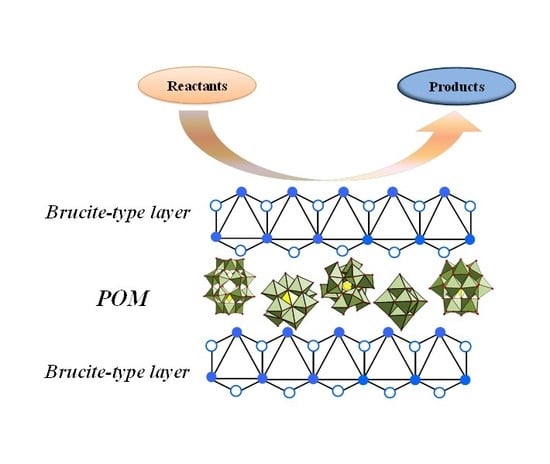
4.2.1. Vanadates
The vanadium-based anions were among the very first polyoxometalates to be successfully intercalated into layered double hydroxides structure. A wide range of polyoxovanadate ions can be inserted in the interlayer space, from discrete to polymeric anions. Although decavanadate is the most used, there are studies that are also devoted to the intercalation of lower oligovanadates. The pH influences the polymerization degree of the oxovanadates. A high nuclearity is associated to a decreased pH value, as seen in
Table 3.
Similar to the chromate containing LDH, the reported methods used to synthesize hydrotalcites with intercalated vanadium-based anions containing different pairs of divalent/trivalent cations in the brucite-like layers are co-precipitation, anion exchange, reconstruction, and hydrothermal method. Vanadium can be incorporated either in one of its anionic forms in the interlayer, mostly depending on the nature of the brucite-like cations and pH value (
Table 4), or can be introduced as a cation, V(III) in the brucite-type layers (
Table 5).
Suitable interlamellar distances are required to favor the intercalation of vanadates with similar hindrance. A good solution to increase the interlayer zone is to use proper-sized anions beforehand to swell the layers of the hydrotalcites, such as terephthalate or dodecyl sulphate. The size of the anions, as well as their charge density and orientation within the interlayer, has proven to have a considerable influence on the catalytic activity of the POM-LDH composites. Layered double hydroxides are known to be bifunctional catalysts, exhibiting neither purely acid nor basic character [
18]. The Brønsted basicity of hydrotalcites is attributed to the existing surface hydroxyls and their combination with vanadium-based anions is believed to provide some Lewis acidity to the LDH. Polyoxometalates that are based on vanadium have often been applied in heterogeneous catalysis because of their interesting redox properties.
Decavanadate-intercalated LDH, having magnesium and aluminum as cations in the brucite-type layers, can be synthesized with great ease by direct ion exchange of a nitrate precursor with an aqueous NaVO
3 solution at an adequate acidic pH value with or without preswelling with terephthalate while following the method that was reported by Villa et al. [
32]. The Raman data are particularly useful for distinguishing between intercalated species, such as [VO
4]
3−, [V
2O
7]
4−, [V
4O
12]
4−, and [V
10O
28]
6− [
33]. The Raman spectrum that was reported by Twu et al. of the LDH containing decavanadate, prepared by direct exchange of an LDH with an acidified V solution, indicates that decavanadate is the dominant anionic species found intercalated, as evidenced by the peaks at 320, 454, 534, 595, 834, 975, and 998 cm
−1 [
34]. Although the signal to noise ratio of the spectrum for the LDH is not as good as that for the [V
10O
28]
6− ions in solution, it is quite clear that all of the characteristic peaks are attributed to decavanadate ions. For the sample that was heated to 160 °C, changes appear in both the XRD and Raman spectrum. These include a new reflection at 4.76 Å and the appearance of a broad band centered at 880 cm
−1 [
34]. The characteristic Raman bands of the decavanadate are still present, as are the XRD reflections that are characteristic of the decavanadate-hydrotalcite complex. However, it was observed that the decavanadate species are transforming into other vanadate species at temperatures above 100 °C [
34]. The XRD analysis of the decavanadate-LDH sample indicated a gallery height of 7.1 Å, which also suggested that the [V
10O
28]
6− orientation with the C
2 axis is parallel to the layers [
34]. The novel [V
10O
28]
6−-MgAl-LDH composites are used as catalysts in a variety of electron transfer reactions.
Maciuca et al. [
35] have successfully synthesized a solid containing decavanadate and pyrovanadate anions simultaneously intercalated between the brucite-type layers of a hydrotalcite. The UV–VIS spectrum of the sample exhibits a band at 300–320 nm, which is specific for V
4+ having a pseudo-tetrahedral geometry, [V
2O
7]
4− (representing 30% of V species), and a band around 420–450 nm, characteristic to V
5+ ions with an octahedral arrangement, [V
10O
28]
6− (70% of V species) [
35]. The catalytic activity of the prepared LDH material was evaluated in the oxidation reaction of the tetrahydrothiophene to sulfolane with dilute H
2O
2 aqueous solution as oxidant. It was observed that low conversions were obtained for the vanadium-containing material, probably due to the formation of some complex species, other than [V
10O
28]
6− or [V
2O
7]
4−, which probably do not possess any catalytic activity. Dobrea et al. [
36] have also tried to intercalate decavanadate anions between the brucite-type layers of a MgAl-LDH. In the Raman spectra recorded, they have observed some bands in the regions 800–1000 cm
−1 and 200–370 cm
−1 that belong to two types of anions, namely (VO
3)
nn− and [V
10O
28]
6−. The ratio between these anionic species strongly depends on the vanadium content in the LDH structure. When the V loading increases, the bands that are specific to the (VO
3)
nn− ions decrease until complete disappearance, while the [V
10O
28]
6− anions prevail [
36]. Moreover, the XRD patterns of V-exchanged LDH samples display the same reflections characteristic to the layered materials, but at smaller values of 2θ. There are also some changes in the d
003-value that suggest the increase of the basal spacing and, thus, prove the replacement of smaller nitrate anions by larger species. At low concentrations of vanadate, only a partial exchange has occurred and reflections specific for both nitrate and oxyanions coexist in the XRD patterns. For higher V/Al ratios, the exchange was almost complete and the d
003-value increased up to 10.8 Å for V-LDH. The XRD patterns of the exchanged materials presented broader and lower intensity reflections when compared to the parent LDH accounting for the intercalation of various oxyanions species, which produce a certain level of disorder in the lamellar structure [
36].
