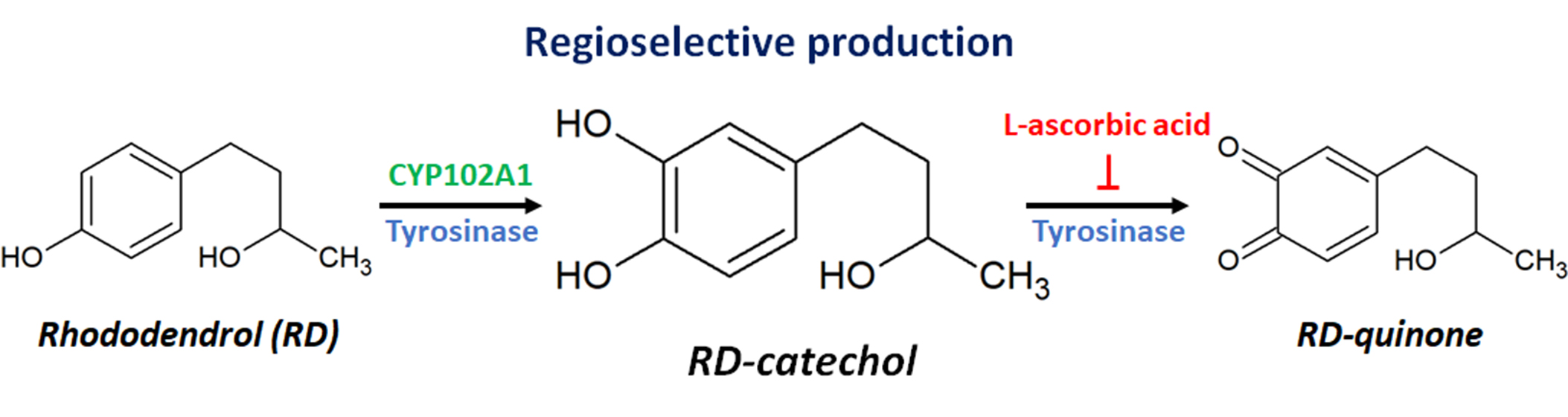
Regioselective Hydroxylation of Rhododendrol by CYP102A1 and Tyrosinase


Abstract
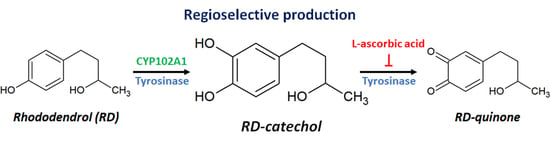
:
1. Introduction
2. Results and Discussion
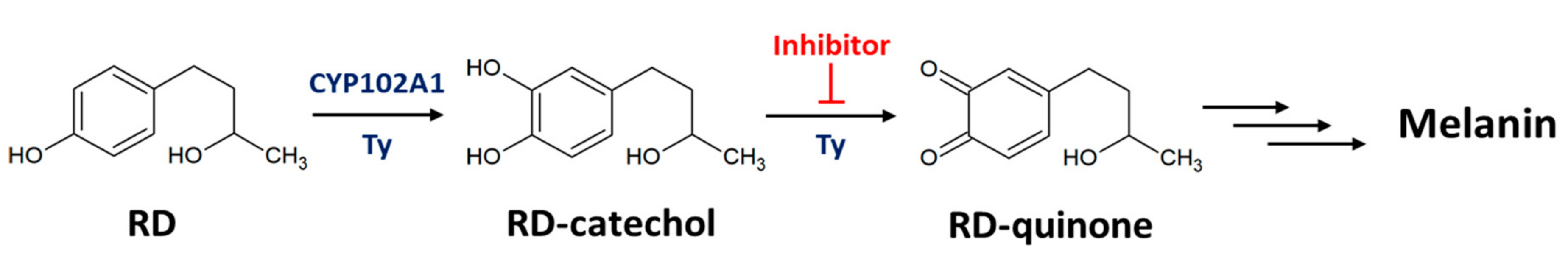
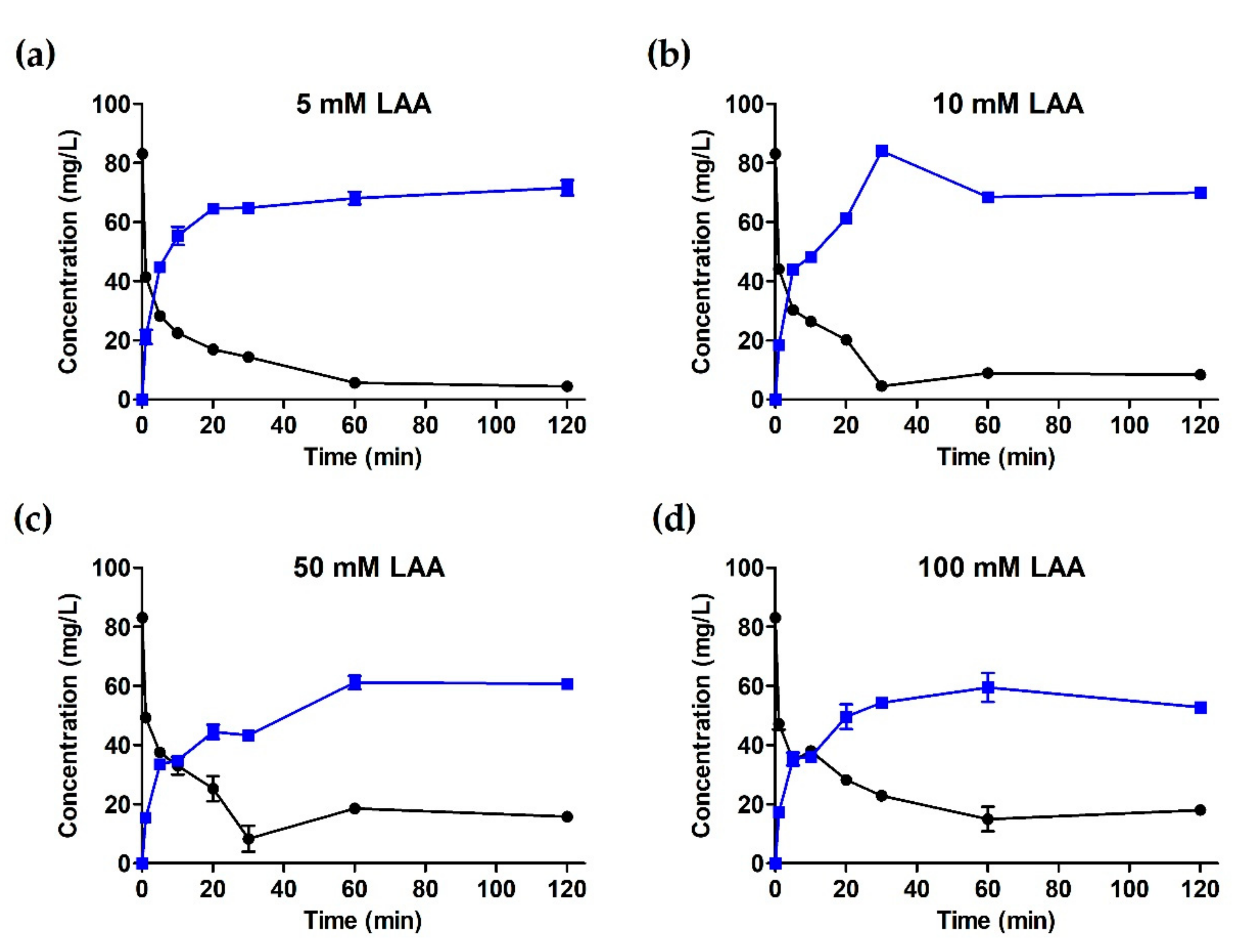
2.1. Hydroxylation of RD by Engineered CYP102A1
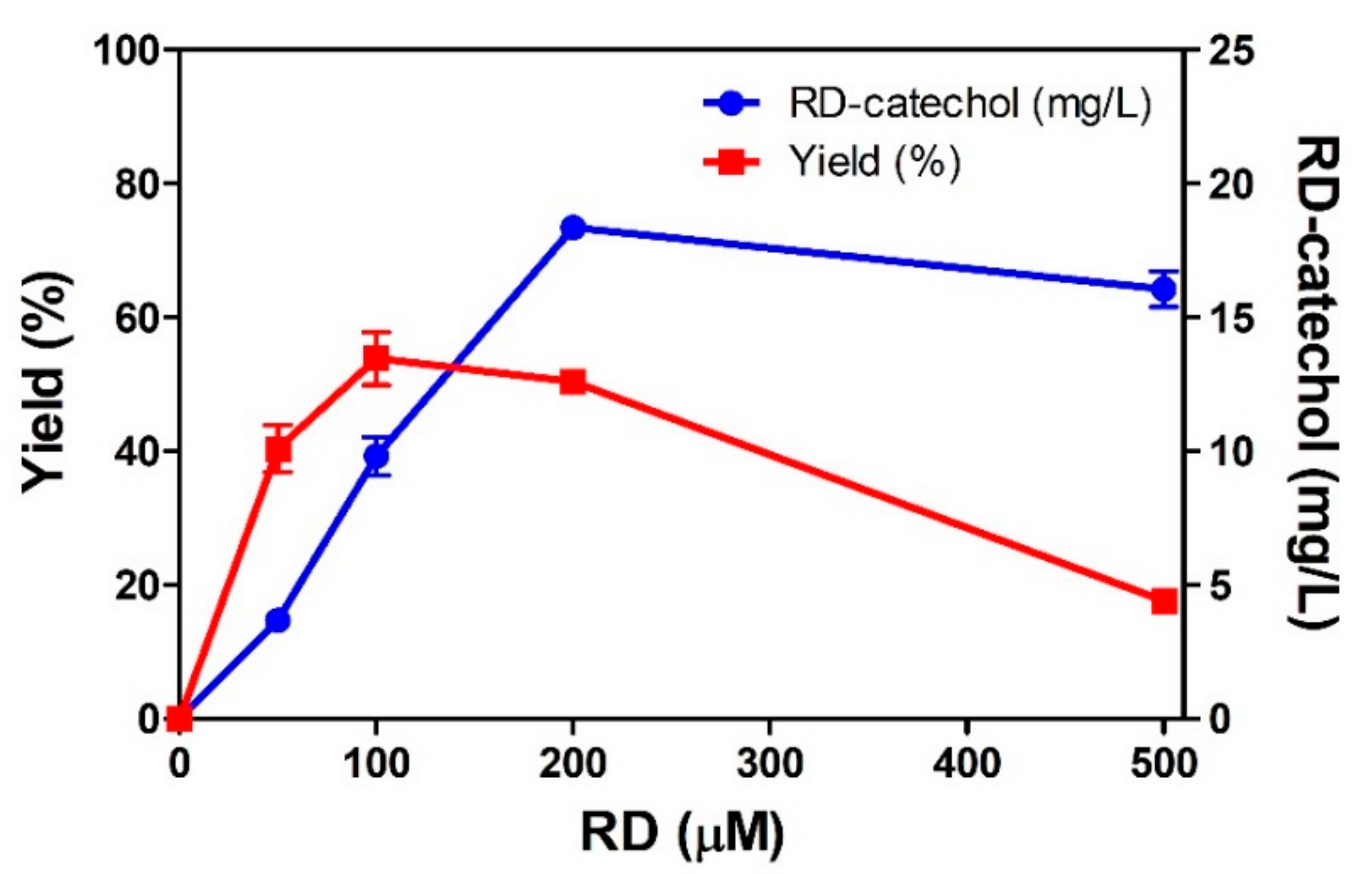
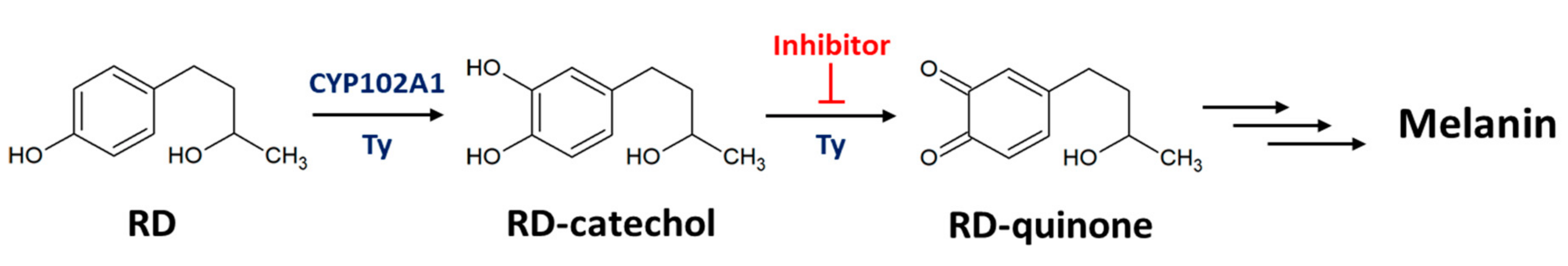
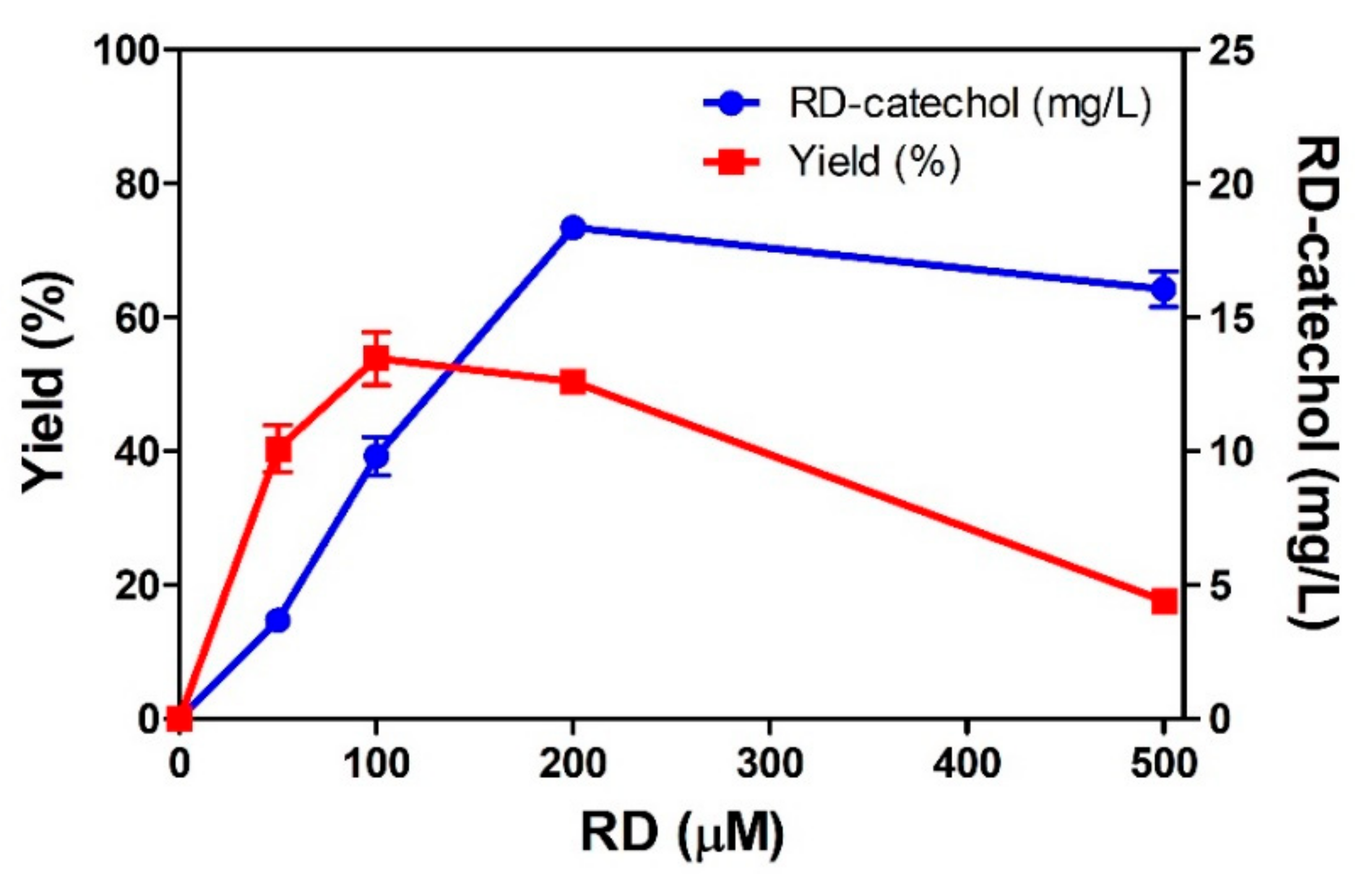
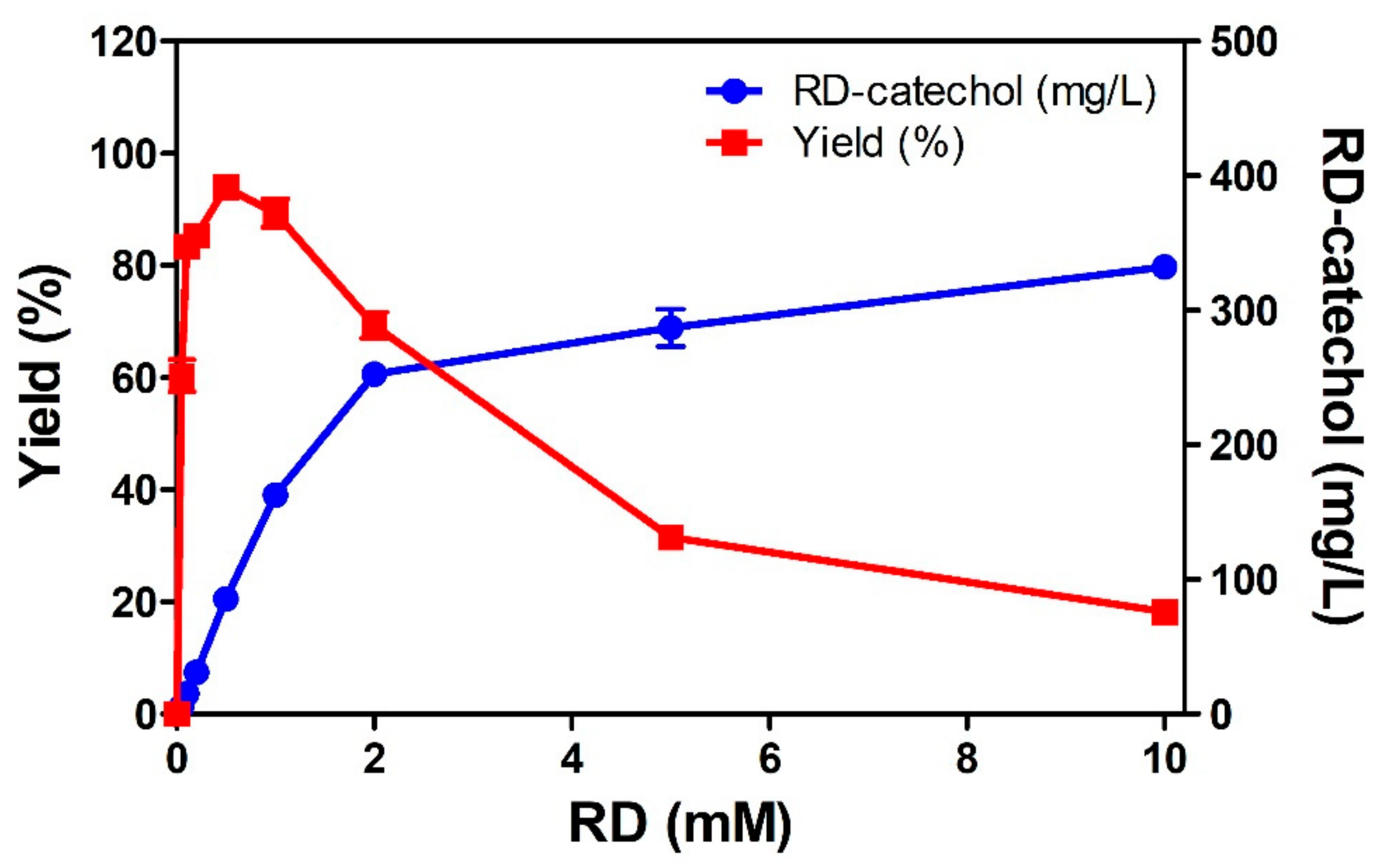
2.2. RD-Catechol Production by Engineered CYP102A1
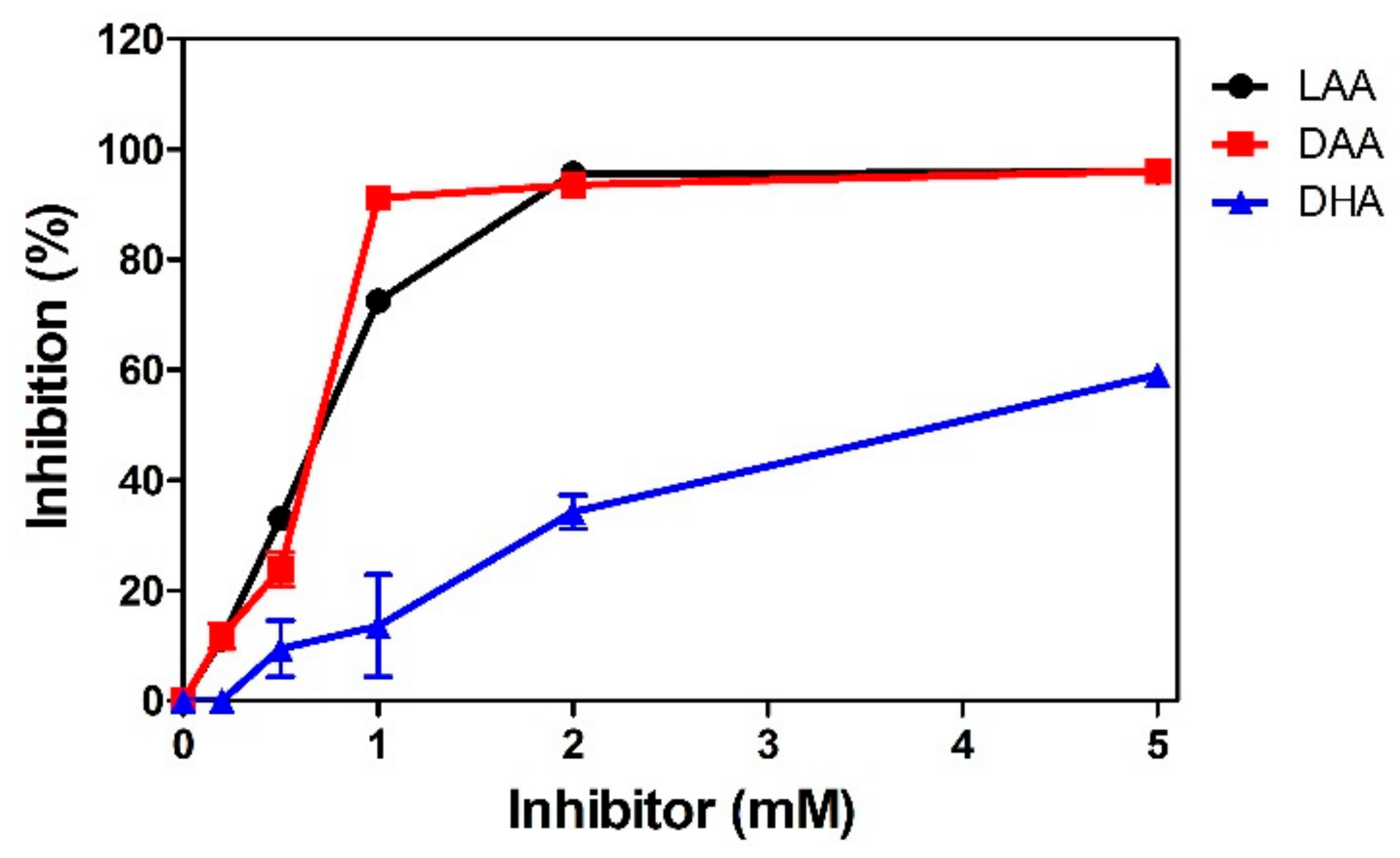
2.3. Inhibition of RD-Quinone Formation by Ascorbic Acid Analogs
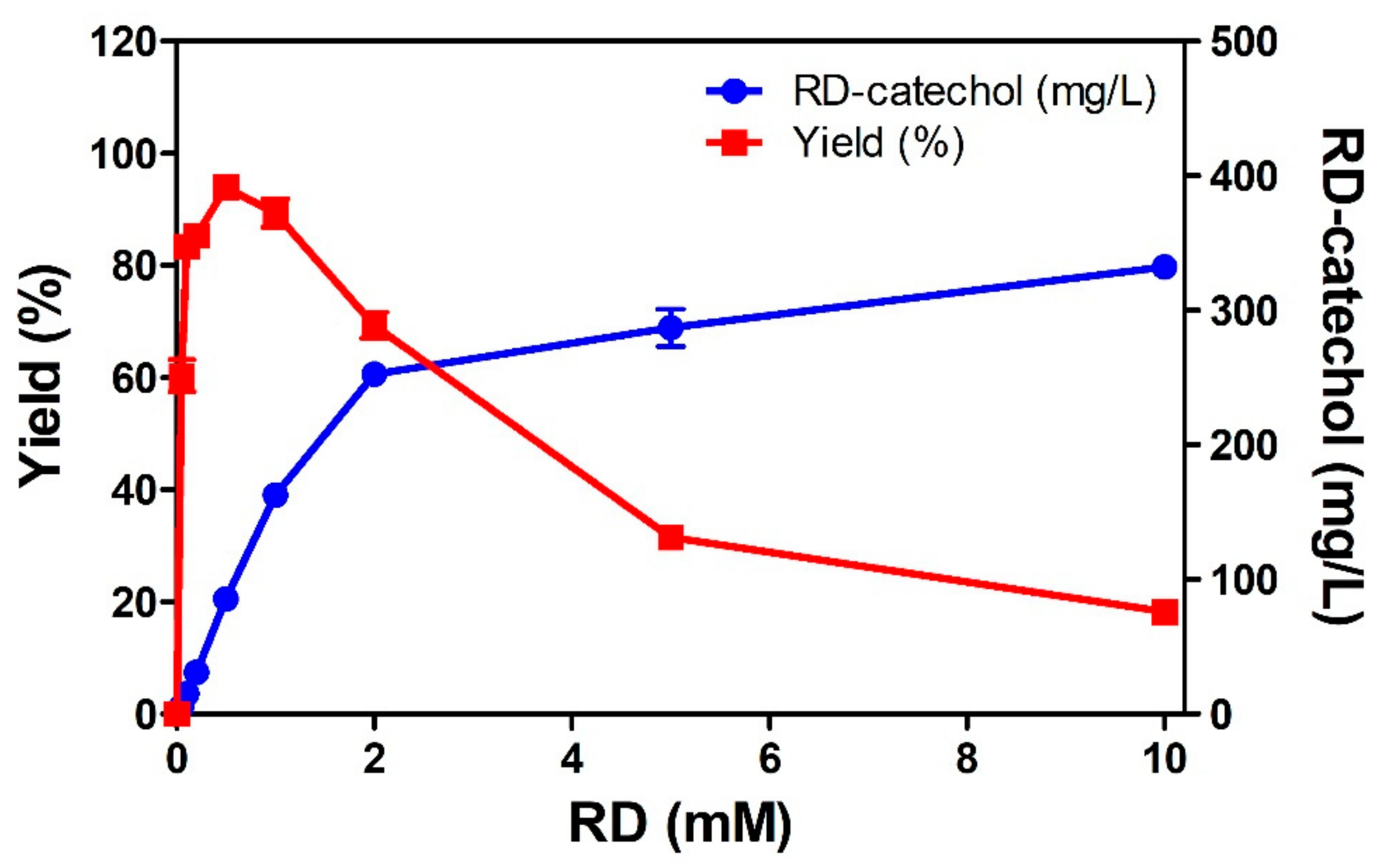
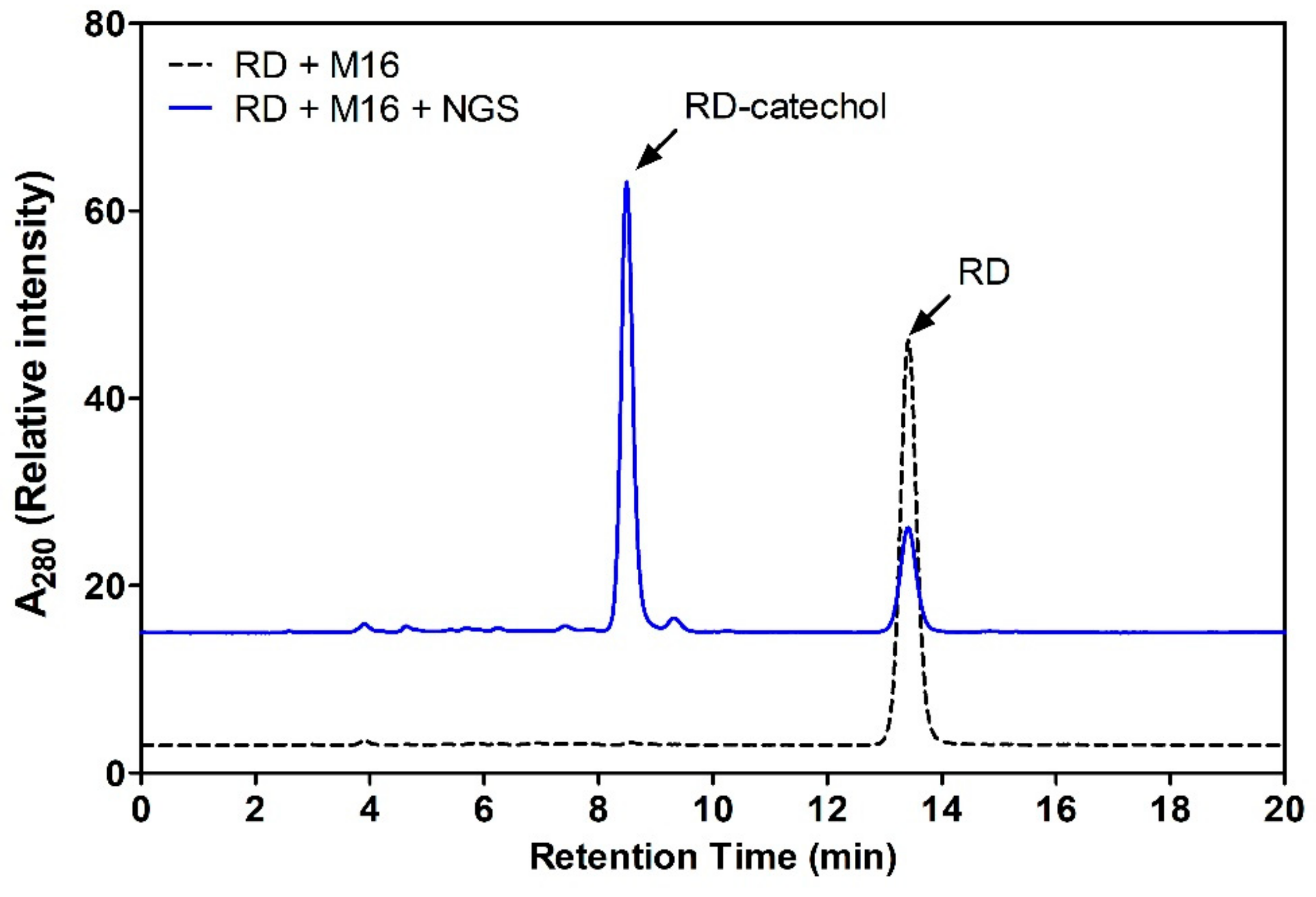
2.4. Regioselective Production of RD-Catechol by Ty
3. Materials and Methods
3.1. Materials
3.2. Preparation of Engineered CYP102A1
3.3. Hydroxylation of RD by Engineered CYP102A1
3.4. Inhibition of RD-Quinone Formation
3.5. Production of RD-Catechol by Tyrosinase
3.6. Identification of RD-Catechol by LC-MS
3.7. Identification of RD-Catechol by NMR Spectroscopy
3.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schmid, A.; Dordick, J.S.; Hauer, B.; Kiener, A.; Wubbolts, M.; Witholt, B. Industrial biocatalysis today and tomorrow. Nature 2001, 409, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.A.; Nogueirab, F.G.E.; Silvac, M.C.; Lopes, J.H.; Tavaresa, T.S.; Ramalho, T.C.; Corrêaa, A.D. Novel eco-friendly biocatalyst: Soybean peroxidase immobilized onto activated carbon obtained from agricultural waste. RSC Adv. 2017, 7, 16460–16466. