Development of Specific Inhibitors for Oncogenic Phosphatase PPM1D by Using Ion-Responsive DNA Aptamer Library

Abstract
:1. Introduction
2. Results
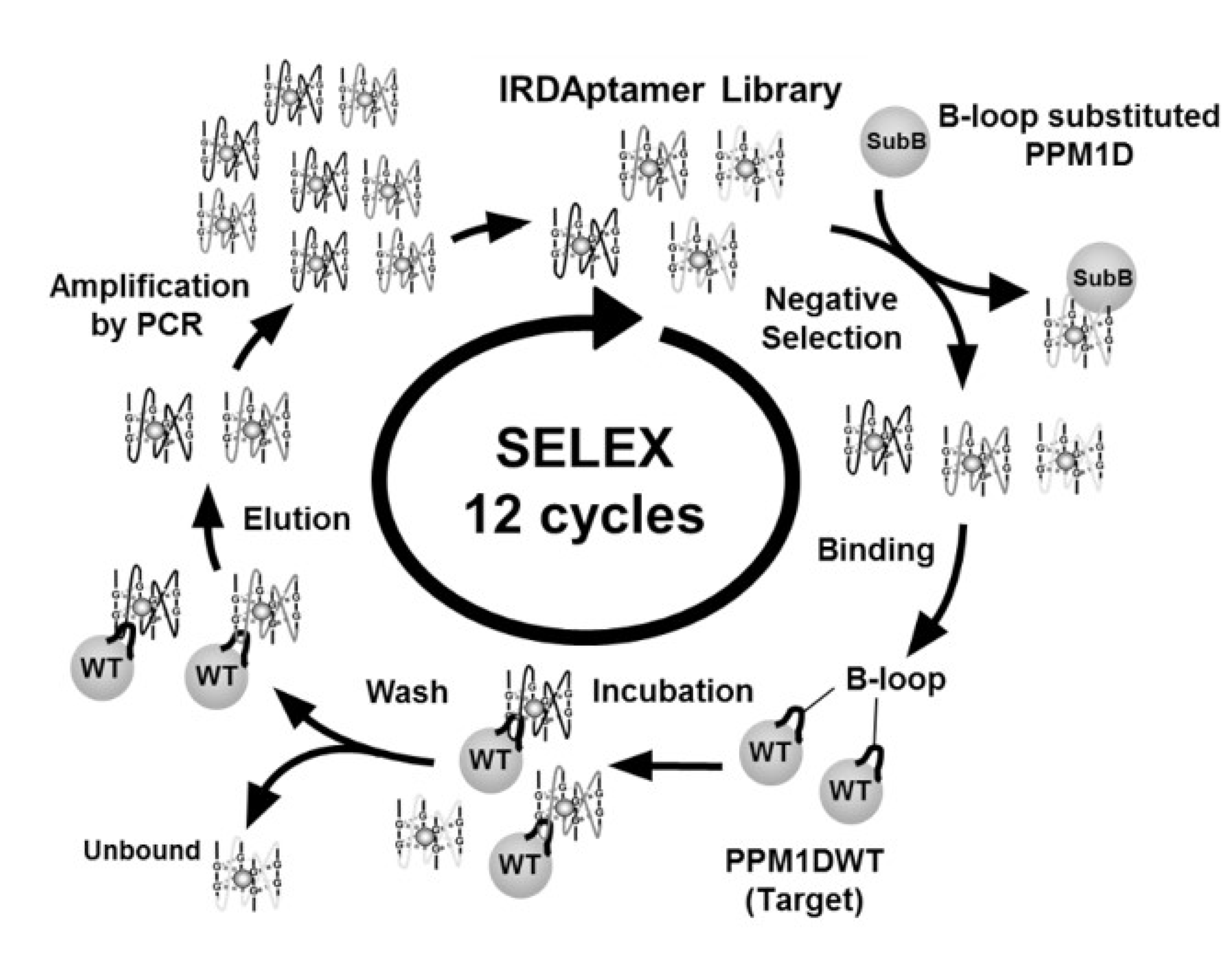
2.1. Screening of Ion-Responsive G-Quadruplex DNA Aptamer Targeting PPM1D
2.2. Binding Property of M1D-Q5F Aptamer on PPM1D Inhibition
2.3. Ion-Responsibility of M1D-Q5F Aptamer in the Conformation and Inhibitory Activity
2.4. Stability of M1D-Q5F Aptamer
2.5. Effect of M1D-Q5F Aptamer on Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Preparation of Recombinant Protein
4.2. DNA Library Design
4.3. Screening of DNA Aptamer Using SELEX Method
4.4. Malachite Green Assay Using Phosphorylated Peptide
4.5. pNPPase Assay
4.6. ELISA Binding Assay
4.7. Intermolecular Interaction Analysis Using BLItzTM System
4.8. Circular Dichroism Spectroscopy
4.9. Serum Stability and Nuclease Resistance of Aptamer
4.10. Cell Lines
4.11. Antibodies and Western Blotting Analysis
4.12. Cell Proliferation Assay
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lu, X.; Nguyen, T.A.; Moon, S.H.; Darlington, Y.; Sommer, M.; Donehower, L.A. The type 2C phosphatase Wip1: An oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008, 27, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambros, M.B.; Natrajan, R.; Geyer, F.C.; Lopez-Garcia, M.A.; Dedes, K.J.; Savage, K.; Lacroix-Triki, M.; Jones, R.L.; Lord, C.J.; Linardopoulos, S.; et al. PPM1D gene amplification and overexpression in breast cancer: A qRT-PCR and chromogenic in situ hybridization study. Mod. Pathol. 2010, 23, 1334–1345. [Google Scholar] [CrossRef] [Green Version]
- Chuman, Y.; Yagi, H.; Fukuda, T.; Nomura, T.; Matsukizono, M.; Shimohigashi, Y.; Sakaguchi, K. Characterization of the active site and a unique uncompetitive inhibitor of the PPM1-type protein phosphatase PPM1D. Protein Pept. Lett. 2008, 15, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Ruark, E.; Snape, K.; Humburg, P.; Loveday, C.; Bajrami, I.; Brough, R.; Rodrigues, D.N.; Renwick, A.; Seal, S.; Ramsay, E.; et al. Breast and Ovarian Cancer Susceptibility Collaboration; Wellcome Trust Case Control Consortium. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature 2013, 493, 406–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuman, Y.; Kurihashi, W.; Mizukami, Y.; Nashimoto, T.; Yagi, H.; Sakaguchi, K. PPM1D430, a novel alternative splicing variant of the human PPM1D, can dephosphorylate p53 and exhibits specific tissue expression. J. Biochem. 2009, 145, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nannenga, B.; Lu, X.; Dumble, M.; Van Maanen, M.; Nguyen, T.A.; Sutton, R.; Kumar, T.R.; Donehower, L.A. Augmented cancer resistance and DNA damage response phenotypes in PPM1D null mice. Mol. Carcinog. 