As previously reported [
18], the overlapping and broadening of some reflections at higher 2θ values can be attributed to the turbostratic effects that are caused by the new intercalated anionic species [
36]. The samples that were prepared by Dobrea et al. were also tested as catalysts for the oxidation of dibenzothiophene (DBT) to dibenzothiophene-sulfone with H
2O
2. It was observed that an increased amount of vanadium-based anions will produce a change of the oxyanion species from monomers (vanadate) to polymeric species (decavanadate). Moreover, the V-LDH samples suffered major modifications during the oxidation reaction, leading to a decreased catalytic activity that is attributed to their low stability [
36].
Villa et al. have reported the obtaining of MgAl-layered double hydroxide pillared with vanadium under mildly acidic conditions, but this time the XRD, Raman, and IR characterization certified the presence in the interlayer region of pure decavanadate ([V
10O
28]
6−) anions [
32]. The compound was used as a catalyst for the selective epoxidation of different allylic and homoallylic alcohols, mostly of terpene origin. The epoxidation of geraniol while using decavanadate-containing hydrotalcite (without or with preswelling) as catalyst, toluene as solvent, and t-BuOOH dissolved in decane as oxidant, led to high conversions of the substrate with minimal formation of geranial, which indicated that an increased effectiveness is obtained by working under anhydrous conditions that are able to prevent the decavanadate anions hydrolysis and stop any possible leaching. Additionally, it was demonstrated that low conversions and selectivities are obtained when vanadium-based anions are introduced in the LDH structure without rigorous pH control, which might lead to the formation of low-nuclearity polymers [
32]. The decavanadate-containing LDH was used as catalyst for the epoxidation of various terpenic allylic alcohols in toluene with t-BuOOH/decane as an oxidant high conversions and epoxide selectivities being obtained for geraniol, nerol, perollyl alcohol, myrtenol, and trans-pinocarveol [
32]. Even homoallylic alcohols (C=C–C–COH), such as isopulegol and nopol [
32], were successfully epoxidized with high selectivity, but a decreased yield. Additionally, a rather low conversion and epoxide selectivity were obtained when β-citronellol was used as a substrate. A possible explanation for these unsatisfactory results can be attributed to the large distance between the alcohol group and the double bond. The allylic alcohols were smoothly epoxidized, except verbenol, whose bulky carbon skeleton did not allow for the coordination of the alcohol to the V [
32]. High conversions with excellent selectivities were also obtained for crotyl alcohol and cinnamyl alcohol [
32]. The results of the epoxidation strongly suggest that the allylic alcohol group of the substrate coordinates on the peroxo-V center [
32] before oxygen transfer takes place. Therefore, it might be said that the active vanadium species participate in the epoxidation reaction with two coordination sites, one that is needed for the peroxide activation, and the other one for the alcohol coordination [
32]. The vanadium atom is initially bound to the pillar with at least five V–O–V bonds [
32], so it is mandatory to break one or two of these bonds to be reassured that the reaction takes place. However, there are still enough V–O–V bonds intact to keep all vanadium within the polyanions [
32].
The alkoxylation of n-butanol with ethylene oxide (EO) to produce butoxy mono-ethylene glycol ether (BMGE) has been also studied on this type of LDH [
37]. Malherbe et al. [
37] used a commercial MgAl-LDH, which was reconstructed with intercalated decavanadate (sample MAV) after calcination, calcined at 450 °C (sample MAVCAL), and then rehydrated in water (MAVHYD) or in a 1 M KOH solution (MAVKOH). The calcination–rehydration procedure was used to observe the influence of rehydration on the reactivity of the mixed oxides. The XRD patterns of the three catalysts used in this study were also analyzed. The parent material used, a calcined hydrotalcite, presents the formation of a periclase MgO phase, while aluminum oxide could not be detected because of its amorphous structure. After the anion exchange and another calcination step, the periclase phase is significantly reduced [
37]. A possible explanation is that a part of the magnesium participates in the formation of a ternary oxide (Mg–Al–V)–O [
37]. The rehydration step decreases, even more, the proportion of the periclase phase in the final material and the XRPD patterns clearly indicate that most of the MgO phase will be transformed into an amorphous hydroxide form [
37]. The catalytic performances of the new catalysts that were obtained after calcination or calcination–rehydration were tested in the synthesis of 2-butoxyethanol and compared to that of the untreated MAV. The conversion of ethylene oxide (EO) in a batch reactor increases with the reaction time in the order MAV < MAVHYD < MAVKOH [
18]. The calcined solid, MAVCAL shows better conversion for low reaction times but it has the huge disadvantage of being deactivated after 20 h reaction. A likely cause of deactivation is associated to the appearance of a significant amount of a polymer type material in the product. The gelatinous solid is thought to contain polyethylene glycols (PEGs) and polyethoxylates, which strongly adsorb on the surface of the catalyst. A good solution is to rehydrate the calcined LDH in a KOH solution to eliminate the formation of unwanted polyethoxylates [
37]. During rehydration, superficial O
2− sites are transformed into hydroxyl groups with lower basicity and high catalytic activity [
37], while the use of KOH might either lead to an increase in the amount of OH groups or might ensure the formation of stronger ones.