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.M.; Han, S.S.; Kim, H.S. Industrial applications of enzyme biocatalysis: Current status and future aspects. Biotechnol. Adv. 2015, 33, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Nitti, A.; Bianchi, G.; Po, R.; Swager, T.M.; Pasini, D. Domino Direct Arylation and Cross-Aldol for Rapid Construction of Extended Polycyclic pi-Scaffolds. J. Am. Chem. Soc. 2017, 139, 8788–8791. [Google Scholar] [CrossRef] [PubMed]
- Osburn, P.L.; Bergbreiter, D.E. Molecular engineering of organic reagents and catalysts using soluble polymers. Prog. Polym. Sci. 2001, 26, 2015–2081. [Google Scholar] [CrossRef]
- Nelson, D.R. The cytochrome p450 homepage. Hum. Genom. 2009, 4, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Munro, A.W. Unusual cytochrome p450 enzymes and reactions. J. Biol. Chem. 2013, 288, 17065–17073. [Google Scholar] [CrossRef] [Green Version]
- Urlacher, V.B.; Girhard, M. Cytochrome P450 Monooxygenases in Biotechnology and Synthetic Biology. Trends Biotechnol. 2019, 37, 882–897. [Google Scholar] [CrossRef]
- Munro, A.W.; Daff, S.; Coggins, J.R.; Lindsay, J.G.; Chapman, S.K. Probing electron transfer in flavocytochrome P-450 BM3 and its component domains. Eur. J. Biochem. 1996, 239, 403–409. [Google Scholar] [CrossRef]
