Ethanol Dehydrogenation: A Reaction Path Study by Means of Temporal Analysis of Products

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Catalyst Characterization
2.1.1. N2 Sorption
2.1.2. Scanning Transmission Electron Microscopy
2.2. Temporal Analysis of Products
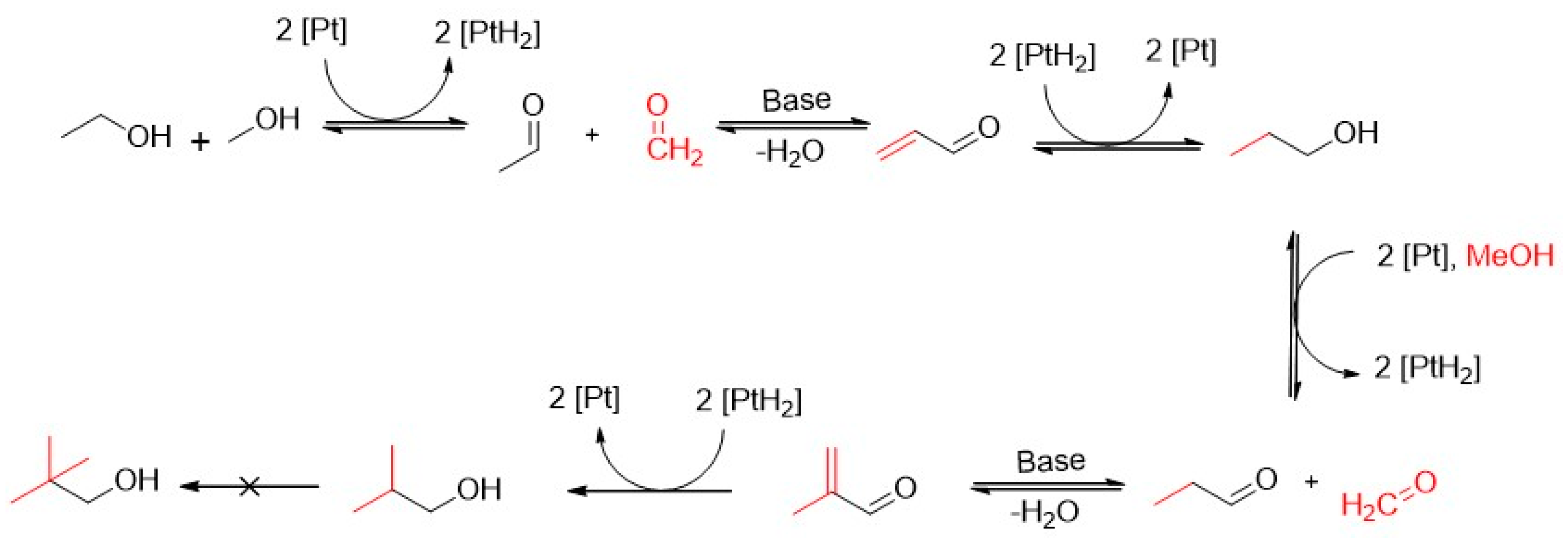
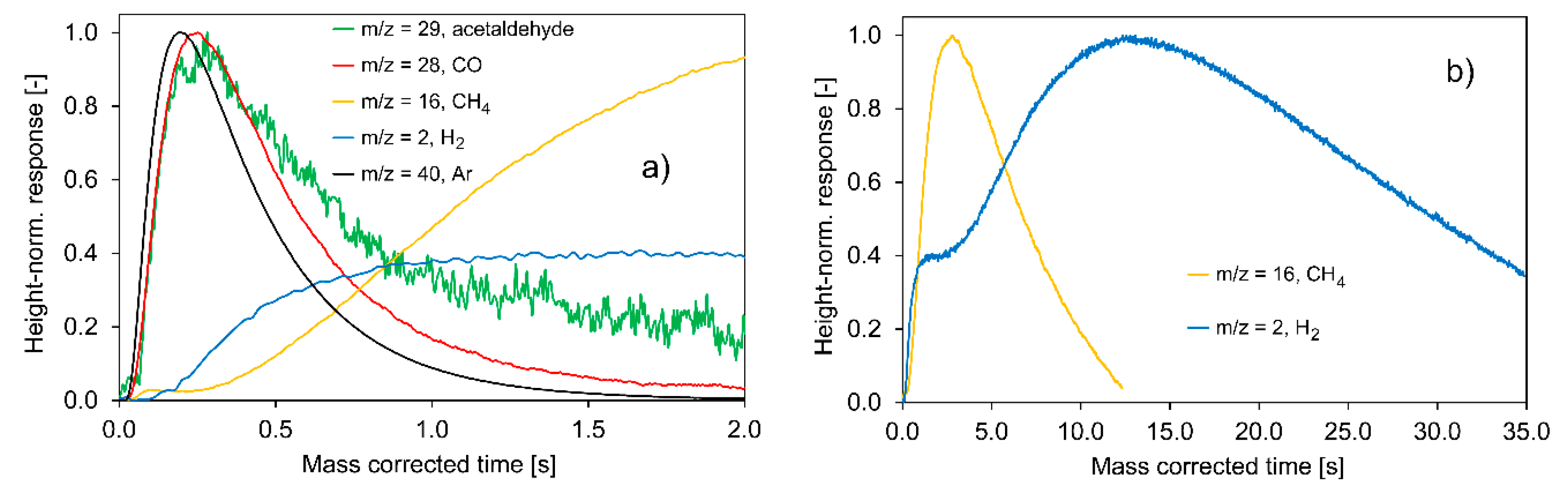
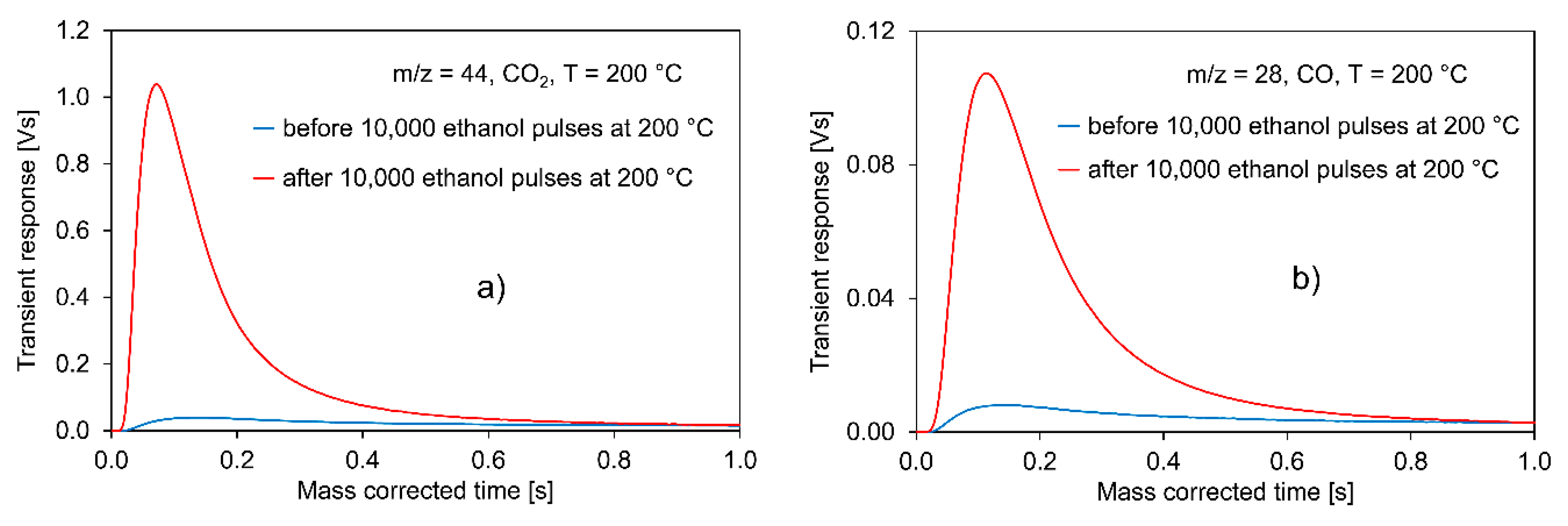
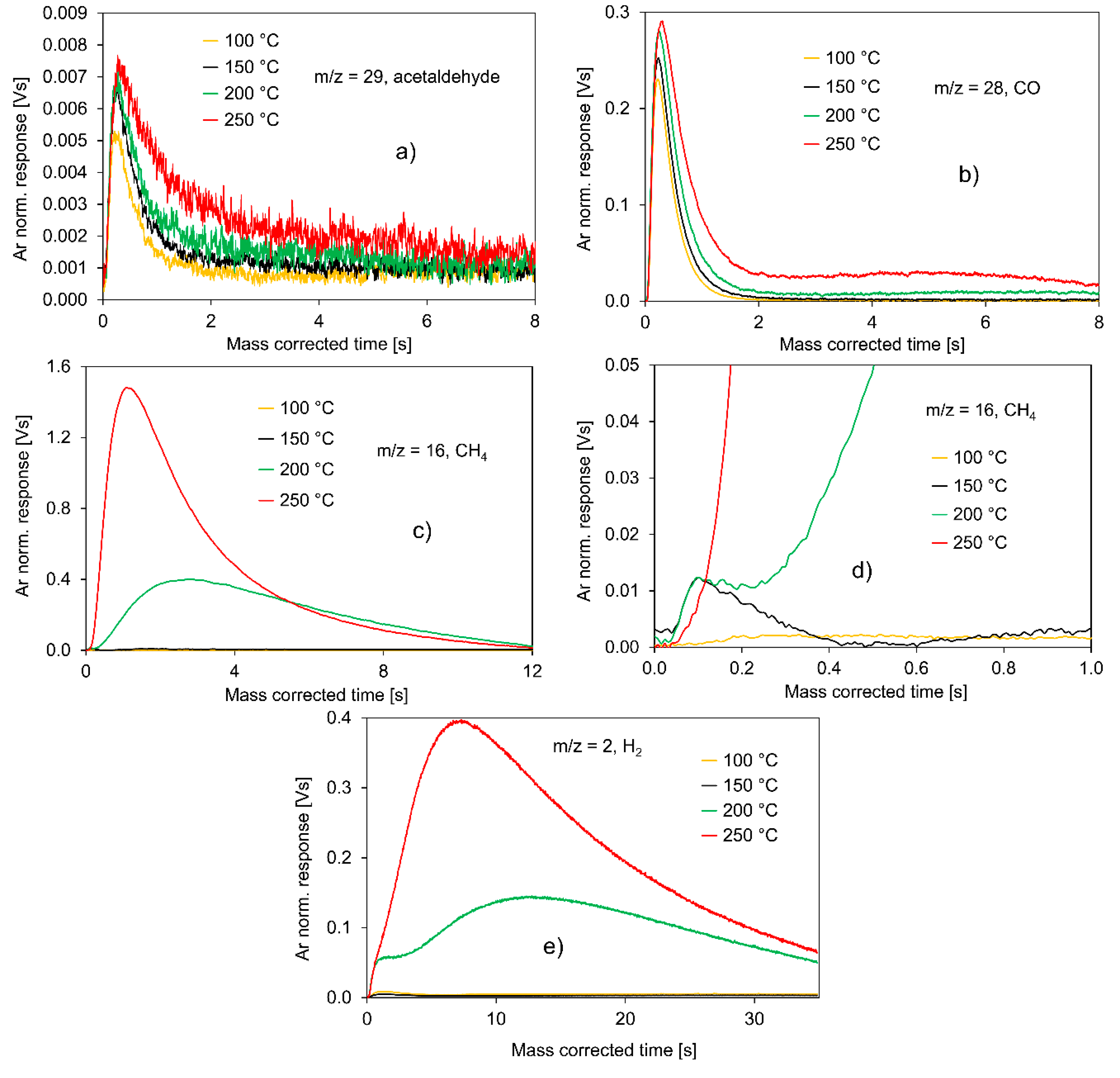
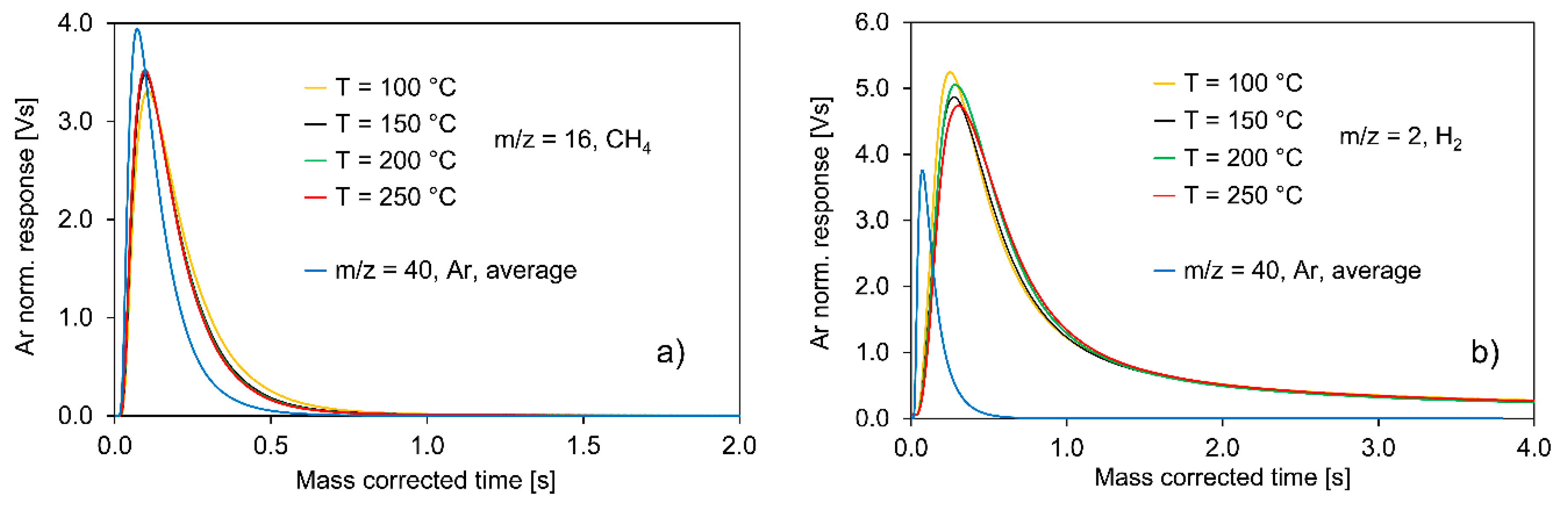
2.2.1. Experiments with Pt/C
- The total consumption of ethanol by the catalyst surface;
- The slow CH4 and H2 evolution curves;
- The relatively fast evolution of CO and acetaldehyde on a comparable time scale;