Malherbe et al. [
20] reported the synthesis of two systems, namely [CuCr–V
2O
7] and [CuCr–V
10O
28], which were also used as catalysts for the alkoxylation of n-butanol with ethylene oxide (EO) [
20]. The parent material, [CuCr–Cl]-LDH was obtained by the co-precipitation method, and then through the appropriate
chimie douce exchange reactions, the original chloride anions were replaced by decavanadate and pyrovanadate species [
20]. From the catalytic tests, it was observed that higher conversions are obtained for the solid modified with decavanadate anions. The increased activity was attributed to a partial reduction of the vanadium species, with the active sites being not V
5+ but V
4+ [
20].
The vanadate-exchanged layered double hydroxides with cations, such as Mg-Al [
38,
41,
42,
45], Ni-Al [
50], or Zn-Al [
52] in the brucite-like layers provide materials that differ in catalytic performance during the oxidative dehydrogenation (ODH) of various substrates (propane, n-butane), mostly depending on the nature of intercalated vanadate and the vanadium content [
52], which can be modified during preparation. Consequently, the applied synthesis methods play a crucial role in the development of compounds with increased catalytic activities for ODH. The most active in the oxidative dehydrogenation of propane were the samples that were derived from the decavanadate-exchanged LDH under their calcined forms, namely their corresponding mixed oxides, while, for n-butane, the highest selectivity to ODH products and lowest selectivity to CO
x were observed for the V-poor solids [
42]. These observations can be correlated with the increased nucleophilic character (basicity) of the oxygen atoms present in Mg–O–V units existing in orthovanadate than in V–O–V units (that lead to total oxidation) existing in α-Mg
2V
2O
7 and MgV
2O
6. At high vanadium loading, the presence of highly crystalline α-Mg
2V
2O
7 accounted for the observed decrease in 1,3-butadiene selectivity [
42]. For ODH, acid and basic sites are both necessary for the reaction to take place, but it was proven that the total selectivity for dehydrogenated products was higher on the catalysts with increased base sites concentration [
52].
Bahranowski et al. [
40] have prepared different decavanadate-exchanged hydrotalcite-like compounds that were catalytically active in the selective oxidation of cyclohexene with dioxygen. Their activities mostly depended on the composition of the brucite layers, and decreased in the order MgAl > ZnAl > ZnCr [
40]. The lowering of the M(II)/M(III) ratio in the brucite layer allowed for an increased loading of decavanadate anions, which eventually led to higher activity. Moreover, a pyrovanadate-exchanged-MgAl-LDH was also synthesized and used as a catalyst in the same reaction. It presented a lack of activity, which was probably due to the small interlayer distance that made the catalytic sites inaccessible. A considerable amount of epoxide was still observed in this case, which indicated the presence of decavanadate group in the interlamellar space [
40]. Recently, Nejati et al. [
59] obtained ZnFe–VO
4-LDH (
Figure 7), which was used as an efficient electrocatalyst for water oxidation in alkali solution. In electrochemical water splitting, ZnFe–VO
4-LDH exhibits a superior OER performance, being expressed as lower onset overpotential, smaller Tafel slope, and larger exchange current density [
59] when compared to the bare glassy carbon electrode. Layered double hydroxides that were intercalated with vanadium-based species were also used as anticorrosive materials. Tedim et al. [
54] successfully reported the preparation of ZnAl–V
2O
7-LDH, which was able to provide efficient active corrosion protection of an aluminum alloy. It was found that the inhibition by vanadates mainly occurs in alkaline solutions, where metavanadates and pyrovanadates are the most abundant. Vanadium can be introduced not only in the interlayer space, but also as a cation in the brucite-like layers (
Table 5). Kooli et al. [
44] obtained hydrotalcite-like materials containing vanadium, as V(III), together with Mg(II) and Al(III), in the layers, and carbonate in the interlayer. It was found that the nature of the precursor influences the type of the Mg-V-O phases formed; thus, when starting from Mg
3(VO
4)
2, the isolated [VO
4] units are formed. Dula et al. [
41] have tried to use this solid as a catalyst for oxidative dehydrogenation of propane (ODH), obtaining high conversions of the substrate, but with low selectivities.
