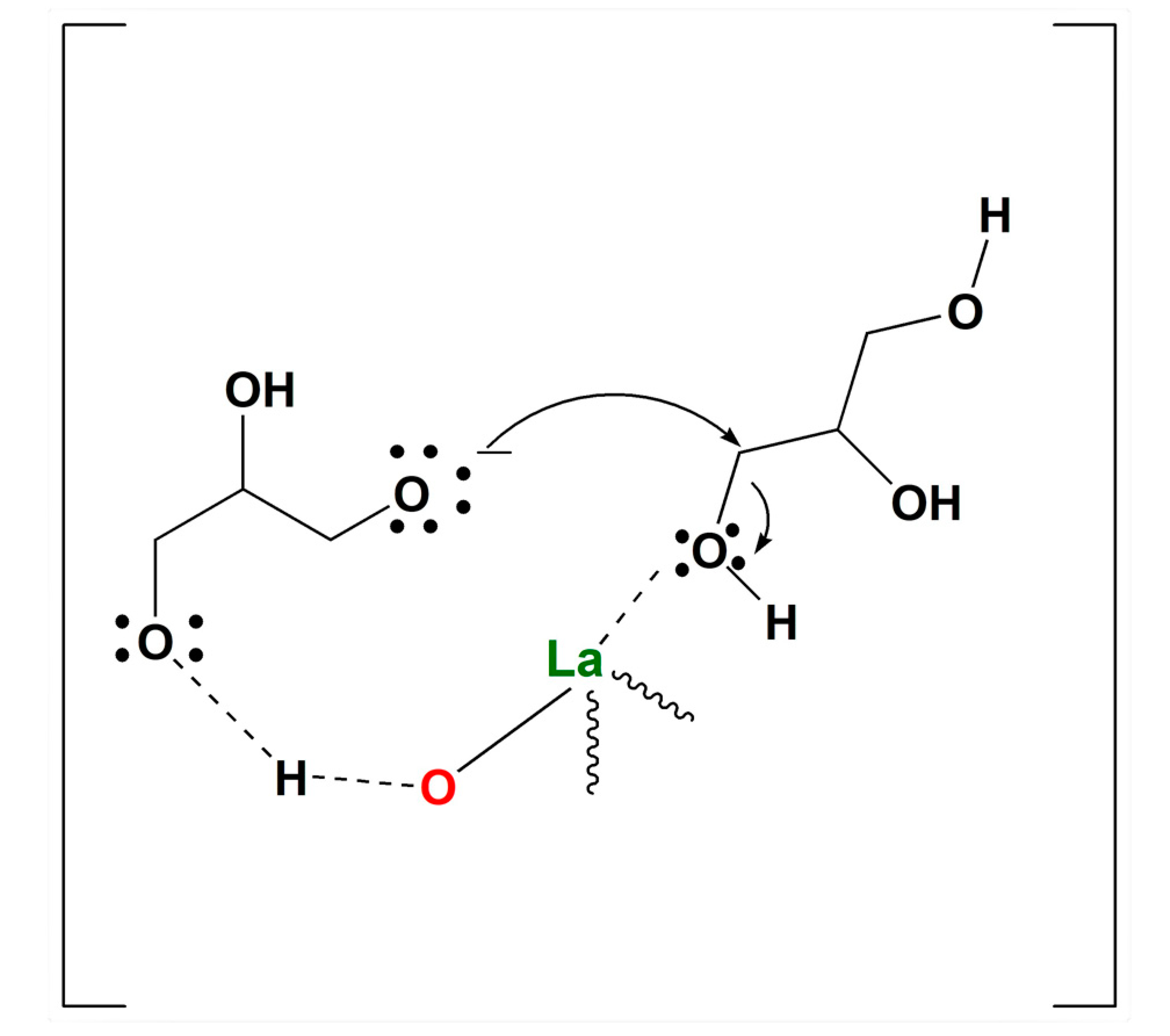
A plausible explanation of the efficiency of catalyst (
2) in the etherification could be found in the amphoteric properties of lanthanum oxide derivatives, through which a bifunctional catalysis phenomenon can be invoked. In particular, as reported in
Figure 10, the basic sites of 2, represented by oxygen atoms linked to lanthanum catalyst, should provide de-protonation of hydroxyl groups of glycerol, thus creating alcoholate functionalities. Simultaneously, the Lewis acid sites, represented by lanthanum, should coordinate glycerol OH groups, favoring the condensation reaction (i.e., elimination of water) (
Figure 10) [
14,
25].