- The strongly increased formation of CO2 and CO due to carbon burn-off on the catalyst surface after the 10,000 pulses of ethanol;
- The transient responses from single CH4/H2 pulsing over the Pt/C sample.
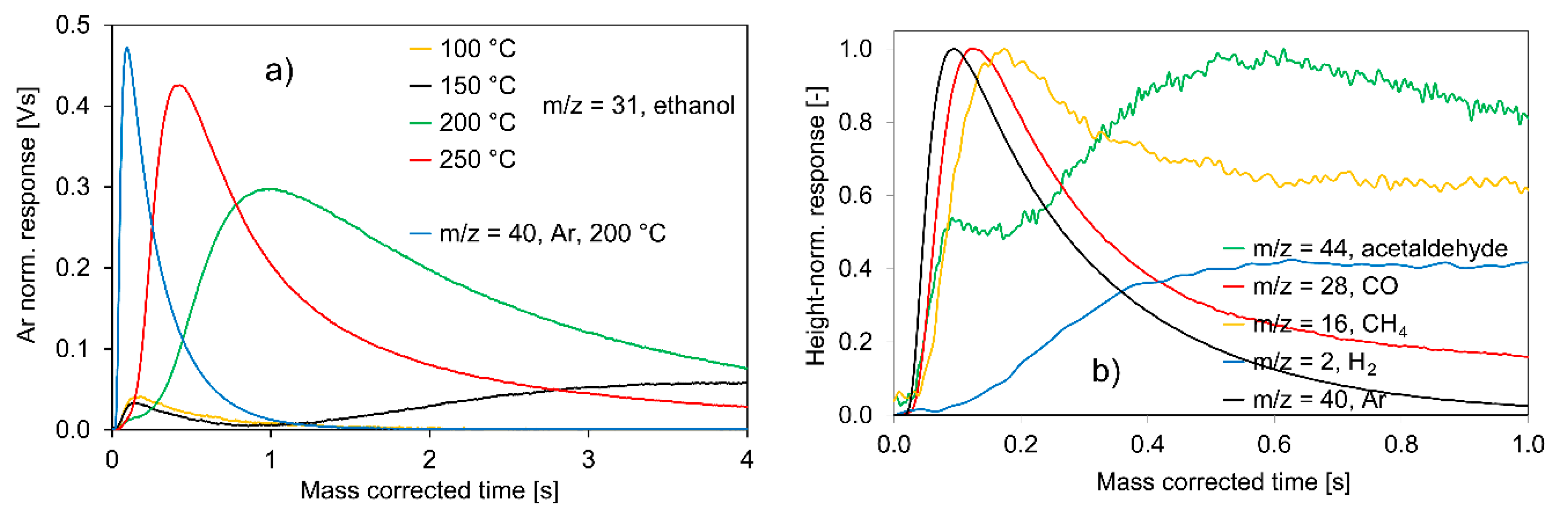
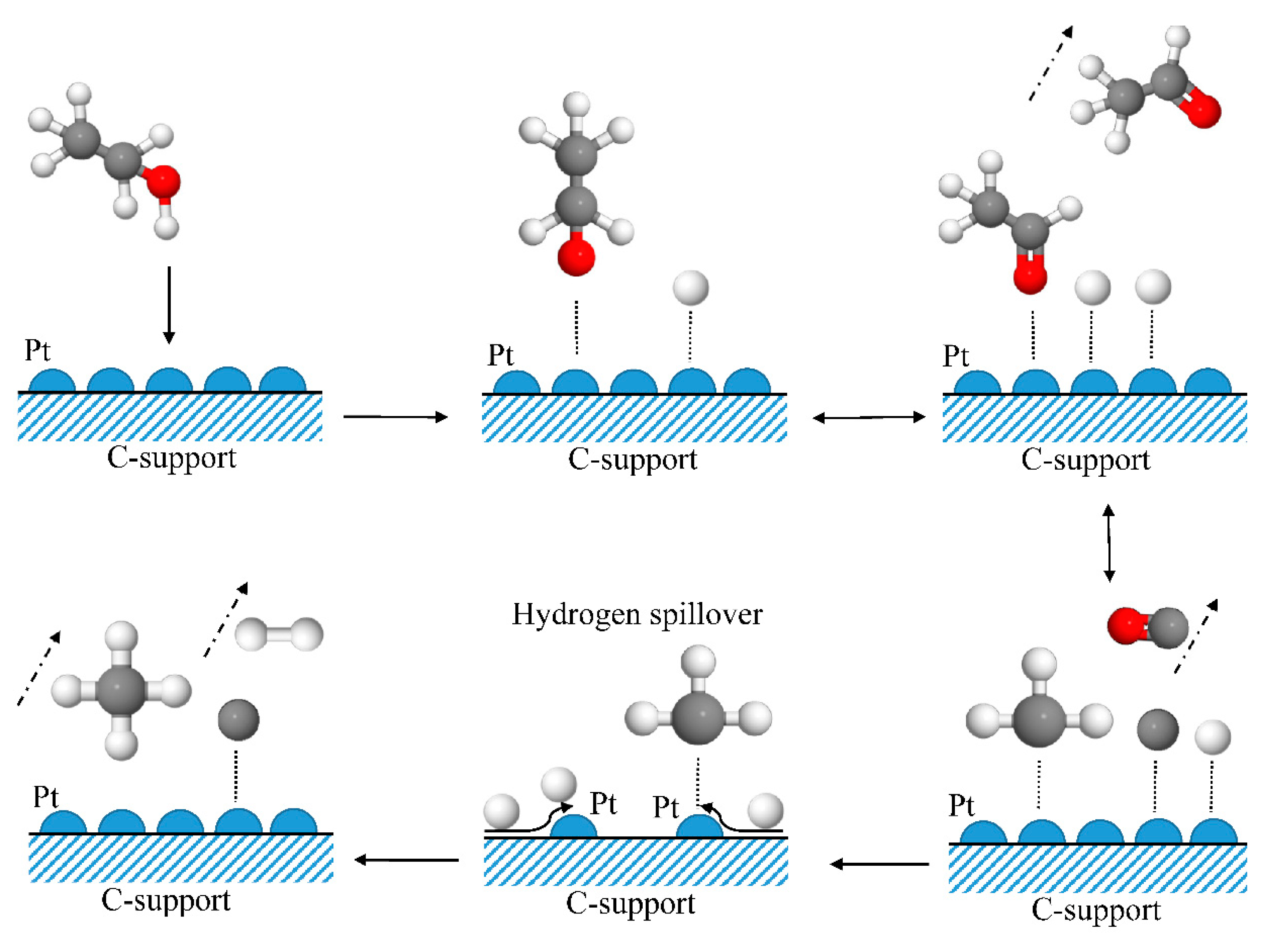
2.2.2. Experiments with Ir/C
- H2 and acetaldehyde as products of ethanol dehydrogenation are formed, together with CO and CH4 from acetaldehyde decomposition. Zhang et al. [67] and Cai et al. [68] investigated steam reforming of ethanol using an Ir/CeO2 catalyst. They first observed ethanol dehydrogenation to acetaldehyde followed by decomposition of acetaldehyde to CH4 and CO. These findings are in good agreement with those presented here.