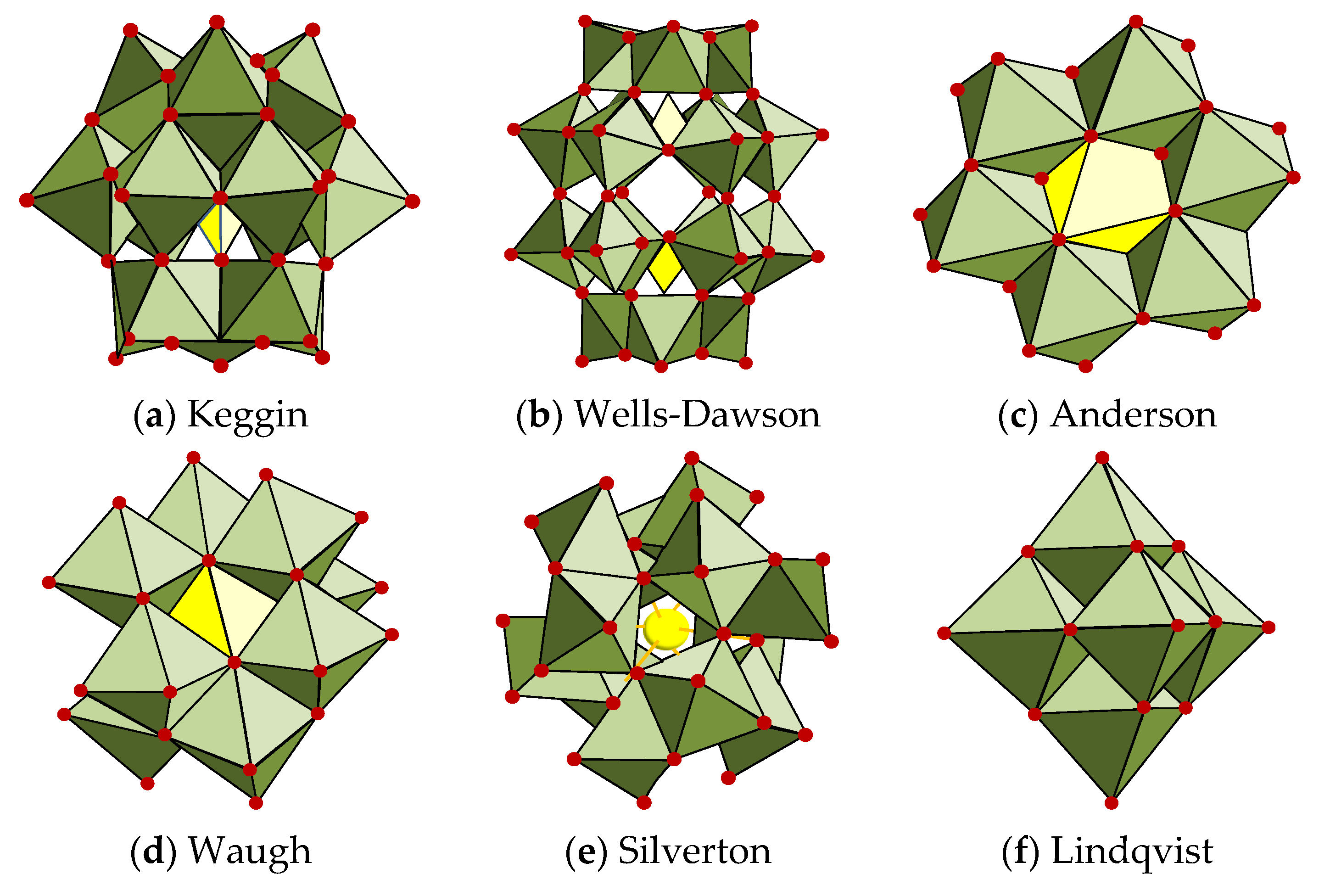
4.2.2. Molybdates
The intercalation of molybdenum-based anions in the structure of layered double hydroxides is a laborious process and it mostly depends on the pH and the Mo(VI) concentration. Moreover, the evolution of Mo(VI) in solutions is enriched, due to the existence of polymers that may lead to the obtaining of a variety of anions, the predominant ones being [MoO
4]
2−, [HMoO
4]
−, [Mo
6O
19]
2−, [Mo
7O
24]
6−, [HMo
7O
24]
5−, [H
2Mo
7O
24]
4−, and [Mo
8O
26]
2− [
60]. The geometry of smaller anions, such as [MoO
4]
2−, is tetrahedral, while larger anions, like [Mo
6O
19]
2− and [Mo
7O
24]
6−, possess octahedral or distorted octahedral configuration. The intercalation of vanadates in the interlayer space of layered double hydroxides has been intensively studied, while that of molybdates has been restricted to the heptamolybdate [Mo
7O
24]
6− ions that can be stabilized at a lower pH and can act as pillars that are used to expand the distance between the brucite-type sheets [
61]. Additionally, Van Laar et al. [
62] discovered that the [MoO
4]
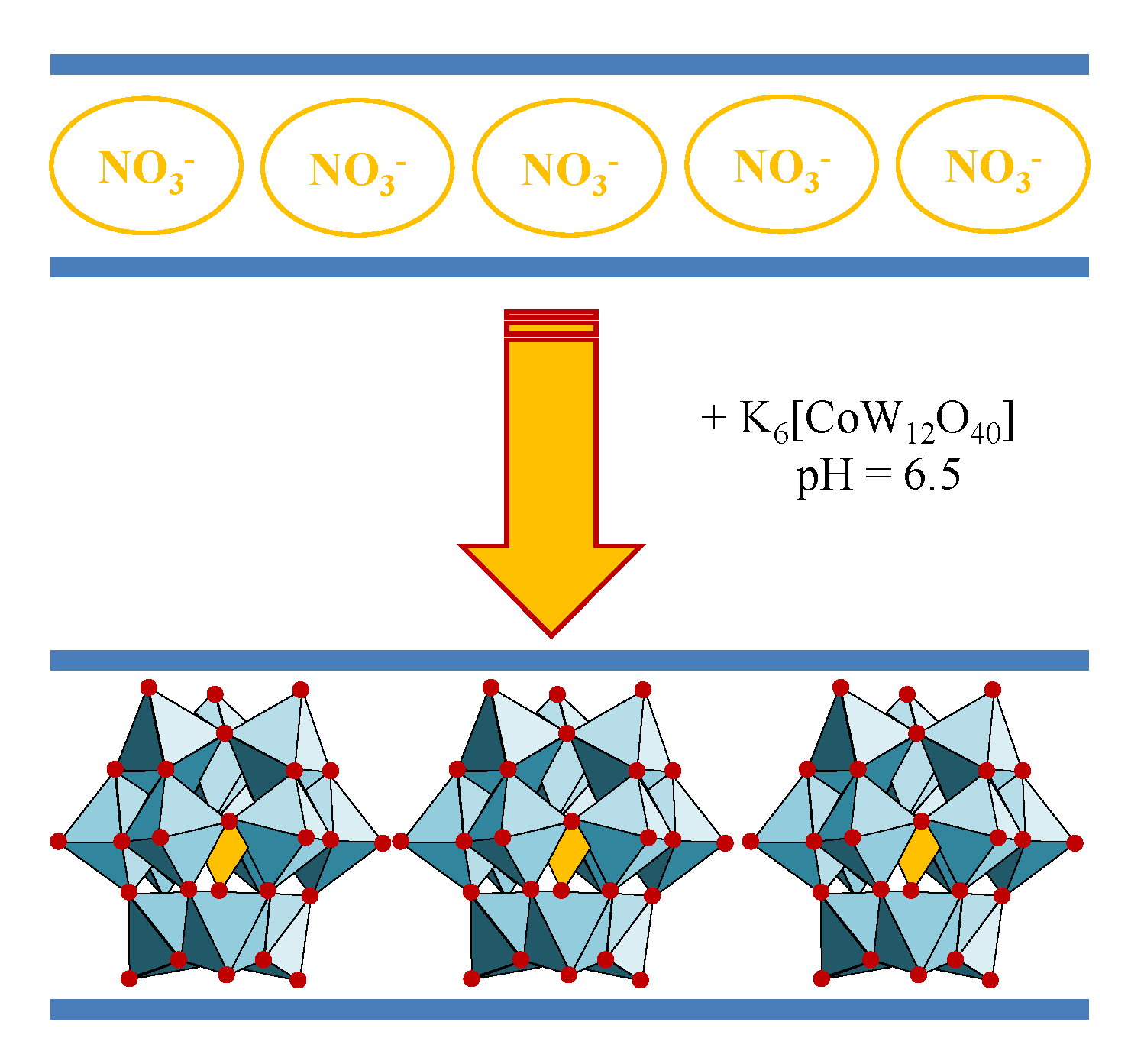
2− hydrolysis at a pH value of 10 inhibits the intercalation in the LDH structure and polymerization of molybdate does not occur until pH < 7 (
Figure 8). The molybdate-containing LDH with slightly different chemical composition could be useful as selective catalysts for different processes. The first studies that were recorded for the characterization of oxomolybdate species at different acidic pH values were based on potentiometric titrations [
63]. The main conclusion was that [MoO
4]
2− ions are stable at pH 6.5, while the heptamolybdate [Mo
7O
24]
6− exists in equilibrium with [MoO
4]
2− at a pH between 4 and 6.5. Further acidification, 1.5 < pH < 2.9, enabled the obtaining of octamolybdate anion [Mo
8O
44]
2−. Larger ionic aggregates are expected to appear at lower pH values, but there are no studies that are related to this statement [