In addition, the mesoporous structure of the support, due to the pore sizes, should be suitable for the accommodation of linear oligomers, thus enhancing the linear selectivity.
2.3. Catalysts Recycle
Catalyst 2 was recycled three times, under optimized conditions. After each run, it was washed with methanol and calcinated at 700 °C to remove the organic phase. In the second and third runs, conversion gradually decreased, while selectivity favored only diglycerol (
3) and triglycerol (
4). (
Table 4)
A plausible explanation of these results could be given assuming that the decrease of pore sizes and rugosity of support, due to catalyst reactivation (by calcination), slows down etherification, which is thought to occur mainly inside them privileging, for steric reasons, the formation of linear oligomers.
2.4. Oligomers Identification
Glycerol oligomers obtained from the catalytic test were identified using HPLC chromatography coupled with ESI-IT-TOF-HR-MS technique (C18 Supelcosil column), exploiting similar studies reported in the literature [
10,
34,
35].
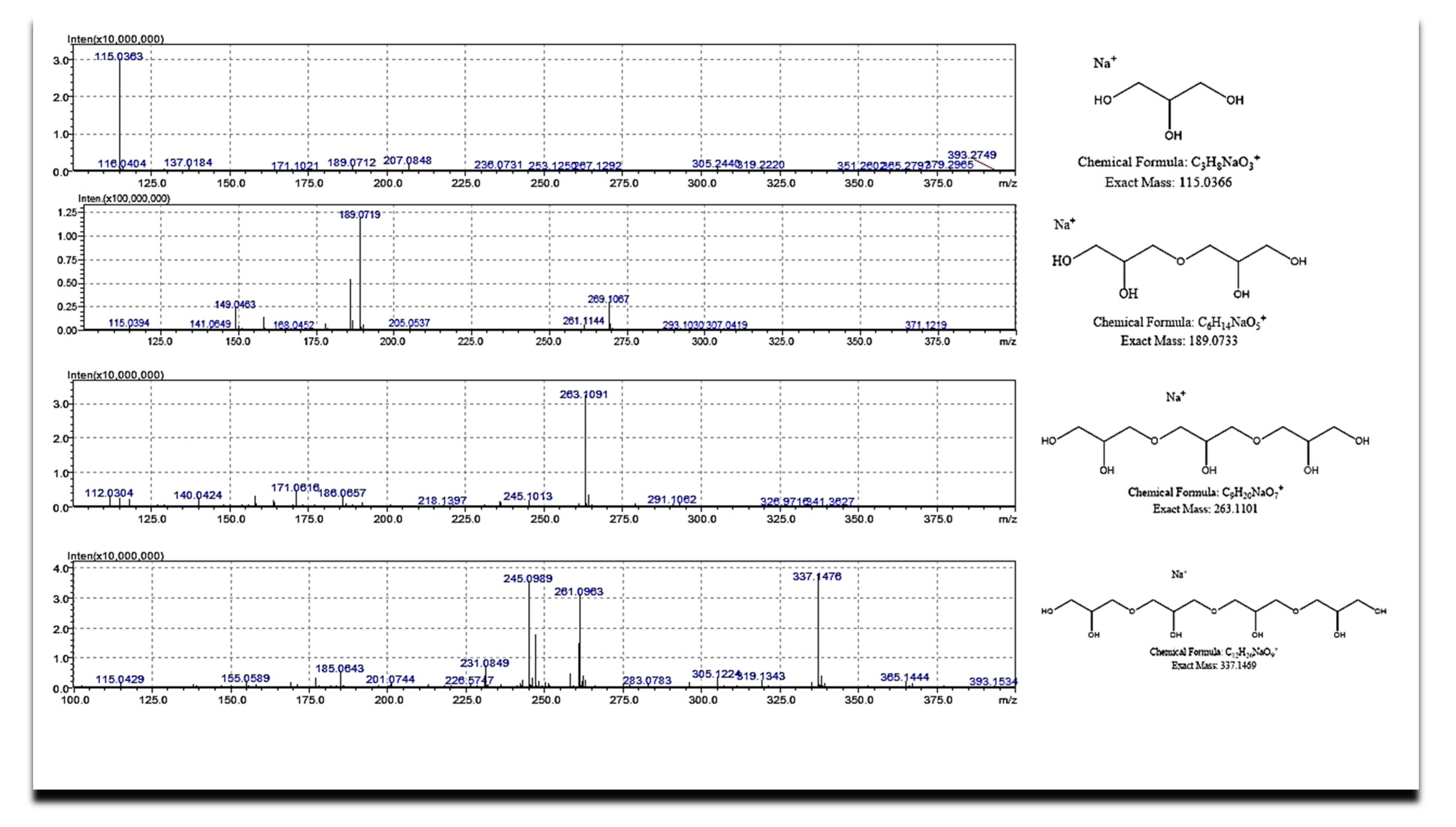
Figure 11 and
Table 5 report ESI(+)-HR-Mass spectra and the peaks assignment of major products, respectively [
35,
36,
37].