- Other reaction products, such as acetic acid (m/z = 60), butanol isomers (m/z = 56), diethyl ether (m/z = 59) or ethyl acetate (m/z = 70), are not detected. Boualouche et al. [66], however, found CuIr alloys being active for the formation of ethyl acetate.
- The evolution curve of CO and that of acetaldehyde at lower residence times are very narrow and only slightly broader than that of Ar, representing the diffusion-only case.
- The transient responses of CO and acetaldehyde coincide in their rising sections at lower residence times. This again means that they desorb on a comparable time scale from the catalyst surface within this time-span and confirms reaction step (6) for the Ir/C catalyst, i.e., the fast CO formation from acetaldehyde.
- The response pulses of H2 and those of CH4 at higher residence times are very broad, again raising the question of whether the recombination steps of long-lived CH3* and H* adsorbates, as per Equations (7) and (8), are responsible or reversible adsorption and spillover processes.
- The acetaldehyde response pulse is much broader at longer residence times, indicating that reaction step (5), i.e., the decomposition of the ethoxy adsorbate into rapidly desorbing acetaldehyde and H* species, should be extended by the formation of a long-lived CH3CHO* adsorbate.
- The evolution curve for CH4 reveals a fairly narrow section at shorter residence times. On Ir/C, the decomposition of acetaldehyde, according to reaction step (6), directly yields a small fraction of rapidly desorbing CH4 molecules.