- Lussenburg, B.M.A.; Babel, L.C.; Vermeulen, N.P.E.; Commandeur, J.N.M. Evaluation of alkoxyresorufins as fluorescent substrates for cytochrome P450 BM3 and site-directed mutants. Anal. Biochem. 2005, 341, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.H.; Kim, K.H.; Kim, D.H.; Jung, H.C.; Pan, J.G. The bacterial P450 BM3: A prototype for a biocatalyst with human P450 activities. Trends Biotechnol. 2007, 25, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, C.J.; Bell, S.G.; Wong, L.L. P450 (BM3) (CYP102A1): Connecting the dots. Chem. Soc. Rev. 2012, 41, 1218–1260. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.Y.; Ryu, S.H.; Park, S.H.; Cha, G.S.; Kim, D.H.; Kim, K.H.; Hong, A.W.; Ahn, T.; Pan, J.G.; Joung, Y.H.; et al. Chimeric cytochromes P450 engineered by domain swapping and random mutagenesis for producing human metabolites of drugs. Biotechnol. Bioeng. 2014, 111, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Urlacher, V.B.; Eiben, S. Cytochrome P450 monooxygenases: Perspectives for synthetic application. Trends Biotechnol. 2006, 24, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Ismaya, W.T.; Rozeboom, H.J.; Weijn, A.; Mes, J.J.; Fusetti, F.; Wichers, H.J.; Dijkstra, B.W. Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry 2011, 50, 5477–5486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsden, C.A.; Riley, P.A. Tyrosinase: The four oxidation states of the active site and their relevance to enzymatic activation, oxidation and inactivation. Bioorg. Med. Chem. 2014, 22, 2388–2395. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Namasivayam, V.; Manickam, M.; Jung, S.H. Inhibitors of Melanogenesis: An Updated Review. J. Med. Chem. 2018, 61, 7395–7418. [Google Scholar] [CrossRef]