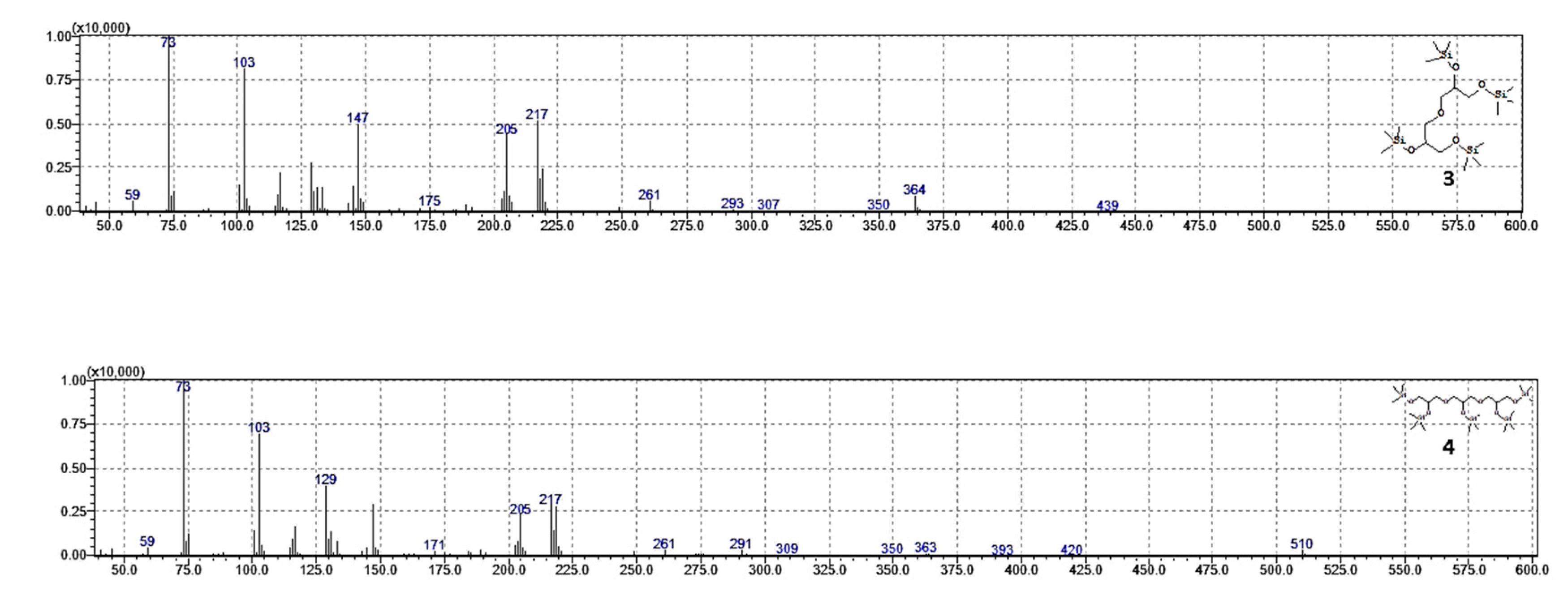
In addition, dimer and trimer were also investigated by GC-MS analyses of silanized [
38] products in EI mode (70 eV), confirming the linear structure of the oligomers (
Figure 12).
