The Preparation and Characterization of Co–Ni Nanoparticles and the Testing of a Heterogenized Co–Ni/Alumina Catalyst for CO Hydrogenation

 , ,
, ,  ,
, 

Abstract
1. Introduction
2. Results
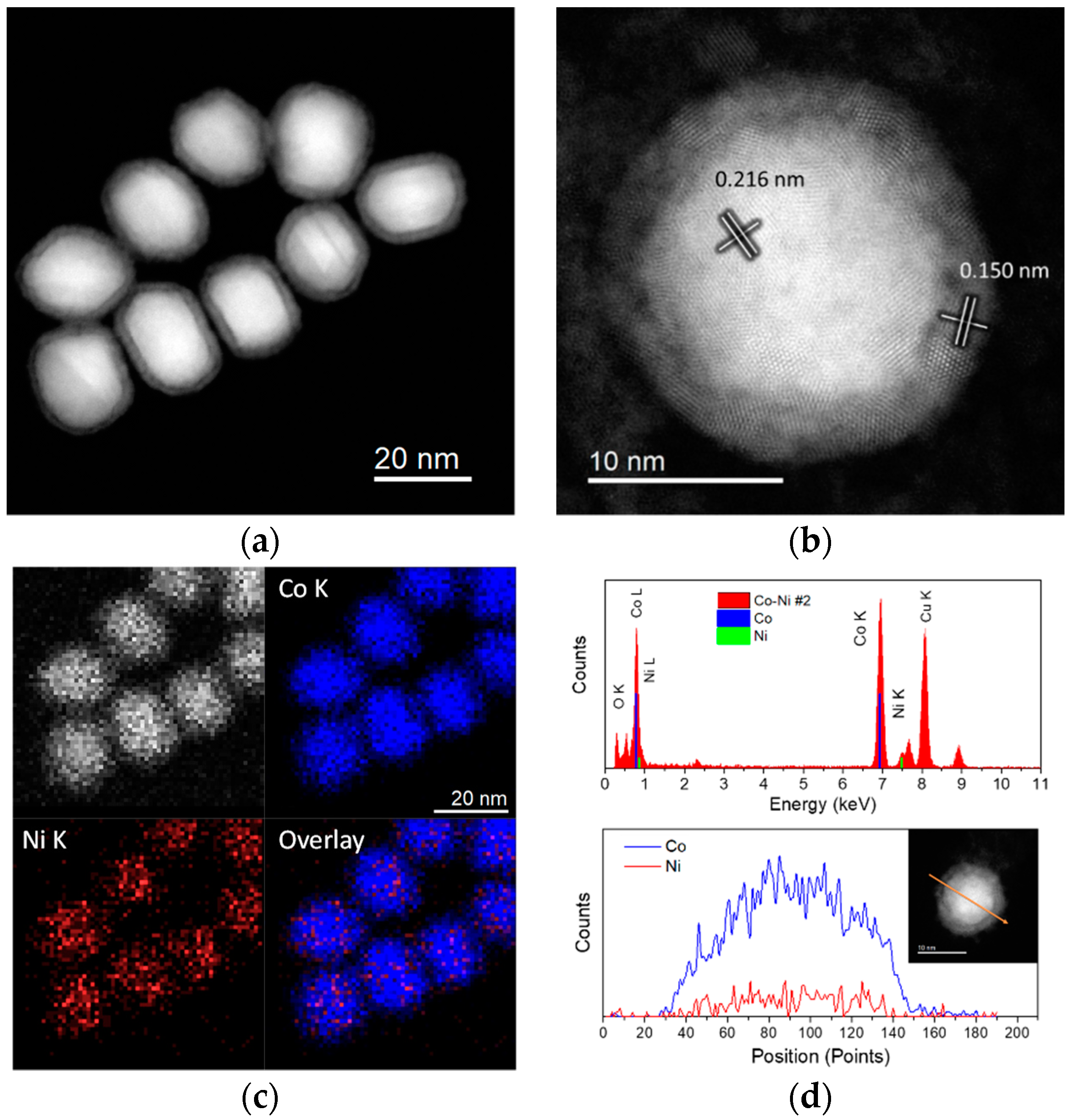
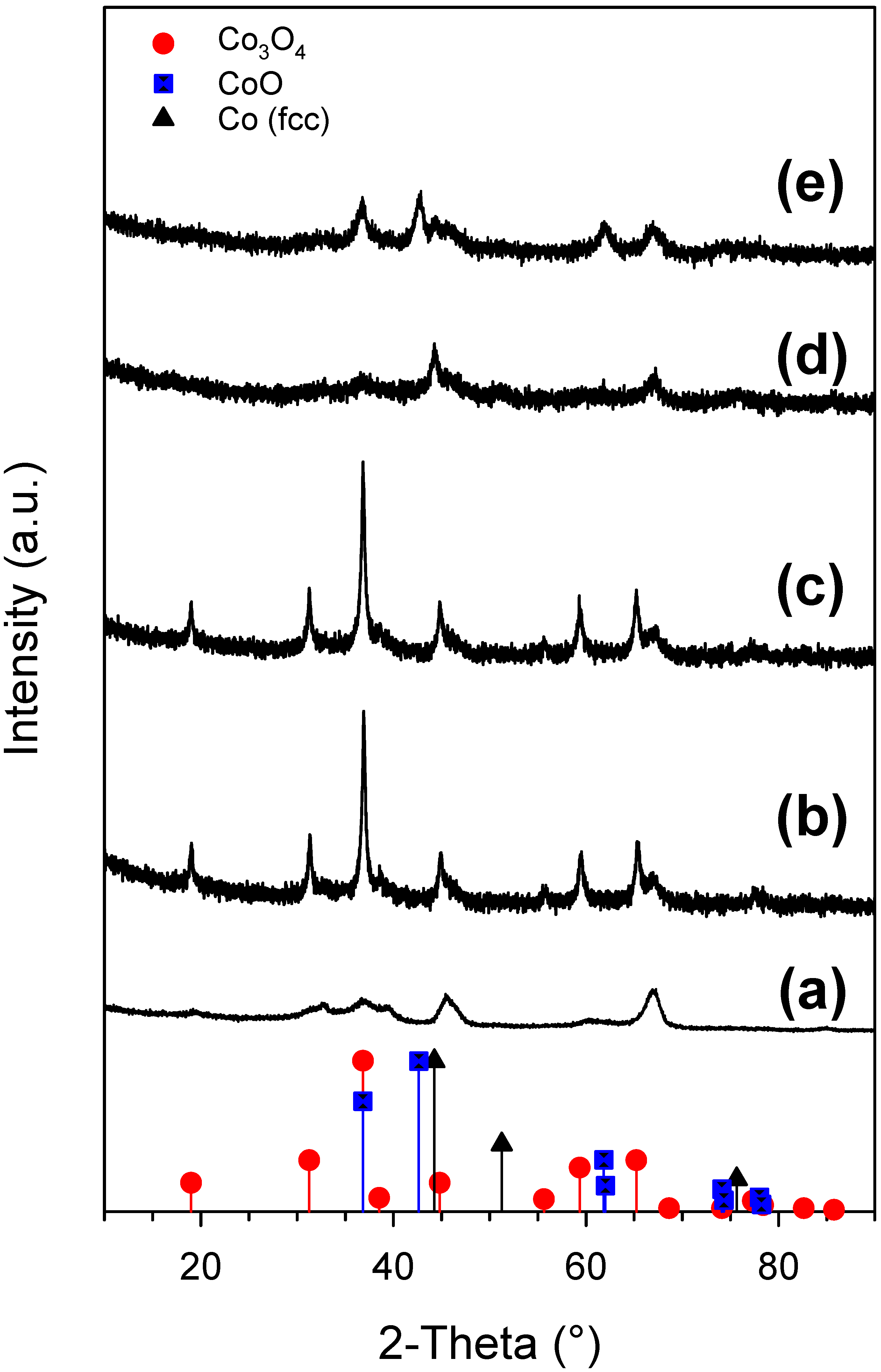
2.1. Cobalt–Nickel Nanoparticles
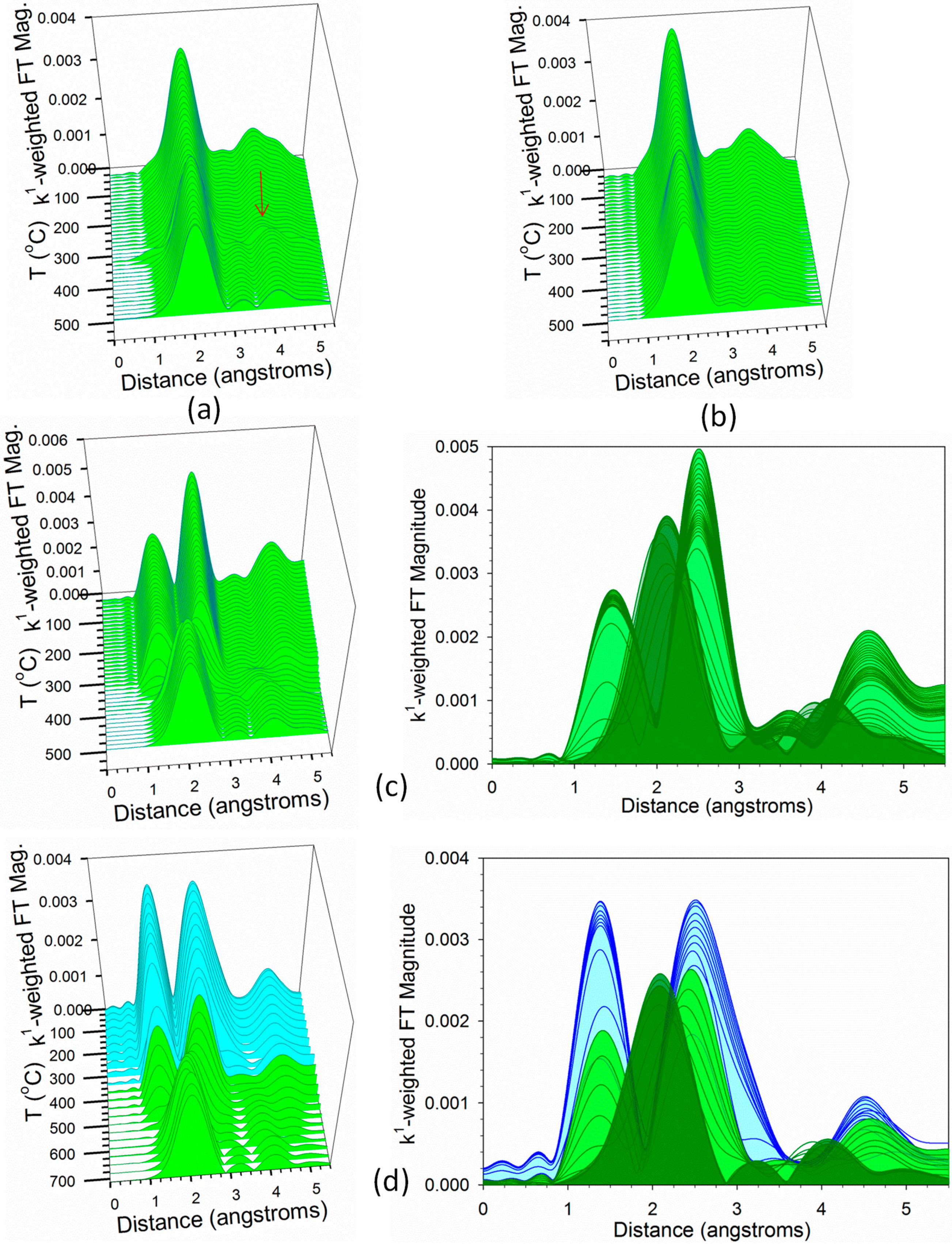
2.2. Heterogenized Catalysts
2.3. Activity Data
3. Discussion
4. Materials and Methods
4.1. Sample Denomination
- Sample #1 20% metal (98.4% at. Co/1.6% at. Ni)/Al2O3
- Sample #2 20% metal (92.7% at. Co/7.3% at. Ni)/Al2O3
- Sample #3 20% metal (89.7% at. Co/10.3% at. Ni)/Al2O3
- Sample #4 25% metal (90% at. Co/10% at. Ni)/Al2O3 calcined 350 °C
- Sample #5 25% Co/Al2O3 calcined 350 °C
4.2. Reagents
4.3. Preparation of Ni and Co Precursors
4.4. Preparation of Ni–Co Nanoparticles
- (a)
