Properties of Iron-Modified-by-Silver Supported on Mordenite as Catalysts for NOx Reduction

 ,
,  ,
,  ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
- The radius of micropores must be comparable to the ionic radius of the target cation.
- The Si/Al ratio should not be very high; moderate ratios are preferred; in such case, the zeolite framework becomes more negatively charged, and then the cation adsorption capacity is enhanced.
- A uniform distribution of ion-exchange centers (i.e., Al atoms) around channels and cavities is required [56].
3. Materials and Methods
3.1. Samples Preparation
3.2. Characterization Methods
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, J.; Wang, Y.; Qu, H. Dependence of Summertime Surface Ozone on NOx and VOC Emissions over the United States: Peak Time and Value. Geophys. Res. Lett. 2019, 46, 3540–3550. [Google Scholar] [CrossRef] [Green Version]
- Sillman, S. 11.11-Tropospheric Ozone and Photochemical Smog. In Treatise on Geochemistry, 2nd ed.; Holland, H.D., Turekian, K.K., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 11, pp. 415–437. [Google Scholar]
- De Richter, R.; Caillol, S. Fighting global warming: The potential of photocat alysis against CO2, CH4, N2O, CFCs, tropospheric O3, BC and other major contributors to climate change. J. Photochem. Photobiol. C Photochem. Rev. 2011, 12, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.V. Vehicular Emissions in Review. SAE Int. J. Engines 2012, 5, 216–234. [Google Scholar] [CrossRef] [Green Version]
- Leinert, S.; Daly, H.; Hyde, B.; Gallachóir, B.Ó. Co-Benefits? Not always: Quantifying the negative effect of a CO2-reducing car taxation policy on NOx emissions. Energy Policy 2013, 63, 1151–1159. [Google Scholar] [CrossRef]
- Oliva, P. Environmental Regulations and Corruption: Automobile Emissions in Mexico City. J. Polit. Econ. 2015, 123, 686–723. [Google Scholar] [CrossRef] [Green Version]
- Environmental Protection Agency. Inventory of U.S. Greenhouse Gas Emissions and Sinks: 1990–2014. Available online: https://www.epa.gov/ghgemissions/inventory-us-greenhouse-gas-emissions-and-sinks-1990-2014 (accessed on 26 August 2020).
- Ramos, A.; Muñoz, J.; Andrés, F.; Armas, O. NOx emissions from diesel light duty vehicle tested under NEDC and real-word driving conditions. Transp. Res. Part D Transp. Environ. 2018, 63, 37–48. [Google Scholar] [CrossRef]