2006, 45, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Nannenga, B.; Demidov, O.N.; Bulavin, D.V.; Cooney, A.; Brayton, C.; Zhang, Y.; Mbawuike, I.N.; Bradley, A.; Appella, E.; et al. Mice deficient for the wild-type p53-induced phosphatase gene (Wip1) exhibit defects in reproductive organs, immune function, and cell cycle control. Mol. Cell Biol. 2002, 22, 1094–1105. [Google Scholar] [CrossRef] [Green Version]
- Yagi, H.; Chuman, Y.; Kozakai, Y.; Imagawa, T.; Takahashi, Y.; Yoshimura, F.; Tanino, K.; Sakaguchi, K. A small molecule inhibitor of p53-inducible protein phosphatase PPM1D. Bioorg. Med. Chem. Lett. 2012, 22, 729–732. [Google Scholar] [CrossRef]
- Ogasawara, S.; Kiyota, Y.; Chuman, Y.; Kowata, A.; Yoshimura, F.; Tanino, K.; Kamada, R.; Sakaguchi, K. Novel inhibitors targeting PPM1D phosphatase potently suppress cancer cell proliferation. Bioorg. Med. Chem. 2015, 23, 6246–6249. [Google Scholar] [CrossRef]
- Hayashi, R.; Tanoue, K.; Durell, S.R.; Chatterjee, D.K.; Jenkins, L.M.; Appella, D.H.; Appella, E. Optimization of a cyclic peptide inhibitor of Ser/Thr phosphatase PPM1D (Wip1). Biochemistry 2011, 50, 4537–4549. [Google Scholar] [CrossRef] [Green Version]
- Gilmartin, A.G.; Faitg, T.H.; Richter, M.; Groy, A.; Seefeld, M.A.; Darcy, M.G.; Peng, X.; Federowicz, K.; Yang, J.; Zhang, S.Y.; et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat. Chem. Biol. 2014, 10, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Guo, D. Targeted Therapy for Hepatocellular Carcinoma: Co-Delivery of Sorafenib and Curcumin Using Lactosylated pH-Responsive Nanoparticles. Drug Des. Devel. Ther. 2020, 14, 647–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, P.; Gustafson, J.A.; MacKay, J.A. Genetically engineered nanocarriers for drug delivery. Int. J. Nanomed. 2014, 9, 1617–1626. [Google Scholar]
- Deiana, M.; Pokladek, Z.; Olesiak-Banska, J.; Młynarz, P.; Samoc, M.; Matczyszyn, K. Photochromic switching of the DNA helicity induced by azobenzene derivatives. Sci. Rep. 2016, 6, 28605. [Google Scholar] [CrossRef] [Green Version]
- Gold, L. Oligonucleotides as research, diagnostic, and therapeutic agents. J. Biol. Chem. 1995, 270, 13581–13584. [Google Scholar] [CrossRef] [Green Version]
- Antipova, O.M.; Zavyalova, E.G.; Golovin, A.V.; Pavlova, G.V.; Kopylov, A.M.; Reshetnikov, R.V. Advances in the application of modified nucleotides in SELEX technology. Biochemistry 2018, 83, 1161–1172. [Google Scholar] [CrossRef]
- Bayat, P.; Nosrati, R.; Alibolandi, M.; Rafatpanah, H.; Abnous, K.; Khedri, M.; Ramezani, M. SELEX methods on the road to protein targeting with nucleic acid aptamers. Biochimie 2018, 154, 132–155. [Google Scholar] [CrossRef]
- Wu, Y.X.; Kwon, Y.J. Aptamers: The “evolution” of SELEX. Methods 2016, 106, 21–28. [Google Scholar] [CrossRef]