2.2.3. Experiments with Cu/C
3. Materials and Methods
3.1. Catalysts
3.2. N2 Sorption
3.3. Scanning Electron Microscopy
3.4. Temporal Analysis of Products
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, D.; Liu, Z.; Liu, Q. Efficient conversion of ethanol to 1-butanol and C5-C9 alcohols over calcium carbide. RSC Adv. 2019, 9, 18941–18948. [Google Scholar] [CrossRef] [Green Version]
- Hui, X.; Niemeyer, K.E.; Brady, K.B.; Sung, C.J. Reduced chemistry for butanol isomers at engine-relevant conditions. Energy Fuels 2017, 31, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Quesada, J.; Faba, L.; Díaz, E.; Ordóñez, S. Copper-Basic Sites Synergic Effect on the Ethanol Dehydrogenation and Condensation Reactions. ChemCatChem 2018, 10, 3583–3592. [Google Scholar] [CrossRef]
- Tayrabekova, S.; Mäki-Arvela, P.; Peurla, M.; Paturi, P.; Eränen, K.; Ergazieva, G.E.; Aho, A.; Murzin, D.Y.; Dossumov, K. Catalytic dehydrogenation of ethanol into acetaldehyde and isobutanol using mono- and multicomponent copper catalysts. Comptes Rendus Chim. 2018, 21, 194–209. [Google Scholar] [CrossRef]
- Ochoa, J.V.; Farci, E.; Cavani, F.; Sinisi, F.; Artiglia, L.; Agnoli, S.; Granozzi, G.; Paganini, M.C.; Malfatti, L. CeOx/TiO2 (Rutile) Nanocomposites for the Low-Temperature Dehydrogenation of Ethanol to Acetaldehyde: A Diffuse Reflectance Infrared Fourier Transform Spectroscopy-Mass Spectrometry Study. ACS Appl. Nano Mater. 2019, 2, 3434–3443. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, K.; An, Z.; Zhu, Y.; Shu, X.; Song, H.; Xiang, X.; He, J. Acid-Base Promoted Dehydrogenation Coupling of Ethanol on Supported Ag Particles. Ind. Eng. Chem. Res. 2020, 59, 3342–3350. [Google Scholar] [CrossRef]
- Schemme, S.; Breuer, J.L.; Samsun, R.C.; Peters, R.; Stolten, D. Promising catalytic synthesis pathways towards higher alcohols as suitable transport fuels based on H2 and CO2. J. CO2 Util. 2018, 27, 223–237. [Google Scholar] [CrossRef]
- Wouters, C.; Lehrheuer, B.; Heuser, B.; Pischinger, S. Ottomischkraftstoffe mit Methanol, Ethanol und Butanol. MTZ—Mot. Z. 2020, 81, 16–23. [Google Scholar] [CrossRef]
- Singh, P.; Hui, X.; Sung, C.J. Soot formation in non-premixed counterflow flames of butane and butanol isomers. Combust. Flame 2016, 164, 167–182. [Google Scholar] [CrossRef] [Green Version]
- Siddiki, S.M.A.H.; Touchy, A.S.; Jamil, M.A.R.; Toyao, T.; Shimizu, K.-i. C-Methylation of Alcohols, Ketones, and Indoles with Methanol Using Heterogeneous Platinum Catalysts. ACS Catal. 2018, 8, 3091–3103. [Google Scholar] [CrossRef]
- Pellow, K.J.; Wingad, R.L.; Wass, D.F. Towards the upgrading of fermentation broths to advanced biofuels: A water tolerant catalyst for the conversion of ethanol to isobutanol. Catal. Sci. Technol. 2017, 7, 5128–5134. [Google Scholar] [CrossRef] [Green Version]
- Häusler, J.; Pasel, J.; Woltmann, F.; Peters, R.; Stolten, D. How the D-Band Center Determines the Catalytic Activity in the Synthesis of Carbon Neutral Fuels; EFCATS Summer School: Portoroze, Slovenia, 2020. [Google Scholar]
- Ashok, A.; Kumar, A.; Bhosale, R.; Saad, M.A.S.; AlMomani, F.; Tarlochan, F. Study of ethanol dehydrogenation reaction mechanism for hydrogen production on combustion synthesized cobalt catalyst. Int. J. Hydrog. Energy 2017, 42, 23464–23473. [Google Scholar] [CrossRef]
- Kim, Y.K.; Ryu, S. Evidences for Different Reaction Sites for Dehydrogenation and Dehydration of Ethanol over Vanadia Supported on Titania. Bull. Korean Chem. Soc. 2019, 40, 489–495. [Google Scholar] [CrossRef]
- Krutpijit, C.; Tian, W.; Jongsomjit, B.; Pjontek, D.; Herrera, J.E. Lithium promotion in ethanol oxidative dehydrogenation over Al- modified Ag/Montmorillonite clays. Mol. Catal. 2020, 483, 110717. [Google Scholar] [CrossRef]
- Campisano, I.S.P.; Rodella, C.B.; Sousa, Z.S.B.; Henriques, C.A.; da Silva, V.T. Influence of thermal treatment conditions on the characteristics of Cu-based metal oxides derived from hydrotalcite-like compounds and their performance in bio-ethanol dehydrogenation to acetaldehyde. Catal. Today 2018, 306, 111–120. [Google Scholar] [CrossRef]
- Garbarino, G.; Riani, P.; García, M.V.; Finocchio, E.; Escribano, V.S.; Busca, G. A study of ethanol dehydrogenation to acetaldehyde over copper/zinc aluminate catalysts. Catal. Today 2019. [Google Scholar] [CrossRef]
- Kumar, A.; Ashok, A.; Bhosale, R.R.; al Momani, F. Ethanol dehydrogenation mechanism on cuni catalysts for hydrogen production. Adv. Mater.–TechConnect Briefs 2016, 2016, 4–7. [Google Scholar]
- Li, M.Y.; Lu, W.D.; He, L.; Schüth, F.; Lu, A.H. Tailoring the Surface Structure of Silicon Carbide Support for Copper Catalyzed Ethanol Dehydrogenation. ChemCatChem 2019, 11, 481–487. [Google Scholar] [CrossRef]