64]. Similar to the chromate- and vanadate-containing LDH, the reported methods that are used to prepare hydrotalcites with intercalated molybdenum-based anions containing different pairs of divalent/trivalent cations in the brucite-like layers are co-precipitation, anion exchange, and hydrothermal method. The studies showed that molybdenum can be introduced either as one of its anionic forms in the interlayer (
Table 6) or as a cation in the brucite-type layers (
Table 7).
The most used method for molybdate incorporation in MgAl-LDH structure is the anionic exchange. However, Drezdon et al. [
66] reported a new technique derived from the classical ionic exchange that primarily involves the synthesis of an organic anion-pillared precursor, also called swelling agent, which is subsequently exchanged with an adequate polyoxometalate anion under acidic conditions [
66]. In this case, the organic anion needs to have slightly larger dimensions than the polyoxometalate to be incorporated. By creating an acidic medium, the organic anion already incorporated in the LDH will be protonated, which weakens the electrostatic interaction between the cationic layers and the intercalated organic species and, thus, allowing for the partial exchange of the organically-pillared hydrotalcite with the polyoxometalate [
66]. Moreover, the anion affinity in LDH interlayers depends on the size of the ion and its charge [
67]. The monovalent anions have lower affinities and, hence, their participation in anion exchange reactions is favored when compared to divalent anions [
67]. Based on the XRD results, Davantes et al. [
68] were able to create a representative scheme that describes the interlayer exchange of different anions (
Figure 9). The affinity series decreases in the order [Mo
7O
24]
6− > CO
32− > [MoO
4]
2− > SO
42− [
60]. The diffraction patterns of the Mo-intercalated samples that were obtained by Carriazo et al. [
69] are characteristic to hydrotalcite-like structure, indicating a rhombohedral packing of the layers. The powder XRD patterns of layered double hydroxides usually present a decrease in the intensities of the (00l) lines, while the value of
l increases, but, in this situation, it was observed that the intensity of the (006) and (009) lines are larger than that of the (003) line.