- Hori, S.; Herrera, A.; Rossi, J.J.; Zhou, J. Current Advances in Aptamers for Cancer Diagnosis and Therapy. Cancers 2018, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Chandola, C.; Kalme, S.; Casteleijn, M.G.; Urtti, A.; Neerathilingam, M. Application of aptamers in diagnostics, drug-delivery and imaging. J. Biosci. 2016, 41, 535–561. [Google Scholar] [CrossRef] [Green Version]
- Ng, E.W.; Shima, D.T.; Calias, P.; Cunningham, E.T., Jr.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.J.; Reyes-Reyes, E.M.; Malik, M.T.; Murphy, E.M.; O’Toole, M.G.; Trent, J.O. G-quadruplex oligonucleotide as a cancer-targeting agent: Uses and mechanisms. Biochimica Biophysica Acta 2017, 1861, 1414–1428. [Google Scholar] [CrossRef] [PubMed]
- Nabavinia, M.S.; Gholoobi, A.; Charbgoo, F.; Nabavinia, M.; Ramezani, M.; Abnous, K. Anti-MUC1 aptamer: A potential opportunity for cancer treatment. Med. Res. Rev. 2017, 37, 1518–1539. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Liu, G.; Kai, M. DNA Aptamers in the Diagnosis and Treatment of Human Diseases. Molecules 2015, 20, 20979–20997. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, H.; Jain, A.; Zhang, L.; Liu, C.; Cheng, K. Discovery of Aptamer Ligands for Hepatic Stellate Cells Using SELEX. Theranostics 2017, 7, 2982–2995. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Wang, Z.; Chen, Z.; Pan, L. Switching the activity of Taq polymerase using clamp-like triplex aptamer structure. Nucleic Acids Res. 2020, 48, 8591–8600. [Google Scholar] [CrossRef]
- Catherine, A.T.; Shishido, S.N.; Robbins-Welty, G.A.; Diegelman-Parente, A. Rational design of a structure-switching DNA aptamer for potassium ions. FEBS Open Bio. 2014, 4, 788–795. [Google Scholar] [CrossRef] [Green Version]
- Lech, C.J.; Heddi, B.; Phan, A.T. Guanine base stacking in G-quadruplex nucleic acids. Nucleic Acids Res. 2013, 41, 2034–2046. [Google Scholar] [CrossRef] [Green Version]
- Roxo, C.; Kotkowiak, W.; Pasternak, A. G-Quadruplex-Forming Aptamers-Characteristics, Applications, and Perspectives. Molecules 2019, 24, 3781. [Google Scholar] [CrossRef] [Green Version]
- Kwok, C.K.; Merrick, C.J. G-Quadruplexes: Prediction, characterization, and biological application. Trends Biotechnol. 2017, 35, 997–1013. [Google Scholar] [CrossRef]
- Takenaka, S.; Juskowiak, B. Fluorescence detection of potassium ion using the G-quadruplex structure. Anal. Sci. 2011, 27, 1167–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, T.; Podbevšek, P.; Plavec, J.; Sugimoto, N. Effects of metal ions and cosolutes on G-quadruplex topology. J. Inorg. Biochem. 