- Wei, H.; Liu, W.; Zhang, J.; Qin, Z. Effects of simulated acid rain on soil fauna community composition and their ecological niches. Environ. Pollut. 2017, 220, 460–468. [Google Scholar] [CrossRef]
- Gurjar, B.R.; Jain, A.; Sharma, A.; Agarwal, A.; Gupta, P.; Nagpure, A.S.; Lelieveld, J. Human health risks in megacities due to air pollution. Atmos. Environ. 2010, 44, 4606–4613. [Google Scholar] [CrossRef]
- Piumetti, M.; Bensaid, S.; Fino, D.; Russo, N. Catalysis in Diesel engine NOx after treatment: A review. Catal. Struct. React. 2015, 1, 155–173. [Google Scholar] [CrossRef]
- Xin, Y.; Li, Q.; Zhang, Z. Zeolitic Materials for DeNOx Selective Catalytic Reduction. ChemCatChem 2018, 10, 29–41. [Google Scholar] [CrossRef]
- Mrad, R.; Aissat, A.; Cousin, R.; Courcot, D.; Siffert, S. Catalysts for NOx selective catalytic reduction by hydrocarbons (HC-SCR). Appl. Catal. A Gen. 2015, 504, 542–548. [Google Scholar] [CrossRef]
- Traa, Y.; Burger, B.; Weitkamp, J. Zeolite-based materials for the selective catalytic reduction of NOx with hydrocarbons. Microporous Mesoporous Mater. 1999, 30, 3–41. [Google Scholar] [CrossRef]
- Cant, N.W.; Liu, I.O.Y. Mechanism of the selective reduction of nitrogen oxides by hydrocarbons on zeolite catalysts. Catal. Today 2000, 63, 133–146. [Google Scholar] [CrossRef]
- Bartolomeu, R.; Azambre, B.; Westermann, A.; Fernandes, A.; Bértolo, R.; Hamoud, H.I.; Henriques, C.; Da Costa, P.; Ribeiro, F. Investigation of the nature of silver species on different Ag-containing NOx reduction catalysts: On the effect of the support. Appl. Catal. B Environ. 2014, 150–151, 204–217. [Google Scholar] [CrossRef]
- Castellanos, I.; Marie, O. An operando FT-IR study of the NOx SCR over Co-HFER and Fe-HFER using acetylene as a reducing agent. Catal. Today 2017, 283, 54–65. [Google Scholar] [CrossRef]
- Čapek, L.; Kreibich, V.; Dedeček, J.; Grygar, T.; Wichterlová, B.; Sobalík, Z.; Martens, J.A.; Brosius, R.; Tokarová, V. Analysis of Fe species in zeolites by UV-VIS-NIR, IR spectra and voltammetry. Effect of preparation, Fe loading and zeolite type. Microporous Mesoporous Mater. 2005, 80, 279–289. [Google Scholar] [CrossRef]