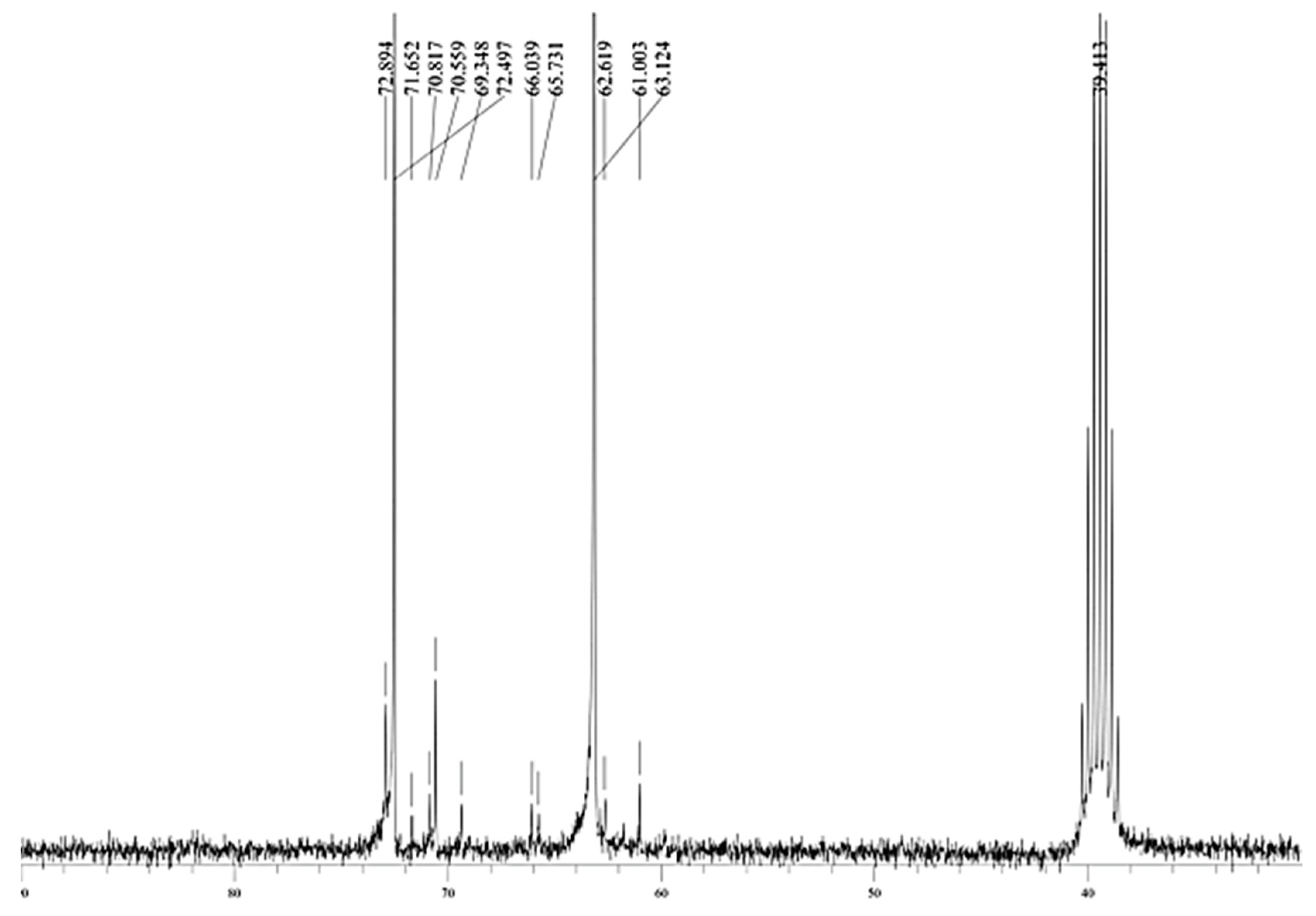
A further confirmation of the presence of linear polymerization products is given by the NMR analysis, which shows, as reported in
Figure 13, only the typical chemical shifts of linear oligomers in the 60–75 ppm zone [
9] (see also the Supplemental Materials for complete NMR analysis).