- Goldfeder, M.; Kanteev, M.; Adir, N.; Fishman, A. Influencing the monophenolase/diphenolase activity ratio in tyrosinase. Biochim. Biophys. Acta 2013, 1834, 629–633. [Google Scholar] [CrossRef]
- Halaouli, S.; Asther, M.; Sigoillot, J.C.; Hamdi, M.; Lomascolo, A. Fungal tyrosinases: New prospects in molecular characteristics, bioengineering and biotechnological applications. J. Appl. Microbiol. 2006, 100, 219–232. [Google Scholar] [CrossRef]
- Hernandez-Romero, D.; Sanchez-Amat, A.; Solano, F. A tyrosinase with an abnormally high tyrosine hydroxylase/dopa oxidase ratio. FEBS J. 2006, 273, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Shuster Ben-Yosef, V.; Sendovski, M.; Fishman, A. Directed evolution of tyrosinase for enhanced monophenolase/diphenolase activity ratio. Enzym. Microb. Technol. 2010, 47, 372–376. [Google Scholar] [CrossRef]
- Molloy, S.; Nikodinovic-Runic, J.; Martin, L.B.; Hartmann, H.; Solano, F.; Decker, H.; O’Connor, K.E. Engineering of a bacterial tyrosinase for improved catalytic efficiency towards D-tyrosine using random and site directed mutagenesis approaches. Biotechnol. Bioeng. 2013, 110, 1849–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, N.; Kim, E.J.; Kim, B.G. Regioselective hydroxylation of trans-resveratrol via inhibition of tyrosinase from Streptomyces avermitilis MA4680. ACS Chem. Biol. 2012, 7, 1687–1692. [Google Scholar] [CrossRef]