- Lee, K.; Kosaka, H.; Sato, S.; Yokoi, T.; Choi, B.; Kim, D. Effects of Cu loading and zeolite topology on the selective catalytic reduction with C3H6 over Cu/zeolite catalysts. J. Ind. Eng. Chem. 2019, 72, 73–86. [Google Scholar] [CrossRef]
- Chen, H.Y.; Wang, X.; Sachtler, W.M.H. Reduction of NOx over various Fe/zeolite catalysts. Appl. Catal. A Gen. 2000, 194, 159–168. [Google Scholar] [CrossRef]
- Bin Lim, J.; Shin, J.; Ahn, N.H.; Heo, I.; Hong, S.B. Selective catalytic reduction of NO with CH4 over cobalt-exchanged cage-based, small-pore zeolites with different framework structures. Appl. Catal. B Environ. 2020, 267, 118710. [Google Scholar]
- Shibata, J.; Takada, Y.; Shichi, A.; Satokawa, S.; Satsuma, A.; Hattori, T. Influence of zeolite support on activity enhancement by addition of hydrogen for SCR of NO by propane over Ag-zeolites. Appl. Catal. B Environ. 2004, 54, 137–144. [Google Scholar] [CrossRef]
- Hamoud, H.I.; Valtchev, V.; Daturi, M. Selective catalytic reduction of NOx over Cu- and Fe-exchanged zeolites and their mechanical mixture. Appl. Catal. B Environ. 2019, 250, 419–428. [Google Scholar] [CrossRef]
- Dorado, F.; De Lucas, A.; García, P.B.; Romero, A.; Valverde, J.L.; Asencio, I. SCR of NO by propene on monometallic (Co or Ni) and bimetallic (Co/Ag or Ni/Ag) mordenite-based catalysts. Ind. Eng. Chem. Res. 2005, 44, 8988–8996. [Google Scholar] [CrossRef]
- Lee, K.; Choi, B.; Lee, C.; Oh, K. Effects of SiO2/Al2O3 ratio, reaction atmosphere and metal additive on de-NOx performance of HC-SCR over Cu-based ZSM-5. J. Ind. Eng. Chem. 2020, 90, 132–144. [Google Scholar] [CrossRef]
- Serra, R.M.; Aspromonte, S.G.; Miró, E.E.; Boix, A.V. Hydrocarbon adsorption and NOx-SCR on (Cs, Co) mordenite. Appl. Catal. B Environ. 2015, 166–167, 592–602. [Google Scholar] [CrossRef]
- Cheng, X.; Su, D.; Wang, Z.; Ma, C.; Wang, M. Catalytic reduction of nitrogen oxide by carbon monoxide, methane and hydrogen over transition metals supported on BEA zeolites. Int. J. Hydrog. Energy 2018, 43, 21969–21981. [Google Scholar] [CrossRef]
- Abu-Zied, B.M.; Schwieger, W.; Unger, A. Nitrous oxide decomposition over transition metal exchanged ZSM-5 zeolites prepared by the solid-state ion-exchange method. Appl. Catal. B Environ. 2008, 84, 277–288. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, H.; Haller, G.; Li, Y. Recent advances in the selective catalytic reduction of NOx with NH3 on Cu-Chabazite catalysts. Appl. Catal. B Environ. 2017, 202, 346–354. [Google Scholar] [CrossRef]