3. Materials and Methods
Surfactant P123, n-butanol, HCl (37 wt %), tetraethylorthosilicate (TEOS), lanthanum nitrate hexahydrate (La(NO3)3·6H2O), ethanol, citric acid, methanol, and water LC/MS degree were purchased from Sigma-Aldrich, (MERCK, MI, Italy) and used as received, without any further treatment. Glycerol resulting from FAMEs synthesis was gift of Greenswitch industry (MT; Matera, Italy).
GC/MS analyses were run on a Shimadzu GLC 17-A instrument (Shimadzu, MI, Italy) connected with a Shimadzu QP5050A selective mass detector (Shimadzu, MI, Italy) using a SLB-5MS column (30 m × 0.25 mm id, film thickness 0.25 μm). Mass spectra were performed in EI mode (70 eV), and conversions were determined using 2,5-dimethylanisole as an external standard. The identification of glycerol oligomers and determination of selectivities were performed by HPLC with a C18 Column Supelcosil (15 cm × 4.6 mm id), a solvent program starting with H2O to methanol for 40 min, and a flow rate of 1.0 mL/min, with an IT-TOF detector (Shimadzu, MI, Italy) High-resolution mass spectra (HRMS) were obtained using a Shimadzu LCMS-IT-TOF instrument (Shimadzu, MI, Italy). with the following settings: mass range 50–1000 m/z, ionization system electrospray ion source in positive ion mode, nebulizer gas nitrogen at 1.5 L/min, dry gas nitrogen at 102 MPa and 250 °C, collision gas argon.
ATR-FTIR spectra of mesoporous silica and catalyst were recorded in the range of 400–4000 cm−1 on a Perkin Elmer spectrometer instrument (Perkin Elmer, MI, Italy ).
NMR spectra were recorded on an Agilent Technologies 300 MHz spectrometer (Agilent Scientific Instrumets Santa Clara, CA, USA); the 1H NMR spectra were referenced to residual isotopic impurity of DMSO-d6 (2.50 ppm); the 13C NMR spectra (75 MHz) were referenced to DMSO-d6 (39.52 ppm).
Elemental analyses and XPS spectra were obtained by X-ray photoelectron spectroscopy (XPS) on a Scanning XPS Microprobe (PHI 5000 Versa Probe II, Physical Electronics) (Physical Electronic, Monterotondo, RM, Italy).equipped with a monochromatic Al Kα X-ray source (1486.6 eV), operating at 15 kV and 24.8 W, with a spot of 100 µm. Survey (0–1200 eV) and high-resolution spectra (C1s, O1s, Si2p and La3d5/2) were recorded in FAT (Fixed Analyzer Transmission) mode at a pass energy of 117.40 and 29.35 eV, respectively. The analyzer energy resolution, evaluated on the FWHM Ag 3d5/2 photoemission line, was 0.7 eV for a pass energy of 29.35 eV. Surface charging was compensated using a dual beam charge neutralization, with a flux of low energy electrons (≈1 eV) combined with very low energy positive Ar
+ ions (10 eV). The hydrocarbon component of the C1s spectrum was used as internal standard for charging correction, and it was fixed at 285.0 eV [
39]. All spectra were collected at an angle of 45° with respect to the sample surface. The best fitting of the C1s signal was carried out with MultiPak (Physical Electronics) data processing software (Version 9.0 Physical Electronic, Monterotondo, RM, Italy). Atomic concentrations were determined from the high-resolution spectra (Version 9.0 Physical Electronic, Monterotondo, RM, Italy) after subtracting a Shirley-type background.
BET (Brunauer–Emmett–Teller) specific surface area was obtained by the N2 adsorption–desorption method on powder samples using an Autos orb IQ Chemo TCD (Quantachrome Instruments, Boynton Beach, FL, USA). Samples (500 mg) were pre-treated at 77 K for 2 h before N2 adsorption.