- Lee, N.; Lee, S.H.; Baek, K.; Kim, B.G. Heterologous expression of tyrosinase (MelC2) from Streptomyces avermitilis MA4680 in E. coli and its application for ortho-hydroxylation of resveratrol to produce piceatannol. Appl. Microbiol. Biotechnol. 2015, 99, 7915–7924. [Google Scholar] [CrossRef]
- Lee, S.H.; Baek, K.; Lee, J.E.; Kim, B.G. Using tyrosinase as a monophenol monooxygenase: A combined strategy for effective inhibition of melanin formation. Biotechnol. Bioeng. 2016, 113, 735–743. [Google Scholar] [CrossRef]
- Tallent, W.H. d-Betuligenol from Rhododendron maximum L. J. Org. Chem. 1964, 29, 988–989. [Google Scholar] [CrossRef]
- Fujita, T.; Hatamoto, H.; Iwasaki, T.; Takafuji, S.I. Bioconversion of rhododendrol by Acer nikoense. Phytochemistry 1995, 39, 1085–1089. [Google Scholar] [CrossRef]
- Pan, H.; Lundgren, L.N. Rhododendrol glycosides and phenyl glucoside esters from inner bark of Betula pubescens. Phytochemistry 1994, 36, 79–83. [Google Scholar] [CrossRef]
- Ito, S.; Wakamatsu, K. Biochemical Mechanism of Rhododendrol-Induced Leukoderma. Int. J. Mol. Sci. 2018, 19, 552. [Google Scholar]
- Ito, S.; Ojika, M.; Yamashita, T.; Wakamatsu, K. Tyrosinase-catalyzed oxidation of rhododendrol produces 2-methylchromane-6,7-dione, the putative ultimate toxic metabolite: Implications for melanocyte toxicity. Pigment Cell Melanoma Res. 2014, 27, 744–753. [Google Scholar] [CrossRef]