- Ponomareva, E.A.; Krasnikova, I.V.; Egorova, E.V.; Mishakov, I.V.; Vedyagin, A.A. Ethanol dehydrogenation over copper supported on carbon macrofibers. Mendeleev Commun. 2017, 27, 210–212. [Google Scholar] [CrossRef]
- Ponomareva, E.A.; Krasnikova, I.V.; Egorova, E.V.; Mishakov, I.V.; Vedyagin, A.A. Dehydrogenation of ethanol over carbon-supported Cu–Co catalysts modified by catalytic chemical vapor deposition. React. Kinet. Mech. Catal. 2017, 122, 399–408. [Google Scholar] [CrossRef]
- Ponomareva, E.A.; Shkinev, V.M.; Zaglyadova, S.V.; Krasnikova, I.V.; Egorova, E.V. Copper Catalysts based on carbon–carbon fiburous materials for ethanol dehydrogenation. Russ. J. Appl. Chem. 2016, 89, 598–602. [Google Scholar] [CrossRef]
- Ponomareva, Е.A.; Oladapo, T.O.; Egorova, Е.V. Bicomponent Catalysts for Ethanol Dehydrogenation. Russ. J. Gen. Chem. 2017, 87, 3093–3096. [Google Scholar] [CrossRef]
- Wang, Q.N.; Shi, L.; Li, W.; Li, W.C.; Si, R.; Schüth, F.; Lu, A.H. Cu supported on thin carbon layer-coated porous SiO2 for efficient ethanol dehydrogenation. Catal. Sci. Technol. 2018, 8, 472–479. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, Q.N.; Yang, X.; Wang, D.; Li, W.C.; Zheng, Y.; Chen, M.; Lu, A.H. A Highly Porous Carbon Support Rich in Graphitic-N Stabilizes Copper Nanocatalysts for Efficient Ethanol Dehydrogenation. ChemCatChem 2017, 9, 505–510. [Google Scholar] [CrossRef]
- Chuklina, S.G.; Pylinina, A.I.; Podzorova, L.I.; Mikhailina, N.A.; Mikhalenko, I.I. Ethanol dehydrogenation on copper catalysts with ytterbium stabilized tetragonal ZrO2 support. Russ. J. Phys. Chem. A 2016, 90, 2370–2376. [Google Scholar] [CrossRef]
- Hanukovich, S.; Dang, A.; Christopher, P. Influence of Metal Oxide Support Acid Sites on Cu-Catalyzed Nonoxidative Dehydrogenation of Ethanol to Acetaldehyde. ACS Catal. 2019, 9, 3537–3550. [Google Scholar] [CrossRef]
- De Waele, J.; Galvita, V.V.; Poelman, H.; Detavernier, C.; Thybaut, J.W. Formation and stability of an active PdZn nanoparticle catalyst on a hydrotalcite-based support for ethanol dehydrogenation. Catal. Sci. Technol. 2017, 7, 3715–3727. [Google Scholar] [CrossRef] [Green Version]
- Conesa, J.M.; Morales, M.V.; López-Olmos, C.; Rodríguez-Ramos, I.; Guerrero-Ruiz, A. Comparative study of Cu, Ag and Ag-Cu catalysts over graphite in the ethanol dehydrogenation reaction: Catalytic activity, deactivation and regeneration. Appl. Catal. A Gen. 2019, 576, 54–64. [Google Scholar] [CrossRef]
- Yu, D.; Dai, W.; Wu, G.; Guan, N.; Li, L. Stabilizing copper species using zeolite for ethanol catalytic dehydrogenation to acetaldehyde. Chin. J. Catal. 2019, 40, 1375–1384. [Google Scholar] [CrossRef]
- De Waele, J.; Galvita, V.V.; Poelman, H.; Gabrovska, M.; Nikolova, D.; Damyanova, S.; Thybaut, J.W. Ethanol dehydrogenation over Cu catalysts promoted with Ni: Stability control. Appl. Catal. A Gen. 2020, 591, 117401. [Google Scholar] [CrossRef]
- Rosset, M.; Perez-Lopez, O.W. Catalytic properties of Cu–Mg–Al hydrotalcites, their oxides and reduced phases for ethanol dehydrogenation, Reaction Kinetics. Mech. Catal. 2018, 123, 689–705. [Google Scholar]
- Rosset, M.; Perez-Lopez, O.W. Cu–Ca–Al catalysts derived from hydrocalumite and their application to ethanol dehydrogenation, Reaction Kinetics. Mech. Catal. 2019, 126, 497–511. [Google Scholar]
- Ohira, M.; Liu, H.; He, D.; Hirata, Y.; Sano, M.; Suzuki, T.; Miyake, T. Catalytic performance and reaction pathways of Cu/SiO2 and ZnO/SiO2 for dehydrogenation of ethanol to acetaldehyde. J. Japan Pet. Inst. 2018, 61, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Pinthong, P.; Praserthdam, P.; Jongsomjit, B. Oxidative dehydrogenation of ethanol over vanadium-and molybdenum-modified mg-al mixed oxide derived from hydrotalcite. J. Oleo Sci. 2019, 68, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Pinthong, P.; Praserthdam, P.; Jongsomjit, B. Effect of calcination temperature on mg-al layered double hydroxides (LDH) as promising catalysts in oxidative dehydrogenation of ethanol to acetaldehyde. J. Oleo Sci. 2019, 68, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, E.J.; Li, H.; Yu, W.Y.; Mullen, G.M.; Henkelman, G.; Mullins, C.B. Mechanistic insights on ethanol dehydrogenation on Pd-Au model catalysts: A combined experimental and DFT study. Phys. Chem. Chem. Phys. 2017, 19, 30578–30589. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Li, Y.; Liang, C. Dehydrogenation Mechanism of Ethanol on Co(111) Surface: A First-principles Study. Acta Chim. Sin. 2019, 77, 559–568. [Google Scholar] [CrossRef]