2017, 166, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, G.N.; Lee, M.P.; Neidle, S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 2002, 417, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.S.; Yoon, H.J.; Kim, B.; Yim, Y.H.; So, H.Y.; Shin, S.K. Mass spectrometric studies of alkali metal ion binding on thrombin-binding aptamer DNA. J. Am. Soc. Mass. Spectrom. 2010, 21, 1245–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, B.; Lin, Y.; Wang, C.; Li, F.; Wang, Z.; Zhang, H.; Li, X.F.; Le, X.C. Aptamer binding assays for proteins: The thrombin example–A review. Analytica Chimica Acta 2014, 837, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, N.; Ellington, A.; Stanton, M. Aptamer beacons for the direct detection of proteins. Anal. Biochem. 2001, 294, 126–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Fleming, A.M.; Averill, A.M.; Burrows, C.J.; Wallace, S.S. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 2015, 43, 4039–4054. [Google Scholar] [CrossRef] [Green Version]
- Riccardi, C.; Meyer, A.; Vasseur, J.; Krauss, I.R.; Paduano, L.; Morvan, F.; Montesarchio, D. Fine-tuning the properties of the thrombin binding aptamer through cyclization: Effect of the 5′-3′ connecting linker on the aptamer stability and anticoagulant activity. Bioorg. Chem. 2020, 94, 103379. [Google Scholar] [CrossRef]
- Hud, N.V.; Smith, F.W.; Anet, F.A.; Feigon, J. The selectivity for K+ versus Na+ in DNA quadruplexes is dominated by relative free energies of hydration: A thermodynamic analysis by 1H NMR. Biochemistry 1996, 35, 15383–15390. [Google Scholar] [CrossRef]
- Lewis, D.L.; Lechleiter, J.D.; Kim, D.; Nanavati, C.; Clapham, D.E. Intracellular regulation of ion channels in cell membranes. Mayo. Clin. Proc. 1990, 65, 1127–1143. [Google Scholar] [CrossRef] [Green Version]
- Harrison, M.; Li, J.; Degenhardt, Y.; Hoey, T.; Powers, S. Wip1-deficient mice are resistant to common cancer genes. Trends Mol. Med. 2004, 10, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Garrigos, M.; Orlowski, S.; Mir, L.M. Competitive and non-competitive inhibition of the multidrug-resistance-associated P-glycoprotein ATPase–Further experimental evidence for a multisite model. Eur. J. Biochem. 1997, 244, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Hu, X.; Chen, Z.; Liu, L.; Tian, Y.; Chen, H.; Cong, Y.S.; Yang, F.; Zhang, L.; Rudolph, K.L.; et al. Phosphatase Wip1 controls antigen-independent B-cell development in a p53-dependent manner. Blood 2015, 126, 620–628. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Demidov, O.N.; Goh, A.M.; Virshup, D.M.; Lane, D.P.; Bulavin, D.V. Phosphatase WIP1 regulates adult neurogenesis and WNT signaling during aging. J. Clin. Invest. 2014, 124, 3263–3273. [Google Scholar] [CrossRef]