- Yang, E.J.; An, J.H.; Son, Y.K.; Yeo, J.H.; Song, K.S. The Cytotoxic Constituents of Betula platyphylla and their Effects on Human Lung A549 Cancer Cells. Nat. Prod. Sci. 2018, 24, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Yang, F.; Wataya-Kaneda, M.; Tanemura, A.; Tsuruta, D.; Katayama, I. 4-(4-Hydroroxyphenyl)-2-butanol (rhododendrol) activates the autophagy-lysosome pathway in melanocytes: Insights into the mechanisms of rhododendrol-induced leukoderma. J. Dermatol. Sci. 2015, 77, 182–185. [Google Scholar] [CrossRef]
- Kasamatsu, S.; Hachiya, A.; Nakamura, S.; Yasuda, Y.; Fujimori, T.; Takano, K.; Moriwaki, S.; Hase, T.; Suzuki, T.; Matsunaga, K. Depigmentation caused by application of the active brightening material, rhododendrol, is related to tyrosinase activity at a certain threshold. J. Dermatol. Sci. 2014, 76, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Munoz, J.L.; Garcia-Molina, F.; García-Ruiz, P.A.; Varon, R.; Tudela, J.; García-Cánovas, F.; Rodriguez-Lopez, J.N. Stereospecific inactivation of tyrosinase by l- and d-ascorbic acid. Biochim. Biophys. Acta. Proteins Proteom. 2009, 1794, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.L.; Rusli, R.A.; Ollis, D.L. Directed Evolution of Enzymes for Industrial Biocatalysis. ChemBioChem 2016, 17, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Julsing, M.K.; Cornelissen, S.; Buhler, B.; Schmid, A. Heme-iron oxygenases: Powerful industrial biocatalysts? Curr. Opin. Chem. Biol. 2008, 12, 177–186. [Google Scholar] [CrossRef]