- Jalid, F.; Khan, T.S.; Haider, M.A. In-silico screening of Pt-based bimetallic alloy catalysts using ab initio microkinetic modeling for non-oxidative dehydrogenation of ethanol to produce acetaldehyde. MRS Commun. 2019, 9, 107–113. [Google Scholar] [CrossRef]
- Khan, T.S.; Jalid, F.; Haider, M.A. First-Principle Microkinetic Modeling of Ethanol Dehydrogenation on Metal Catalyst Surfaces in Non-oxidative Environment: Design of Bimetallic Alloys. Top. Catal. 2018, 61, 1820–1831. [Google Scholar] [CrossRef]
- Wang, Z.T.; Hoyt, R.A.; El-Soda, M.; Madix, R.J.; Kaxiras, E.; Sykes, E.C.H. Dry Dehydrogenation of Ethanol on Pt–Cu Single Atom Alloys. Top. Catal. 2018, 61, 328–335. [Google Scholar] [CrossRef]
- Autthanit, C.; Chatkaew, W.; Praserthdam, P.; Jongsomjit, B. Effect of different phase composition in titania on catalytic behaviors of AgLi/TiO2 catalysts via ethanol dehydrogenation. J. Environ. Chem. Eng. 2019, 8, 103547. [Google Scholar] [CrossRef]
- Autthanit, C.; Praserthdam, P.; Jongsomjit, B. Oxidative and non-oxidative dehydrogenation of ethanol to acetaldehyde over different VOx/SBA-15 catalysts. J. Environ. Chem. Eng. 2018, 6, 6516–6529. [Google Scholar] [CrossRef]
- Mamontov, G.V.; Grabchenko, M.V.; Sobolev, V.I.; Zaikovskii, V.I.; Vodyankina, O.V. Ethanol dehydrogenation over Ag-CeO2/SiO2 catalyst: Role of Ag-CeO2 interface. Appl. Catal. A Gen. 2016, 528, 161–167. [Google Scholar] [CrossRef]
- Dutov, V.V.; Mamontov, G.V.; Sobolev, V.I.; Vodyankina, O.V. Silica-supported silver-containing OMS-2 catalysts for ethanol oxidative dehydrogenation. Catal. Today 2016, 278, 164–173. [Google Scholar] [CrossRef]
- Giannakakis, G.; Trimpalis, A.; Shan, J.; Qi, Z.; Cao, S.; Liu, J.; Ye, J.; Biener, J.; Flytzani-Stephanopoulos, M. NiAu Single Atom Alloys for the Non-oxidative Dehydrogenation of Ethanol to Acetaldehyde and Hydrogen. Top. Catal. 2018, 61, 475–486. [Google Scholar] [CrossRef]
- Shan, J.; Janvelyan, N.; Li, H.; Liu, J.; Egle, T.M.; Ye, J.; Biener, M.M.; Biener, J.; Friend, C.M.; Flytzani-Stephanopoulos, M. Selective non-oxidative dehydrogenation of ethanol to acetaldehyde and hydrogen on highly dilute NiCu alloys. Appl. Catal. B Environ. 2017, 205, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Shan, J.; Liu, J.; Li, M.; Lustig, S.; Lee, S.; Flytzani-Stephanopoulos, M. NiCu single atom alloys catalyze the C–H bond activation in the selective non-oxidative ethanol dehydrogenation reaction. Appl. Catal. B Environ. 2018, 226, 534–543. [Google Scholar] [CrossRef]
- Li, S.; Wang, W.; Liu, X.; Zeng, X.; Li, W.; Tsubaki, N.; Yu, S. Nitrogen-doped graphene nanosheets as metal-free catalysts for dehydrogenation reaction of ethanol. RSC Adv. 2016, 6, 13450–13455. [Google Scholar] [CrossRef]
- Ob-Eye, J.; Jongsomjit, B. Dehydrogenation of ethanol to acetaldehyde over Co/C catalysts. Eng. J. 2019, 23, 1–13. [Google Scholar] [CrossRef]
- Ob-Eye, J.; Praserthdam, P.; Jongsomjit, B. Ethanol dehydrogenation to acetaldehyde over activated carbons-derived from coffee residue. Bull. Chem. React. Eng. Catal. 2019, 14, 268–282. [Google Scholar] [CrossRef] [Green Version]
- Ob-Eye, J.; Praserthdam, P.; Jongsomjit, B. Dehydrogenation of ethanol to acetaldehyde over different metals supported on carbon catalysts. Catalysts 2019, 9, 66. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Garbarino, G.; Allard, L.F.; Wilson, F.; Busca, G.; Flytzani-Stephanopoulos, M. Low-Temperature Dehydrogenation of Ethanol on Atomically Dispersed Gold Supported on ZnZrOx. ACS Catal. 2016, 6, 210–218. [Google Scholar] [CrossRef]
- Mitran, G.; Mieritz, D.G.; Seo, D.-K. Highly Selective Solid Acid Catalyst H1−xTi2(PO4)3−x(SO4)x for Non-Oxidative Dehydrogenation of Methanol and Ethanol. Catalysts 2017, 7, 95. [Google Scholar] [CrossRef] [Green Version]
- Edington, J.W. Electron Diffraction in the Electron Microscope, Electron Diffraction in the Electron Microscope; Macmillan Education: London, UK, 1975; pp. 1–77. [Google Scholar]
- Ouyang, M.; Cao, S.; Yang, S.; Li, M.; Flytzani-Stephanopoulos, M. Atomically dispersed pd supported on zinc oxide for selective nonoxidative ethanol dehydrogenation. Ind. Eng. Chem. Res. 2020, 59, 2648–2656. [Google Scholar] [CrossRef]
- Shin, H.; Choi, M.; Kim, H. A mechanistic model for hydrogen activation, spillover, and its chemical reaction in a zeolite-encapsulated Pt catalyst. Phys. Chem. Chem. Phys. 2016, 18, 7035–7041. [Google Scholar] [CrossRef]
- Yodsin, N.; Rungnim, C.; Promarak, V.; Namuangruk, S.; Kungwan, N.; Rattanawan, R.; Jungsuttiwong, S. Influence of hydrogen spillover on Pt-decorated carbon nanocones for enhancing hydrogen storage capacity: A DFT mechanistic study. Phys. Chem. Chem. Phys. 2018, 20, 21194–21203. [Google Scholar] [CrossRef]