The powder X-ray diffraction (PXRD) spectra were acquired at room temperature by depositing ground powders of each sample onto a Si wafer (zero background) which was rotated (0.5 Hz) during spectrum acquisition. The spectra were acquired with a X’PERT-PRO powder diffractometer from Panalytical (Malvern Panalytical Company, Malvern, UK ), using CuKα radiation (λ = 1.54059 Å), a solid state detector (PIXcel), and a parabolic MPD mirror for Cu radiation. The PXRD spectra were recorded in the range 3.99–99.99° (2Θ) applying a step size of 0.0263° and a counting time of 63.24 sec.
SEM-EDS analyses were performed with an electron microscope FESEM-EDX Carl Zeiss Sigma (Zeiss Sigma Company, MI, Italy) 300 VP. The samples were fixed on aluminum stubs and then sputtered with graphite using a Sputter Quorum Q150. Additionally, the chemical composition was determined by EDX under the scanning electron microscope and X-ray diffraction.
The dimension of molecules was performed with Chem3D software (Version 8.0 Ultra, CambridgeSoft Corporation PerkinElmer Informatics).
3.1. Preparation of the Mesoporous Catalyst La(III)O-KIT-6 (2)
Catalyst was prepared according to a known procedure [
24] by the impregnation/annealing of lanthanum nitrate on mesoporous silica gel. In the first instance, silica gel support was prepared by dissolving 900 mg of surfactant P
123 (0.15 mmol, average Mn 5800), 1.11 mL of n-butanol (ρ0.81 g/mL, 12.13 mmol), and 1.43 mL of HCl concentration (37 wt %) in 32.55 mL of distilled water. To this solution, 2.07 mL (ρ0.933 g/mL, 9.27 mmol) of tetraethylorthosilicate (TEOS) were added, and the suspension was stirred at 35 °C for 24 h. The mixture was heated for a further 24 h in autoclave at 100 °C for the hydrothermal treatment, followed by filtration. The solid residue was washed, dried at 80 °C for 12 h, and calcined at 550 °C for 5 h to remove the organic template. The silica gel was labeled as KIT-6.
Then, 250 mg of KIT-6 were suspended into a solution containing 1.52 g of lanthanum nitrate hexahydrate (La(NO3)3·6H2O (3.5 mmol) and 672 mg of citric acid (C6H8O7, 3.5 mmol), as a chelating agent, dissolved in 10 mL of water/ethanol (1:3 v/v). The mixture was heated under reflux for 24 h. Finally, the solvent was evaporated at 100 °C for 12 h, and the solid residue was calcined at 700 °C for 5 h. This synthesized catalyst was labeled as La(III)O-KIT-6 (2).
3.2. Catalytic Tests
First, 5 g of glycerol (1) (54 mmol) and 100 mg of catalyst (
2) (2 wt %) were charged into a batch reactor connected to a Dean–Stark system and kept at 240 °C under N
2 atmosphere. Oligomerization conversions at 8, 16, and 24 h were determined by monitoring the water formation into the distillation trap, [
32,
33] and further confirmed by GC/MS analyses of reaction mixture by means of a preliminary silylation. [
39] In a typical derivatization procedure, 150 mg of reaction mixture was treated into a 10 mL centrifuge tube with excess amounts (1.00 g ca.) of the silylating reagent composed by hexamethyldisi-lazane:trimethylchlorosilane:pyridine in a 3:1:9 weight ratio. The tube was stirred for 5 min and incubated at room temperature for 30 min, causing the precipitation of NH
4Cl as a white solid. Then, 20 µL of 3,5-dimethylanisole were added to the mixture as an external standard. After centrifugation at 2500 rpm for 5 min, the clear supernatant was analyzed by GC–MS. For the reaction at 24 h, the catalyst was separated by centrifugation and washed with methanol. The recovered catalyst was dried, calcined at 700 °C to remove the organic phase from the pores, and re-used for further runs at 24 h. The reaction mixture was analyzed by liquid chromatography coupled with high-resolution mass spectrometry, using an LC-Electrospray ionization Ion Trap-TOF mass spectrometry instrument equipped with a C18 column, using a gradient water–methanol and a mass range of 50−1000 m/z.


 ,
, 



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