- Kim, M.H.; Nam, I.; Kim, Y.G. Water tolerance of mordenite-type zeolite catalysts for selective reduction of nitric oxide by hydrocarbons. Appl. Catal. B Environ. 1997, 12, 125–145. [Google Scholar] [CrossRef]
- Aspromonte, S.G.; Miró, E.E.; Boix, A.V. FTIR studies of butane, toluene and nitric oxide adsorption on Ag exchanged NaMordenite. Adsorption 2012, 18, 1–12. [Google Scholar] [CrossRef]
- Gurin, V.S.; Petranovskii, V.P.; Hernandez, M.A.; Bogdanchikova, N.E.; Alexeenko, A.A. Silver and copper clusters and small particles stabilized within nanoporous silicate-based materials. Mater. Sci. Eng. A 2005, 391, 71–76. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Gurgul, J.; Łątka, K.; Sánchez-López, P.; Bogdanov, D.; Kotolevich, Y.; Petranovskii, V.; Fuentes, S. Mechanism of formation of framework Fe3+ in bimetallic Ag-Fe mordenites—Effective catalytic centers for deNOx reaction. Microporous Mesoporous Mater. 2020, 299, 109841. [Google Scholar] [CrossRef]
- Qian, W.; Su, Y.; Yang, X.; Yuan, M.; Deng, W.; Zhao, B. Experimental study on selective catalytic reduction of NO with propene over iron based catalysts supported on aluminum pillared clays. J. Fuel Chem. Technol. 2017, 45, 1499–1507. [Google Scholar] [CrossRef]
- Zhang, X.; Su, Y.; Cheng, J.; Lin, R.; Wen, N.; Deng, W.; Zhou, H. Effect of Ag on deNOx performance of SCR-C3H6 over Fe/Al-PILC catalysts. J. Fuel Chem. Technol. 2019, 47, 1368–1378. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, Y.; Zhu, Y.; Chen, M.; Zhang, Z.; Shangguan, W. Efficient Fe-ZSM-5 catalyst with wide active temperature window for NH3 selective catalytic reduction of NO: Synergistic effect of isolated Fe3+ and Fe2O3. J. Catal. 2019, 378, 17–27. [Google Scholar] [CrossRef]
- Dzwigaj, S.; Janas, J.; Rojek, W.; Stievano, L.; Wagner, F.E.; Averseng, F.; Che, M. Effect of iron impurities on the catalytic activity of BEA, MOR and MFI zeolites in the SCR of NO by ethanol. Appl. Catal. B Environ. 2009, 86, 45–52. [Google Scholar] [CrossRef]