- Zhang, H.; Meng, Y.; Song, G.; Li, F. Effect of Hydrogen Spillover to the Hydrogenation of Benzene over Pt/NaA Catalysts, Synthesis and Reactivity in Inorganic. Metal-Org. Nano-Metal Chem. 2016, 46, 940–944. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, J.; Ji, D.; Yuan, A.; Shen, X. Effect of catalyst loading on hydrogen storage capacity of ZIF-8/graphene oxide doped with Pt or Pd via spillover. Microporous Mesoporous Mater. 2016, 229, 68–75. [Google Scholar] [CrossRef]
- Xiang, N.; Xu, P.; Ran, N.; Ye, T. Production of acetic acid from ethanol over CuCr catalysts: Via dehydrogenation-(aldehyde-water shift) reaction. RSC Adv. 2017, 7, 38586–38593. [Google Scholar] [CrossRef] [Green Version]
- Batchu, R.; Galvita, V.V.; Alexopoulos, K.; Glazneva, T.S.; Poelman, H.; Reyniers, M.-F.; Marin, G.B. Ethanol dehydration pathways in H-ZSM-5: Insights from temporal analysis of products. Catal. Today 2019. [Google Scholar] [CrossRef]
- Liang, Z.; Song, L.; Deng, S.; Zhu, Y.; Stavitski, E.; Adzic, R.R.; Chen, J.; Wang, J.X. Direct 12-Electron Oxidation of Ethanol on a Ternary Au(core)-PtIr(Shell) Electrocatalyst. J. Am. Chem. Soc. 2019, 141, 9629–9636. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chai, W.; Henkelman, G. Selectivity for ethanol partial oxidation: The unique chemistry of single-atom alloy catalysts on Au, Ag, and Cu(111). J. Mater. Chem. A 2019. [Google Scholar] [CrossRef]
- Sheng, T.; Lin, W.F.; Hardacre, C.; Hu, P. Significance of β-dehydrogenation in ethanol electro-oxidation on platinum doped with Ru, Rh, Pd, Os and Ir. Phys. Chem. Chem. Phys. 2014, 16, 13248–13254. [Google Scholar] [CrossRef] [PubMed]
- Boualouache, A.; Boucenna, A.; Otmanine, G. Interaction of intermediates with transition metal surfaces in the dehydrogenation of ethanol to ethyl acetate: A theoretical investigation. Progress React. Kinet. Mech. 2019. [Google Scholar] [CrossRef]
- Zhang, B.; Cai, W.; Li, Y.; Xu, Y.; Shen, W. Hydrogen production by steam reforming of ethanol over an Ir/CeO2 catalyst: Reaction mechanism and stability of the catalyst. Int. J. Hydrog. Energy 2008, 33, 4377–4386. [Google Scholar] [CrossRef]
- Cai, W.; Zhang, B.; Li, Y.; Xu, Y.; Shen, W. Hydrogen production by oxidative steam reforming of ethanol over an Ir/CeO2 catalyst. Catalysis Commun. 2007, 8, 1588–1594. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Cranston, R.W.; Inkley, F.A. 17 The Determination of Pore Structures from Nitrogen Adsorption Isotherms. In Advances in Catalysis; Farkas, A., Ed.; Academic Press: London, UK, 1957; pp. 143–154. [Google Scholar]
- Kovacs, A.; Schierholz, R.; Tillmann, K. FEI Titan G2 80-200 CREWLEY. J. Large-Scale Res. Facilities JLSFR 2016, 2, 43. [Google Scholar] [CrossRef] [Green Version]
- Pasel, J.; Schmitt, D.; Hartmann, H.; Besmehn, A.; Dornseiffer, J.; Werner, J.; Mayer, J.; Peters, R. Combined near-ambient pressure photoelectron spectroscopy and temporal analysis of products study of CH4 oxidation on Pd/γ-Al2O3 catalysts. Catal. Today 2019. [Google Scholar] [CrossRef]
- Yaws, C.L.; Satyro, M.A. Chapter 1—Vapor Pressure—Organic Compounds. In The Yaws Handbook of Vapor Pressure, 2nd ed.; Yaws, C.L., Ed.; Gulf Professional Publishing: Houston, TX, USA, 2015; pp. 1–314. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Specific Surface Area m2 g−1 |
|---|---|
| 5 wt % Pt/C | 805 |
| 1 wt % Ir/C | 1013 |
| 3 wt % Cu/C | 1271 |
| Catalyst | Specific Surface Area m2 g−1 | Calculated Ethanol Conversions at 200 °C % |
|---|---|---|
| 5 wt % Pt/C | 805 | ~100 |
| 1 wt % Ir/C | 1013 | ~95 |
| 3 wt % Cu/C | 1271 | ~85 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasel, J.; Häusler, J.; Schmitt, D.; Valencia, H.; Meledina, M.; Mayer, J.; Peters, R. Ethanol Dehydrogenation: A Reaction Path Study by Means of Temporal Analysis of Products. Catalysts 2020, 10, 1151. https://doi.org/10.3390/catal10101151
Pasel J, Häusler J, Schmitt D, Valencia H, Meledina M, Mayer J, Peters R. Ethanol Dehydrogenation: A Reaction Path Study by Means of Temporal Analysis of Products. Catalysts. 2020; 10(10):1151. https://doi.org/10.3390/catal10101151
Chicago/Turabian StylePasel, Joachim, Johannes Häusler, Dirk Schmitt, Helen Valencia, Maria Meledina, Joachim Mayer, and Ralf Peters. 2020. "Ethanol Dehydrogenation: A Reaction Path Study by Means of Temporal Analysis of Products" Catalysts 10, no. 10: 1151. https://doi.org/10.3390/catal10101151
APA StylePasel, J., Häusler, J., Schmitt, D., Valencia, H., Meledina, M., Mayer, J., & Peters, R. (2020). Ethanol Dehydrogenation: A Reaction Path Study by Means of Temporal Analysis of Products. Catalysts, 10(10), 1151. https://doi.org/10.3390/catal10101151