This abnormal behavior is specific for LDH intercalated with different POM and it appears to be due to the large atomic scattering factor of the interlamellar species [
69]. The ZnAl-LDH containing heptamolybdate in the interlayer presents sharp and very symmetric lines probably due to its ordered packing of the layers. Additionally, the value of the interlayer distance was similar to the height of the heptamolybdate (7.2 Å) with its C
2 axis being perpendicular to the layers. This orientation favors the formation of a strong interaction between the brucite-like layers and the intercalated anion [
69]. For the samples MgAl–Mo and NiAl–Mo, a broad reflection that is close to 11 Å is observed, which suggests the presence of a low-ordered phase of Mg-POM [
94] or a novel phase with the anion grafted to the hydroxyl layer [
69]. Raman studies [
95] indicated that this is due to a small amount of [MoO
4]
2− anions that resulted from the polyanion hydrolysis [
69]. Dobrea et al. [
36] have also tried to intercalate molybdenum-based anions between the brucite-type layers of a MgAl-LDH with different Mg/Al ratios. In the Raman spectra that were recorded for the Mo-LDH samples, it was observed that the most important bands are present in the regions 890–950 and 300–360 cm
−1, and they are most likely attributed to the main types of molybdenum-based anions that can appear within the hydrotalcite structure: [MoO
4]
2−, [Mo
2O
7]
2−, and [Mo
7O
24]
6− [
36].
At low Mo loading, the Raman spectra display bands at about 895 and 320 cm
−1, which clearly belong to the monomer [MoO
4]
2− species. In these spectra, there are also some bands at 1050 and 709 cm
−1 that are probably attributed to the residual NO
3− anions [
36]. By increasing the Mo loading, a more complex Raman spectra was obtained, due to the formation of octahedrally coordinated polymolybdate species (i.e., [Mo
2O
7]
2− with band at 920 cm
−1 and [Mo
7O
24]
6− with bands at 947 and 358 cm
−1), while the bands characteristic for the nitrate and molybdate ions decreased in intensity until their complete disappearance. The sample with the highest Mo concentration mainly contained heptamolybdate anions [
36]. It can be stated that different oxyanions are inserted in the structure of hydrotalcites after anion exchange, and their nature mostly depended on the molybdate species loading and, of course, on the pH used by analyzing the wavenumbers that are specific for various Mo species found in literature (
Table 8) and the Raman spectra of the compounds that were prepared by Dobrea et al. [
36]. Infrared spectroscopy appears to be a better technique for analyzing the structure of Mo(VI) species, but only in aqueous solutions, not necessary intercalated within the hydrotalcite materials, even though the Raman measurements have been widely used for the characterization of polymolybdate anions intercalated between the cationic layers of LDH.
However, Yu et al. [
88] were able to record the FT-IR spectrum of [MoO
4]
2−-pillared ZnAl hydrotalcite, which consists of bands attributed to hydroxyl stretching vibrations (3430 cm
−1) and bending modes of interlayer water (1630 cm
−1). There is also a band at 1368 cm
−1 that is specific for CO
32− stretching mode that might indicate the contamination of the material by atmospheric CO
2 [
88]. The band corresponding to the antisymmetric mode of Mo–O–Mo in [MoO
4]
2− is observed at 834 cm
−1. Moreover, the bands at 620, 559, and 428 cm
−1 can be related to the vibrations modes of the oxygen atoms at the layer crystal lattice [
88]. Polyoxometalates that are based on molybdenum have been often applied in heterogeneous catalysis due to their interesting redox properties.
Layered double hydroxides containing heptamolybdate [Mo7O24]6− anions in the interlamellar space that were prepared by anion exchange starting from suitable precursors proved to be effective catalysts in the epoxidation of different substrates.
Carriazo et al. [
72] have prepared, by the ion exchange method, starting from suitable precursors containing terephthalate, three different LDH, combining Al with different divalent cations (Mg, Zn, and Ni) to obtain the brucite-like layers and heptamolybdate in the interlayer [
72]. The compounds were further used as catalysts for the epoxidation of norbornadiene, benzonorbornadiene, and cyclohexene. The hydrotalcite composition and the reaction solvents, i.e. hydrogen peroxide, dioxane, or dioxane–butyl maleate influenced the selectivity to epoxides [
72]. For the hydrotalcites containing NiAl–Mo and ZnAl–Mo, the selectivity in epoxide was very good, approximately 85% for norbornadiene and almost 100% for benzonorbornadiene [
72]. When cyclohexene was used as a substrate, the same tendency as in previously mentioned cases was observed: NiAl–Mo and ZnAl–Mo catalysts led to the highest selectivities in epoxide. As for the solvent, the best results were obtained while using dioxane or acetone. The differences between these two solvents were quite small. However, the replacement of dioxane with dioxane–butyl maleate mixture led to a decrease in the selectivity to epoxide [
72].