- Sazama, P.; Pilar, R.; Mokrzycki, L.; Vondrova, A.; Kaucky, D.; Plsek, J.; Sklenak, S.; Stastny, P.; Klein, P. Remarkably enhanced density and specific activity of active sites in Al-rich Cu-, Fe- and Co-beta zeolites for selective catalytic reduction of NOx. Appl. Catal. B Environ. 2016, 189, 65–74. [Google Scholar] [CrossRef]
- Iwasaki, M.; Yamazaki, K.; Banno, K.; Shinjoh, H. Characterization of Fe/ZSM-5 DeNOx catalysts prepared by different methods: Relationships between active Fe sites and NH3-SCR performance. J. Catal. 2008, 260, 205–216. [Google Scholar] [CrossRef]
- Kumar, M.S.; Schwidder, M.; Grünert, W.; Bentrup, U.; Brückner, A. Selective reduction of NO with Fe-ZSM-5 catalysts of low Fe content: Part II. Assessing the function of different Fe sites by spectroscopic in situ studies. J. Catal. 2006, 239, 173–186. [Google Scholar]
- Heijboer, W.M.; Battiston, A.A.; Knop-Gericke, A.; Hävecker, M.; Bluhm, H.; Weckhuysen, B.M.; Koningsberger, D.C.; de Groot, F.M.F. Redox behaviour of over-exchanged Fe/ZSM5 zeolites studied with in-situ soft X-ray absorption spectroscopy. Phys. Chem. Chem. Phys. 2003, 5, 4484. [Google Scholar] [CrossRef] [Green Version]
- Boroń, P.; Chmielarz, L.; Gurgul, J.; Latka, K.; Gil, B.; Marszalek, B.; Dzwigaj, S. Influence of iron state and acidity of zeolites on the catalytic activity of FeHBEA, FeHZSM-5 and FeHMOR in SCR of NO with NH3 and N2O decomposition. Microporous Mesoporous Mater. 2015, 203, 73–85. [Google Scholar] [CrossRef]
- Shwan, S.; Jansson, J.; Olsson, L.; Skoglundh, M. Effect of post-synthesis hydrogen-treatment on the nature of iron species in Fe-BEA as NH3-SCR catalyst. Catal. Sci. Technol. 2014, 4, 2932. [Google Scholar] [CrossRef]
- Bartolomeu, R.; Mendes, N.; Fernandes, A. NOx SCR with decane using Ag-MFI catalysts: On the effect of silver content and co-cation presence. Catal. Sci. Technol. 2016, 6, 3038–3048. [Google Scholar] [CrossRef]
- Satsuma, A.; Shibata, J.; Shimizu, K.I.; Hattori, T. Ag clusters as active species for HC-SCR over Ag-zeolites. Catal. Surv. Asia 2005, 9, 75–85. [Google Scholar] [CrossRef]
- Shimizu, K.; Sugino, K.; Kato, K.; Yokota, S.; Okumura, K. Reaction Mechanism of H2-Promoted Selective Catalytic Reduction of NO with C3H8 over Ag–MFI Zeolite. J. Phys. Chem. C 2007, 111, 6481–6487. [Google Scholar] [CrossRef]