- Cho, S.J.; Cha, B.S.; Kwon, O.S.; Lim, J.; Shin, D.M.; Han, D.W.; Ishitani, T.; Jho, E.H.; Fornace, A.J.; Cha, H.J. Wip1 directly dephosphorylates NLK and increases Wnt activity during germ cell development. Biochimica Biophysica Acta 2017, 1863, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Torii, S.; Yoshida, T.; Arakawa, S.; Honda, S.; Nakanishi, A.; Shimizu, S. Identification of PPM1D as an essential Ulk1 phosphatase for genotoxic stress-induced autophagy. EMBO Rep. 2016, 17, 1552–1564. [Google Scholar] [CrossRef]
- Li, D.; Zhang, L.; Xu, L.; Liu, L.; He, Y.; Zhang, Y.; Huang, X.; Zhao, T.; Wu, L.; Zhao, Y.; et al. WIP1 phosphatase is a critical regulator of adipogenesis through dephosphorylating PPARγ serine 112. Cell Mol. Life Sci. 2017, 74, 2067–2079. [Google Scholar] [CrossRef]
- Marino, A.A.; Iliev, I.G.; Schwalke, M.A.; Gonzalez, E.; Marler, K.C.; Flanagan, C.A. Association between cell membrane potential and breast cancer. Tumour Biol. 1994, 15, 82–89. [Google Scholar] [CrossRef]
- Frajese, G.V.; Benvenuto, M.; Fantini, M.; Ambrosin, E.; Sacchetti, P.; Masuelli, L.; Giganti, M.G.; Modesti, A.; Bei, R. Potassium increases the antitumor effects of ascorbic acid in breast cancer cell lines in vitro. Oncol. Lett. 2016, 11, 4224–4234. [Google Scholar] [CrossRef] [Green Version]
- Fairhurst, C.; Martin, F.; Watt, I.; Doran, T.; Bland, M.; Brackenbury, W.J. Sodium channel-inhibiting drugs and cancer survival: Protocol for a cohort study using the CPRD primary care database. BMJ Open 2016, 6, e011661. [Google Scholar] [CrossRef] [Green Version]
- Riccardi, C.; Napolitano, E.; Platella, C.; Musumeci, D.; Montesarchio, D. G-quadruplex-based aptamers targeting human thrombin: Discovery, chemical modifications and antithrombotic effects. Pharmacol. Ther. 2020, 7, 107649. [Google Scholar] [CrossRef] [PubMed]
- Odeh, F.; Nsairat, H.; Alshaer, H.; Ismail, M.A.; Esawi, E.; Qaqish, B.; Bawab, A.A.; Ismail, S.I. Aptamers chemistry: Chemical modifications and conjugation strategies. Molecules 2020, 25, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, T.; Nakamura, Y. Aptamers: A Review of Their Chemical Properties and Modifications for Therapeutic Application. Molecules 2019, 24, 4229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, P.J.; Laber, D.A.; Miller, D.M.; Thomas, S.D.; Trent, J.O. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp. Mol. Pathol. 2009, 86, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Bishop, J.S.; Guy-Caffey, J.K.; Ojwang, J.O.; Smith, S.R.; Hogan, M.E.; Cossum, P.A.; Rando, R.F.; Chaudhary, N. Intramolecular G-quartet motifs confer nuclease resistance to a potent anti-HIV oligonucleotide. J. Biol. Chem. 1996, 271, 5698–5703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.K.; Helps, N.R.; Cohen, P.T.; Barford, D. Crystal structure of the protein serine/threonine phosphatase 2C at 2.0 A resolution. EMBO J. 1996, 15, 6798–6809. [Google Scholar] [CrossRef]