- Pollard, D.J.; Woodley, J.M. Biocatalysis for pharmaceutical intermediates: The future is now. Trends Biotechnol. 2007, 25, 66–73. [Google Scholar] [CrossRef]
- Straathof, A.J.J.; Panke, S.; Schmid, A. The production of fine chemicals by biotransformations. Curr. Opin. Biotech. 2002, 13, 548–556. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, K.H.; Kim, D.H.; Liu, K.H.; Jung, H.C.; Pan, J.G.; Yun, C.H. Generation of human metabolites of 7-ethoxycoumarin by bacterial cytochrome P450 BM3. Drug Metab. Dispos. 2008, 36, 2166–2170. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Kim, D.H.; Kim, D.; Kim, D.H.; Jung, H.C.; Pan, J.G.; Ahn, T.; Kim, D.; Yun, C.H. Engineering bacterial cytochrome P450 (P450) BM3 into a prototype with human P450 enzyme activity using indigo formation. Drug Metab. Dispos. 2010, 38, 732–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.H.; Ryu, S.H.; Le, T.K.; Doan, T.T.; Nguyen, T.H.; Park, K.D.; Yim, D.E.; Kim, D.H.; Kang, C.K.; Ahn, T.; et al. Regioselective C-H hydroxylation of omeprazole sulfide by Bacillus megaterium CYP102A1 to produce a human metabolite. Biotechnol. Lett. 2017, 39, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Omura, T.; Sato, R. The Carbon Monoxide-Binding Pigment of Liver Microsomes. II. Solubilization, Purification, and Properties. J. Biol. Chem. 1964, 239, 2379–2385. [Google Scholar] [PubMed]