- Shimizu, K.I.; Satsuma, A. Selective catalytic reduction of NO over supported silver catalysts—Practical and mechanistic aspects. Phys. Chem. Chem. Phys. 2006, 8, 2677–2695. [Google Scholar] [CrossRef]
- More, P.M.; Nguyen, D.L.; Granger, P.; Dujardin, C.; Dongare, M.K.; Umbarkar, S.B. Activation by pretreatment of Ag-Au/Al2O3 bimetallic catalyst to improve low temperature HC-SCR of NOx for lean burn engine exhaust. Appl. Catal. B Environ. 2015, 174–175, 145–156. [Google Scholar] [CrossRef]
- More, P.M.; Nguyen, D.L.; Dongare, M.K.; Umbarkar, S.B.; Nuns, N.; Girardon, J.-S.; Dujardin, C.; Lancelot, C.; Mamede, A.S.; Granger, P. Rational preparation of Ag and Au bimetallic catalysts for the hydrocarbon-SCR of NOx: Sequential deposition vs. coprecipitation method. Appl. Catal. B Environ. 2015, 162, 11–20. [Google Scholar] [CrossRef]
- Sánchez-López, P.; Kotolevich, Y.; Miridonov, S.; Chávez-Rivas, F.; Fuentes, S.; Petranovskii, V. Bimetallic AgFe Systems on Mordenite: Effect of Cation Deposition Order in the NO Reduction with C3H6/CO. Catalysts 2019, 9, 58. [Google Scholar] [CrossRef] [Green Version]
- Motsi, T.; Rowson, N.A.; Simmons, M.J.H. Adsorption of heavy metals from acid mine drainage by natural zeolite. Int. J. Miner. Process. 2009, 92, 42–48. [Google Scholar] [CrossRef]
- Wang, X.S.; Huang, J.; Hu, H.Q.; Wang, J.; Qin, Y. Determination of kinetic and equilibrium parameters of the batch adsorption of Ni (II) from aqueous solutions by Na-mordenite. J. Hazard. Mater. 2007, 142, 468–476. [Google Scholar] [CrossRef]
- Lee, S.H.; Jo, H.Y.; Yun, S.T.; Lee, Y.J. Evaluation of factors affecting performance of a zeolitic rock barrier to remove zinc from water. J. Hazard. Mater. 2010, 175, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Trgo, M.; Perić, J. Interaction of the zeolitic tuff with Zn containing simulated pollutant solutions. J. Colloid Interface Sci. 2003, 260, 166–175. [Google Scholar] [CrossRef]
- Nakamura, H.; Okumura, M.; Machida, M. First-Principles Calculation Study of Mechanism of Cation Adsorption Selectivity of Zeolites: A Guideline for Effective Removal of Radioactive Cesium. J. Phys. Soc. Jpn. 2013, 82, 023801. [Google Scholar] [CrossRef]
- Seliman, A.F.; Borai, E.H. Utilization of natural chabazite and mordenite as a reactive barrier for immobilization of hazardous heavy metals. Environ. Sci. Pollut. Res. 2011, 18, 1098–1107. [Google Scholar] [CrossRef]