Cyclohexene was also used as a substrate for the epoxidation reaction in the work that was reported by Sels et al. [
70]. The catalysts, in this case, were two hydrotalcites containing MgAl or ZnAl as cations in the layers. Both LDH were obtained by co-precipitation and intercalated by the anion exchange method with molybdenum blue, a mixed-valency isopolyacid containing Mo
V and Mo
VI, namely [Mo
4VMo
8VIO
40]
7− [
70]. The exchange within the LDH is rapid because of its high negative charge. However, it was observed that the nature of the cations from the brucite-type layers of the LDH considerably influenced the long-term stability of Mo blue. With Mo blue on MgAl-LDH, the material has changed its color from deep blue to colorless within a few weeks [
70]. This indicates that Mo blue, which is synthesized in a solution at pH 3, is decomposed in time at the surface of MgAl-LDH, which has pronounced basic properties [
70]. Such decomposition problems do not appear with Mo blue that is exchanged on ZnAl-LDH or on DS-ZnAl-LDH (ZnAl-LDH pre-exchanged with dodecyl sulfate) [
70]. The results on epoxidation of cyclohexene with H
2O
2 and with Mo blue exchanged on MgAl-LDH as catalyst indicate a low olefin conversion. Before the addition of H
2O
2 in the reaction medium, the catalyst presents the yellow shade that is specific to the Mo
VI form of the isopolyacid [
70]. The color changed rapidly into red, suggesting the presence of tetraperoxomolybdate [Mo(O
2)
4]
2− indicating the isopolyacid structure destruction [
70]. A slightly higher olefin conversion is obtained with Mo blue that is exchanged on ZnAl-LDH.
Gardner et al. [
68] have also prepared two catalysts that are derived from Mg
2Al and Zn
2Al layered double hydroxides intercalated by heptamolybdate species and then used them for epoxidation of cyclohexene. The catalysts were synthesized by different methods, in situ hydrolysis in the case of Mg
2Al-LDH and anion exchange for the material containing Zn. In the epoxidation reaction, it was observed that all of the heptametallate-intercalated LDH are not stable under the reaction conditions being converted into lower nuclearity species, which migrate to the LDH surface and lead to low conversions and selectivities [
68].
Zavoianu et al. [
79] have studied the effect of the synthesis methods on the physicochemical properties and catalytic performance of MgAl-molybdate-LDH in cyclohexene oxidation with hydrogen peroxide [
79]. Five samples have been prepared by (i) ionic exchange procedure, as described by Kwon et al. [
63], between a hydrotalcite containing carbonate anions previously obtained by co-precipitation at pH = 10 and low supersaturation with aqueous Na
2MoO
4; (ii) competitive ionic exchange at pH 10 while using a hydrotalcite containing carbonate anions, aqueous Na
2MoO
4, and p-toluene sulfonic acid (pTOS) as swelling agent, based on Van Laar studies [
62]; (iii) exchange of the carbonate precursor LDH with pTOS at pH 4.5, followed by the exchange with molybdate at pH 4.5 under a constant stirring for 24 h and a readjustment of the pH to 10 using NaOH; (iv) direct synthesis at high supersaturation and pH 10 [
18]; and, (v) direct synthesis at low supersaturation and pH 10 [
18,
79]. The characterization techniques used (FTIR, Raman, and UV–Vis spectroscopies) indicate the presence of [MoO
4]
2− and smaller amounts of [Mo
7O
24]
6− species in all of the samples. The catalytic activity of the solids is correlated with the basicity, which decreases with the increase in Mo loading [
18,
79]. The samples containing a low Mo amount (about 2.5–2.8%) in the form of [MoO
4]
2− in the interlayer and increased magnesium concentration in the brucite layers have proven to favor the hydroperoxidation reaction of cyclohexene, while the catalysts also containing [Mo
7O
24]
6− species in their structure encouraged the appearance of epoxides [
79].
Van Laar et al. [
62] reported a molybdate-exchanged layered double hydroxide heterogeneous catalyst for the conversion of H
2O
2 into singlet molecular dioxygen (
1O
2). The distinction between epoxidation and
1O
2 oxygenation was studied by using 1-methyl-1-cyclohexene as a substrate. The highest product yield at complete H
2O
2 consumption was obtained by the LDH catalyst containing less Mo amount [
62]. Whalen et al. [
73] have also studied the disproportionation of H
2O
2 into singlet oxygen by using molybdate-exchanged MgAl-LDH with Mg/Al ratio between 1.5 and 5. The chemical trapping (CT) of
1O
2 with β-citronellol performed the production of singlet oxygen. The efficiency of H
2O
2 has been enhanced with the increase of Mg/Al ratio. The yield of the generated
1O
2 was around 30–35%. The rate of H
2O
2 disproportionation sharply decreases when the Mo loading is increased due to the high Mo amount, which reduces the access of β-citronellol to the [MoO
4]
2− anions in the interlayer space, leading to the loss of a part of the
1O
2 produced within the LDH. A few years later, Whalen et al. [
76] discovered a way of increasing the oxidant efficiency of molybdate-containing MgAl-LDHs, which consists in pretreating the catalyst with 1,3-propanediol, glycerol, ethylene glycol, or 1,2-propanediol [
76]. The peroxidation of different olefins with this glycol-modified catalyst led to conversions above 88% and selectivities up to 99% [
76]. Mild oxidation of tetrahydrothiophene (THT) to sulfolane has been studied on molybdenum-containing LDH that was obtained by anionic exchange of a nitrate-LDH precursor with an aqueous solution of Na
2MoO
4·2H
2O at pH 4.5 by Hulea et al. [
35]. The oxidation of tetrahydrothiophene was highly selective towards the sulfoxidation reaction, exclusively resulting in sulfoxide and sulfolane. A conversion of 98% was reached after 45 min. reaction, with a selectivity to sulfolane of 78%.