- Nguyen, N.A.; Jang, J.; Le, T.K.; Nguyen, T.H.H.; Woo, S.M.; Yoo, S.K.; Lee, Y.J.; Park, K.D.; Yeom, S.J.; Kim, G.J.; et al. Biocatalytic Production of a Potent Inhibitor of Adipocyte Differentiation from Phloretin Using Engineered CYP102A1. J. Agric. Food Chem. 2020, 68, 6683–6691. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biocatalyst | Selectivity (%) | Yield (%) | Product Concentration (mg·L−1) | Productivity (mg·L−1·h−1) |
|---|---|---|---|---|
| CYP102A1 1 | 93.5 ± 1.4 | 17.6 ± 0.7 | 16.1 ± 0.7 | 32.1 |
| Ty 2 | 90.4 ± 0.4 | 93.9 ± 1.8 | 85.6 ± 1.6 | 171 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, C.M.; Park, H.S.; Cha, G.S.; Park, K.D.; Yun, C.-H. Regioselective Hydroxylation of Rhododendrol by CYP102A1 and Tyrosinase. Catalysts 2020, 10, 1114. https://doi.org/10.3390/catal10101114
Park CM, Park HS, Cha GS, Park KD, Yun C-H. Regioselective Hydroxylation of Rhododendrol by CYP102A1 and Tyrosinase. Catalysts. 2020; 10(10):1114. https://doi.org/10.3390/catal10101114
Chicago/Turabian StylePark, Chan Mi, Hyun Seo Park, Gun Su Cha, Ki Deok Park, and Chul-Ho Yun. 2020. "Regioselective Hydroxylation of Rhododendrol by CYP102A1 and Tyrosinase" Catalysts 10, no. 10: 1114. https://doi.org/10.3390/catal10101114
APA StylePark, C. M., Park, H. S., Cha, G. S., Park, K. D., & Yun, C.-H. (2020). Regioselective Hydroxylation of Rhododendrol by CYP102A1 and Tyrosinase. Catalysts, 10(10), 1114. https://doi.org/10.3390/catal10101114