- Smith, D.W. Ionic Hydration Enthalpies. J. Chem. Educ. 1977, 54, 540–542. [Google Scholar] [CrossRef]
- Luo, Y.R. Comprehensive Handbook of Chemical Bond. Energies, 1st ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Berlier, G.; Lamberti, C.; Rivallan, M.; Mul, G. Characterization of Fe sites in Fe-zeolites by FTIR spectroscopy of adsorbed NO: Are the spectra obtained in static vacuum and dynamic flow set-ups comparable. Phys. Chem. Chem. Phys. 2010, 12, 358–364. [Google Scholar] [CrossRef]
- Brandenberger, S.; Kröcher, O.; Tissler, A.; Althoff, R. The State of the Art in Selective Catalytic Reduction of NOx by Ammonia Using Metal-Exchanged Zeolite Catalysts. Catal. Rev. 2008, 50, 492–531. [Google Scholar] [CrossRef]
- Trofimova, N.N.; Veligzhanin, A.A.; Murzin, V.Y.; Chernyshov, A.A.; Khramov, E.V.; Zabluda, V.N.; Edel’man, I.S.; Slovokhotov, Y.L.; Zubavichus, Y.V. Structural diagnostics of functional nanomaterials with the use of X-ray synchrotron radiation. Nanotechnol. Russ. 2013, 8, 396–401. [Google Scholar] [CrossRef]
- Ravel, B.; Newville, M. Athena, Artemis, Hephaestus: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 2005, 12, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Newville, M. IFEFFIT: Interactive XAFS analysis and FEFF fitting. J. Synchrotron Radiat. 2001, 8, 322–324. [Google Scholar] [CrossRef]
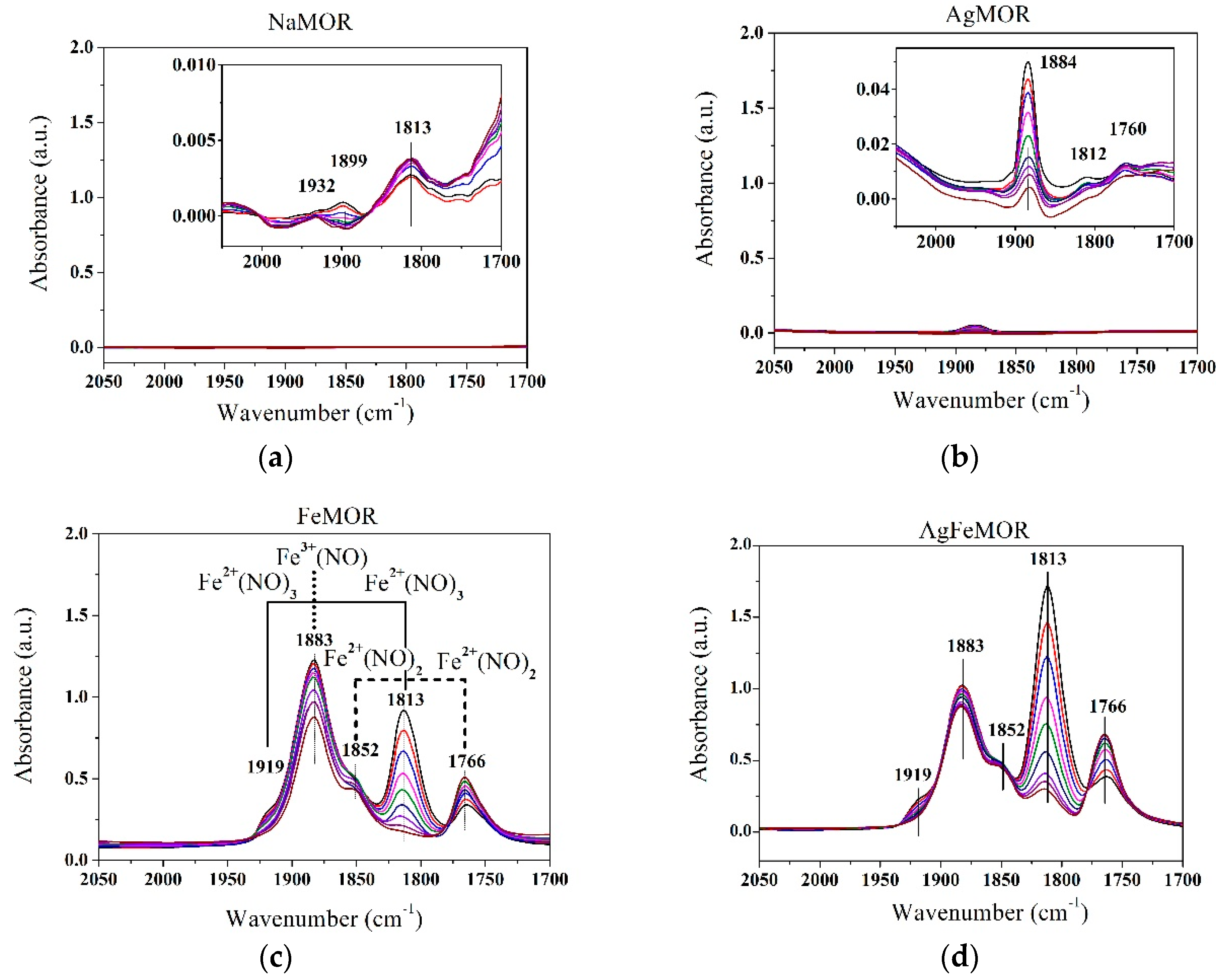




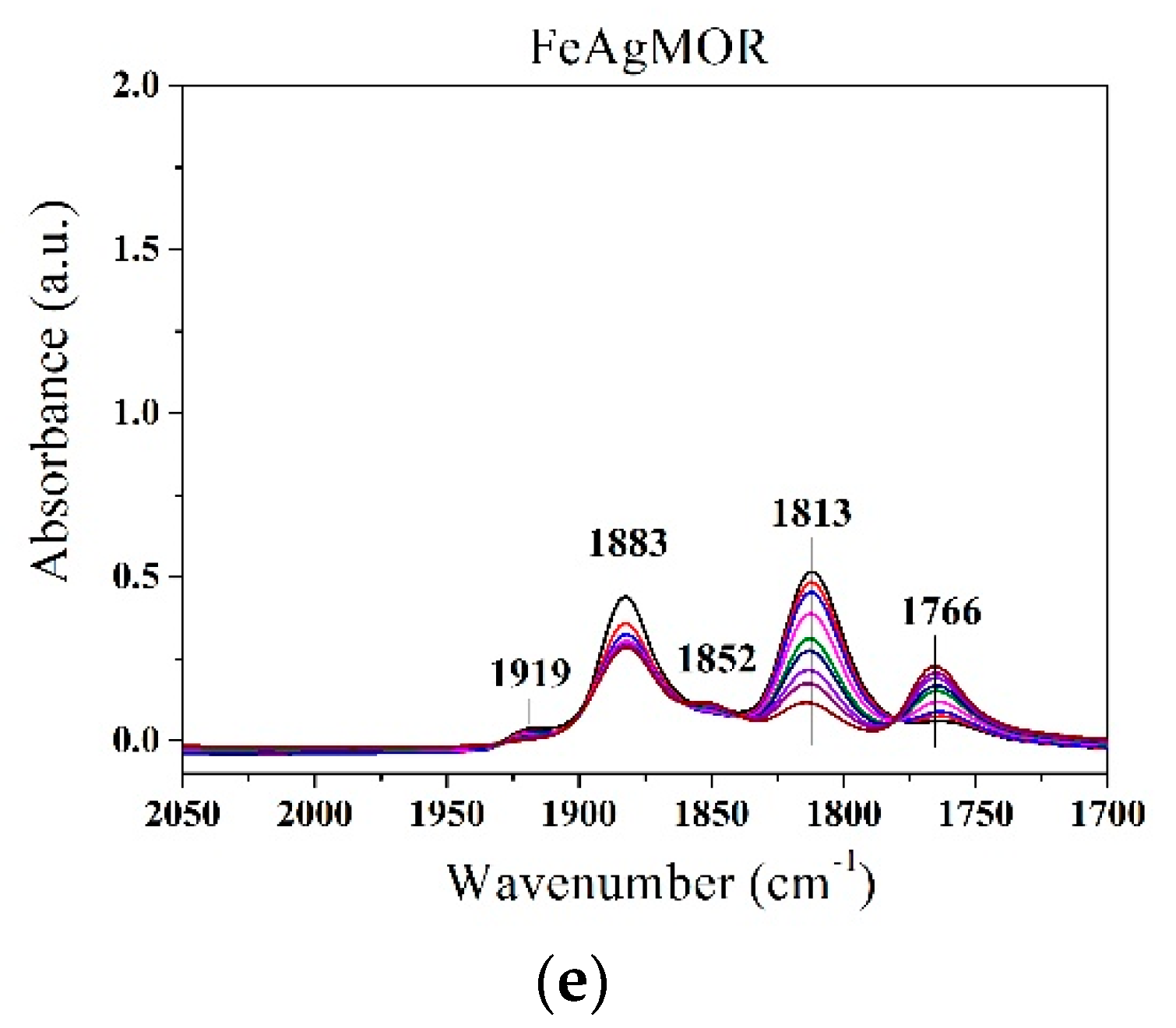
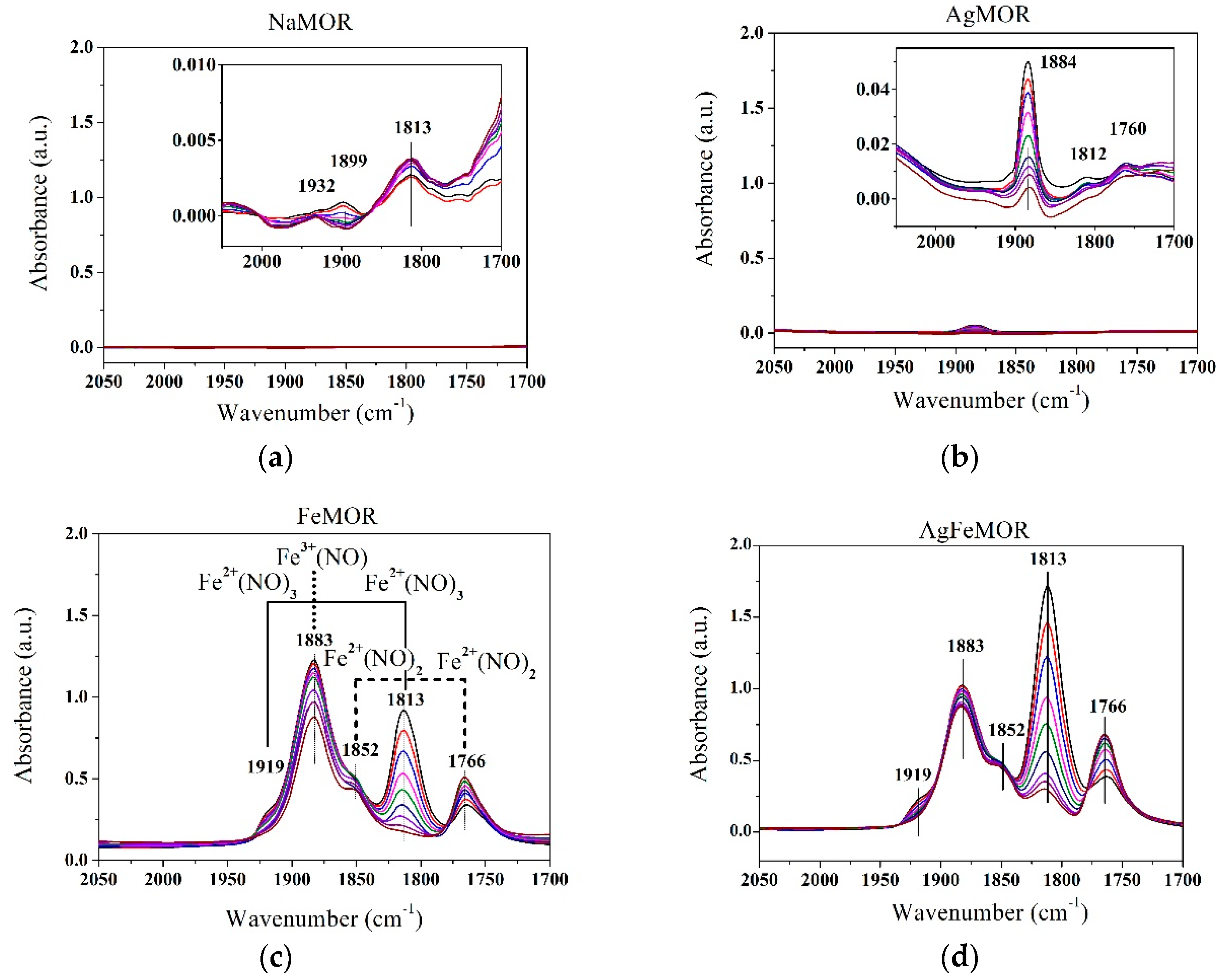{kind=link}
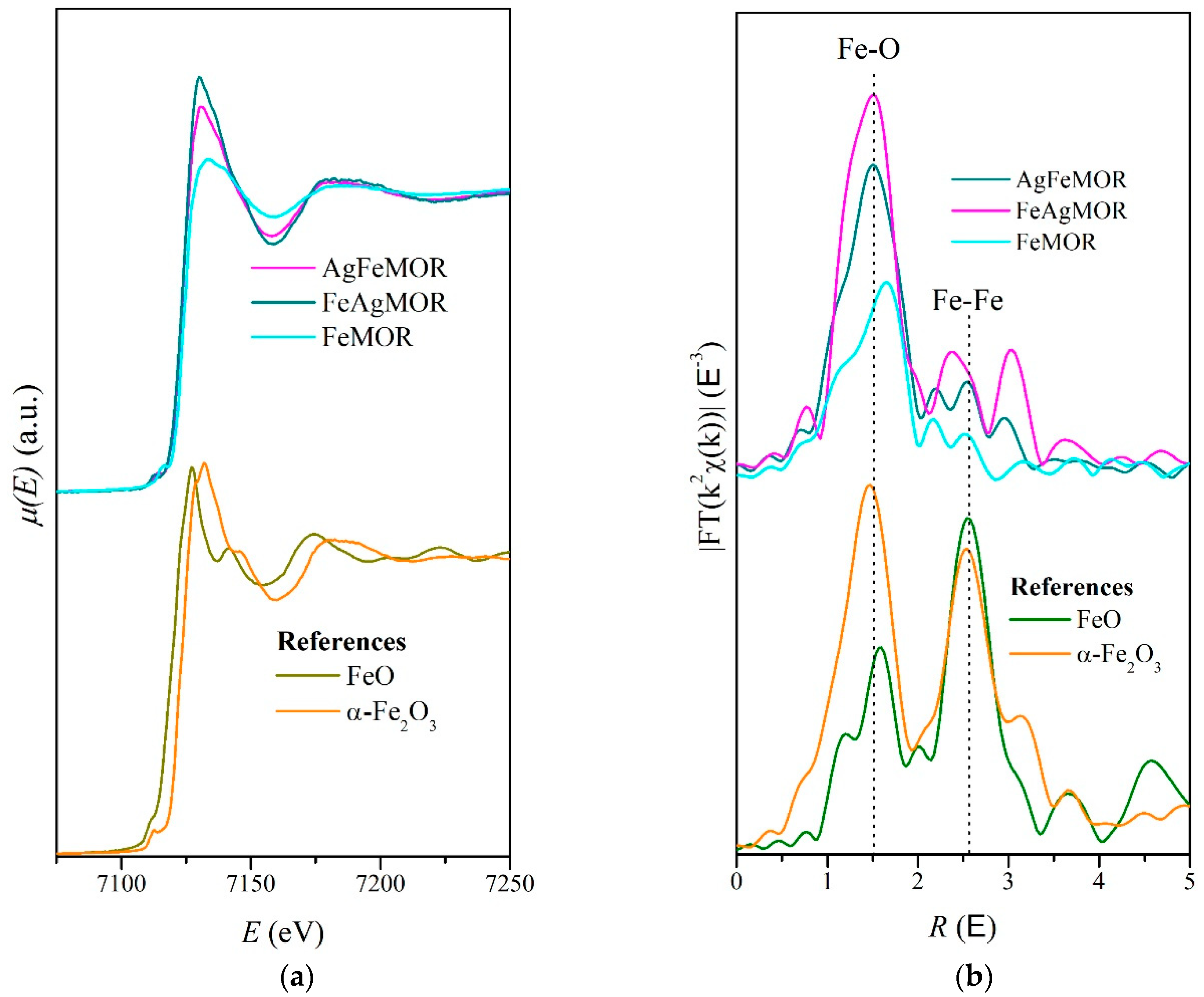
{kind=link}
{kind=link}
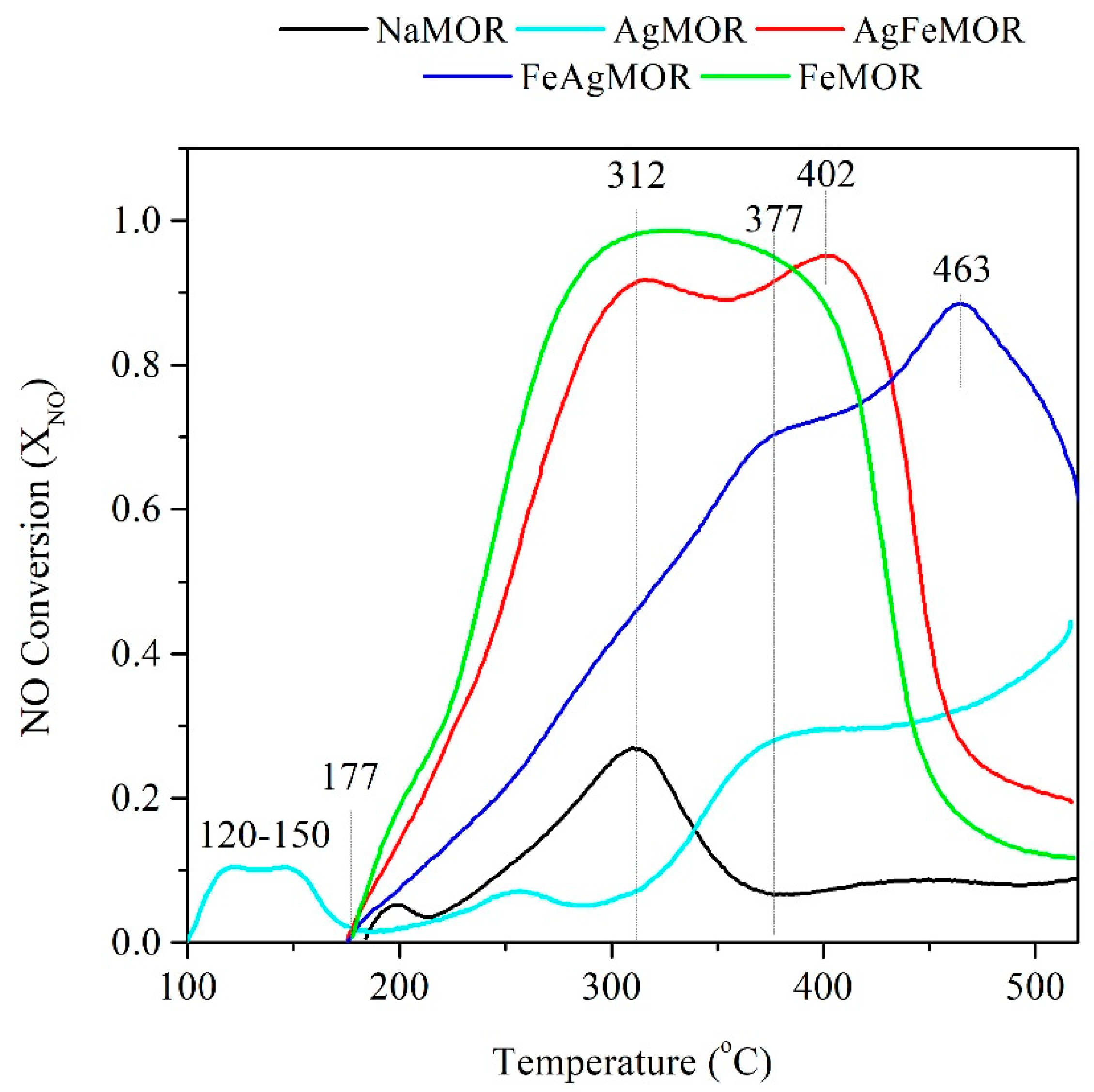{kind=link}
{kind=link}
| Sample | Atomic, % | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Si | Al | O | Ag | Fe | Na | Si/Al | EIEM | ||
| EIEM–Fe2+ | EIEM–Fe3+ | ||||||||
| NaMOR | 48.9 | 7.5 | 33.9 | - | - | 9.7 | 6.5 | 1.29 | |
| AgMOR | 38.1 | 5.8 | 49.9 | 4.6 | - | 1.6 | 6.5 | 1.07 | |
| FeMOR | 44.0 | 6.4 | 45.4 | - | 0.8 | 3.4 | 6.8 | 0.78 | 0.91 |
| FeAgMOR | 37.2 | 5.4 | 51.9 | 3.6 | 0.4 | 1.5 | 6.9 | 1.09 | 1.17 |
| AgFeMOR | 39.7 | 5.9 | 49.7 | 3.1 | 0.9 | 0.7 | 6.7 | 0.95 | 1.10 |