Ciocan et al. [
71] have synthesized two series of Mo-containing hydrotalcites by different techniques: (i) anion exchange at atmospheric pressure, after complete synthesis of LDH, and (ii) anion exchange under hydrothermal conditions during the aging step of LDH synthesis. The influence of the preparation technique on the catalytic properties was investigated in the oxidation of dibenzothiophene (DBT) and anthracene with hydrogen peroxide [
71]. The catalytic tests that were performed under moderate conditions (40–70 °C) indicated that both types of catalysts are efficient, but the solids that were prepared under hydrothermal conditions are more active, probably due to their larger specific surface area, which makes the access to active sites easier [
71]. Dobrea et al. studied the oxidation of dibenzothiophene with H
2O
2 while using as catalyst a MgAl-LDH intercalated with heptamolybdate species [
45]. The catalyst was characterized by Raman spectroscopy before and after the reaction to verify the stability. The DBT conversion strongly increased with the metal content in the Mo-LDH [
45], which was obviously due to the increase in the number of active sites. Large polyoxymolybdate species were formed at high metal loadings, leading to an important increase in the distance between the LDH layers and, hence, to increased accessibility of the large DBT molecule to the catalytic centers [
45]. The Raman spectra recorded confirmed the increased stability of Mo-oxoanions contained in layered double hydroxides, in contrast with the V-oxoanions [
45].
Zavoianu at al. [
74] have studied the oxidation of tert-butanethiol (t-BuSH) while using as catalysts Mo-LDH samples that were obtained from two different molybdenum sources, e.g. Na
2MoO
4 or (NH
4)
6Mo
7O
24 and prepared by two methods: (i) ionic exchange and (ii) co-precipitation at pH 10 under high supersaturation [
74]. When Na
2MoO
4 was used as a molybdenum source, crystalline materials with relatively high basicity and fine dispersion of molybdate species were obtained. These samples showed the best catalytic activities, leading to high conversions of t-BuSH of about 80%. By changing the molybdenum source to (NH
4)
6Mo
7O
24, solids with a lower crystallinity, smaller surface area, lower basicity, and catalytic activity were obtained [
88]. Mitchell and Wass [
75] have synthesized the Mo-LDH catalysts while applying different preparation methods based on the anion exchange technique to be used in propane ODH. The most active and selective catalyst in this reaction was that prepared by classical anion exchange between a calcined commercial hydrotalcite and ammonium dimolybdate with prior impregnation with terephthalic acid and pH adjusted to 4.5 with nitric acid [
18,
75].
Choudary et al. [
80] have reported the use of molybdate-containing MgAl-LDH catalyst for the synthesis of β-bromostyrenes in aqueous medium via the halodecarboxylation reaction of cinnamic acid. The high catalytic activity of the LDH system was attributed to its basic character and the large positive charge of brucite layers which may further lead to the enrichment of bromide ion near the solid surface [
80]. Moreover, the excess of positive charge on the LDH surface shields the negative charge of the peroxomolybdate and bromide, ensuring high halide oxidation rates in this way [
80]. Das and Parida [
86] reported a parallel study of the esterification of acetic acid with n-butanol on ZnAl-LDH intercalated with tungstophosphoric (TPA) and molybdophosphoric (MPA) acid. The conversion of acetic acid was higher for the solid that was prepared with MPA (84.15%), although the selectivity to the ester was 100% for both of the samples. This behavior might be ascribed to the increased surface Brønsted acidity and the specific surface area shown by the Mo-containing sample [
18,
86]. Colombo et al. [
85] prepared at two different pH values a NiZn LDH initially intercalated with acetate ions, which were subsequently replaced with molybdate anions via an ion exchange reaction. The basal spacing in the NiZn-LDH decreased from 13.08 Å to ca. 9.5 Å regardless of the working pH, [
85], which might suggest the intercalation of hydrated molybdate anions. The as-synthesized material and the solid that was obtained by thermal treatment at 250 °C (basal spacing reduced to 7.35 Å due to dehydration) were tested as catalysts for the methyl transesterification of soybean oil [
85]. All of the materials were catalytically active and led to high conversions of the substrate, but poor selectivities [
85].
Klemkaite-Ramanauske et al. [
77] prepared molybdate-containing MgAl layered double hydroxides by using different variations of the co-precipitation method and used them as catalysts for the synthesis of 2-adamantylidene(phenyl)amine Schiff base. All of the synthesized molybdate-containing layered double hydroxides presented similar catalytic activity for the studied reaction, no matter what co-precipitation route was used for preparation [
77]. LDH is usually prepared from divalent and trivalent cations, but Mostafa et al. [
90,
91,
92] succeeded to synthesize by co-precipitation method while using ammonium hydroxide and ammonium carbonate as precipitating agents, a highly crystalline CoMo-LDH, as a new type of M
2+M
6+-LDH. This new type of LDH had shown a highly energetic surface due to the formation of +4 charges between Co
2+ and Mo
6+, as identified by XPS analysis. Muramatsu et al. were also successful in the preparation of a ZnMo-LDH belonging to this new generation of hydrotalcites.
Apart from the crucial role in the conventional catalysis, Mo-containing LDH can also be used as photocatalyst for electrocatalytic water oxidation [
84], as adsorbent [
60,
69,
89], or as an anti-corrosion material [
78,
88].



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}