- Almo, S.C.; Bonanno, J.B.; Sauder, J.M.; Emtage, S.; Dilorenzo, T.P.; Malashkevich, V.; Wasserman, S.R.; Swaminathan, S.; Eswaramoorthy, S.; Agarwal, R.; et al. Structural genomics of protein phosphatases. J. Struct. Funct. Genom. 2007, 8, 121–140. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, S.G.; Choi, H.J.; Rosenbaum, D.M.; Kobilka, T.S.; Thian, F.S.; Edwards, P.C.; Burghammer, M.; Ratnala, V.R.; Sanishvili, R.; Fischetti, R.F.; et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature 2007, 450, 383–387. [Google Scholar] [CrossRef]
- Gelinas, A.D.; Davies, D.R.; Edwards, T.E.; Rohloff, J.C.; Carter, J.D.; Zhang, C.; Gupta, S.; Ishikawa, Y.; Hirota, M.; Nakaishi, Y.; et al. Crystal structure of interleukin-6 in complex with a modified nucleic acid ligand. J. Biol. Chem. 2014, 289, 8720–8734. [Google Scholar] [CrossRef] [Green Version]
- Ahirwar, R.; Nahar, S.; Aggarwal, S.; Ramachandran, S.; Maiti, S.; Nahar, P. In silico selection of an aptamer to estrogen receptor alpha using computational docking employing estrogen response elements as aptamer-alike molecules. Sci. Rep. 2016, 6, 21285. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequences | Frequency |
|---|---|---|
| M1D-Q1F | 5′Primer-AAGGATAGTTTTAGGGTGTGCGTTAGGGTCCGG-TTAGGG-3′Primer | 13 |
| M1D-Q4F | 5′Primer-GAGGAAAAGCTTAGGGTAGACCTTAGG-TGGTTTTTAGGG-3′Primer | 4 |
| M1D-Q5F | 5′Primer-GAGGTAATTGTTAGGGGCGTTGTTAGGGTGGGACTTAGGG-3′Primer | 3 |
| M1D-Q6F | 5′Primer-AAGGGCTTGCTTAGGGAGGTGGTTAGGGAGGTTATTAGGG-3′Primer | 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaneko, A.; Watari, M.; Mizunuma, M.; Saito, H.; Furukawa, K.; Chuman, Y. Development of Specific Inhibitors for Oncogenic Phosphatase PPM1D by Using Ion-Responsive DNA Aptamer Library. Catalysts 2020, 10, 1153. https://doi.org/10.3390/catal10101153
Kaneko A, Watari M, Mizunuma M, Saito H, Furukawa K, Chuman Y. Development of Specific Inhibitors for Oncogenic Phosphatase PPM1D by Using Ion-Responsive DNA Aptamer Library. Catalysts. 2020; 10(10):1153. https://doi.org/10.3390/catal10101153
Chicago/Turabian StyleKaneko, Atsushi, Miyuu Watari, Masataka Mizunuma, Hikaru Saito, Kazuhiro Furukawa, and Yoshiro Chuman. 2020. "Development of Specific Inhibitors for Oncogenic Phosphatase PPM1D by Using Ion-Responsive DNA Aptamer Library" Catalysts 10, no. 10: 1153. https://doi.org/10.3390/catal10101153
APA StyleKaneko, A., Watari, M., Mizunuma, M., Saito, H., Furukawa, K., & Chuman, Y. (2020). Development of Specific Inhibitors for Oncogenic Phosphatase PPM1D by Using Ion-Responsive DNA Aptamer Library. Catalysts, 10(10), 1153. https://doi.org/10.3390/catal10101153