| Target Cation | (Z/r) pm | Cation-oxygen Bond Dissociation Energies, kJ mole−1 | Hydration Enthalpy, kJ Mole−1 | −ΔH° of the Transition of the Cation from the Zeolite-Bound State to the Dissolved State, kJ mole−1 |
|---|---|---|---|---|
| Na+ | 102 | 270 ± 4 | 409 ± 3 | 139 |
| Ag+ | 115 | 221 ± 21 | 473 ± 3 | 252 |
| H+ | 10 | 430 ± 0.29 | 1091 ± 5 | 661 |
| Fe2+ | 78 | 407 ± 1 | 1946 ± 6 | 1539 |
| Fe3+ | 65 | 407 ± 1 | 4430 ± 10 | 4023 |
| Sample | SBET, m2∙g−1 | Vtotal, cm3∙g−1 | Vmicro, cm3∙g−1 | Pore Diameter, Å |
|---|---|---|---|---|
| NaMOR | 338 | 0.19 | 0.16 | 22.8 |
| AgMOR | 322 | 0.19 | 0.14 | 23.2 |
| FeMOR | 398 | 0.23 | 0.17 | 23.3 |
| AgFeMOR | 343 | 0.20 | 0.15 | 23.7 |
| FeAgMOR | 336 | 0.20 | 0.14 | 24.1 |
| Sample | Amount of the Acid Sites, % | Total Acidity, mmol/g | |
|---|---|---|---|
| l-peak | h-peak | ||
| NaMOR | 32 | 68 | 1359 |
| AgMOR | 33 | 67 | 1662 |
| FeMOR | 16 | 84 | 2970 |
| AgFeMOR | 29 | 71 | 2264 |
| FeAgMOR | 19 | 81 | 1251 |
| Sample | Scattering Path | Coordination Number | Interatomic Distance, Å | Debye Factor, Å2 | R-Factor, % |
|---|---|---|---|---|---|
| FeMOR | Fe-O | 0.6 | 1.85 | 0.0061 | 1.7 |
| 1.9 | 2.06 | 0.0061 | |||
| AgFeMOR | Fe-O | 1.6 | 1.94 | 0.0049 | 0.9 |
| 2.0 | 2.10 | 0.0049 | |||
| FeAgMOR | Fe-O | 2.0 | 1.91 | 0.0024 | 1.9 |
| 2.2 | 2.07 | 0.0024 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-López, P.; Kotolevich, Y.; Khramov, E.; Chowdari, R.K.; Estrada, M.A.; Berlier, G.; Zubavichus, Y.; Fuentes, S.; Petranovskii, V.; Chávez-Rivas, F. Properties of Iron-Modified-by-Silver Supported on Mordenite as Catalysts for NOx Reduction. Catalysts 2020, 10, 1156. https://doi.org/10.3390/catal10101156
Sánchez-López P, Kotolevich Y, Khramov E, Chowdari RK, Estrada MA, Berlier G, Zubavichus Y, Fuentes S, Petranovskii V, Chávez-Rivas F. Properties of Iron-Modified-by-Silver Supported on Mordenite as Catalysts for NOx Reduction. Catalysts. 2020; 10(10):1156. https://doi.org/10.3390/catal10101156
Chicago/Turabian StyleSánchez-López, Perla, Yulia Kotolevich, Evgeny Khramov, Ramesh Kumar Chowdari, Miguel Angel Estrada, Gloria Berlier, Yan Zubavichus, Sergio Fuentes, Vitalii Petranovskii, and Fernando Chávez-Rivas. 2020. "Properties of Iron-Modified-by-Silver Supported on Mordenite as Catalysts for NOx Reduction" Catalysts 10, no. 10: 1156. https://doi.org/10.3390/catal10101156