- Co–Ni Sample #1 and #2: The Co–Ni nanoparticles were prepared as follows. 20 mL of oleylamine were previously heated to 120 °C and then the Co and Ni precursors were added to obtain two samples with a Co:Ni desired molar ratio. Then, 30 µL of a 1,2-hexadecanediol (0.3 M) is added and stirring was continued vigorously for 5 min at 170 °C. The temperature was slowly increased to 230 °C and the solution was kept 60 min more at this temperature. The resulting colloid was then cooled slowly to room temperature. Two samples were prepared with an atomic ratio of 98.4/1.6 (Sample #1) and 92.7/7.3 (Sample #2) of Co:Ni respectively. Finally, the nanoparticles were washed by centrifugation several times with a mixture of chloroform and ethanol at 4000 rpm and dried at 120 °C for 4 h.
- (b)
- Co–Ni Sample #3: For this synthesis method of Co–Ni nanoparticles a Co/Ni atomic ratio of 89.7/10.3 was used. Briefly, 1 mmol of CoII (acet) was completely dissolved in 4 mL of oleylamine by using magnetic stirring, and 150 µL of a NiCl2 solution (0.1 M in ethanol) was injected. The mixture was stirred and heated at 100 °C for 30 min. Then, the temperature was raised to 230 °C for 1 h. Finally, the solution was let to cool down to room temperature. The samples were further purified by centrifuging several times, precipitating and redispersing the particles with a mixture of chloroform and ethanol. The final precipitated particles were collected and dried at 120 °C for 4 h.
- (c)
- Co–Ni Sample #4: Sample #4 was prepared by a conventional slurry impregnation method and contained 25% metal by weight, with an atomic ratio of 90/10 of Co/Ni. Co/Alumina-Catalox 150 γ-alumina (150 m2/g) was used as a support. Cobalt nitrate (Alfa Aesar) served as the precursor to load the cobalt onto the Al2O3 support. In this method, which follows a Sasol patent [1], the ratio of the volume of solution used to the weight of alumina was 1:1, such that approximately 2.5 times the pore volume of solution was used to prepare the loading solution. Two impregnation steps were used to load 12.5% of metal by weight for each step. Between each step, the catalyst was dried under vacuum in a rotary evaporator at 60 °C, and the temperature was slowly increased to 100 °C. After the second impregnation/drying step, the catalyst was calcined in air at 350 °C for 4 h.
- (d)
- (d) Co Sample #5: Sample #5 was prepared in the same way as Sample #4, except no Ni was included. The weight was 25% Co.
4.5. Characterization
4.6. Reaction Testing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Espinoza, R.L.; Visagie, J.L.; van Berge, P.J.; Bolder, F.H. Fischer-Tropsch catalysts containing iron and cobalt. U.S. Patent 5,733,839, 31 March 1998. [Google Scholar]
- van Berge, P.J.; Barradas, S.; van de Loosdrecht, J.; Visagie, J.L. Advances in the cobalt catalyzed Fischer-Tropsch synthesis. Erdoel Erdgas Kohle 2001, 117, 138–142. [Google Scholar]
- Glacier Resource Management Group. InvestmentMine. Available online: http://www.infomine.com/investment/ (accessed on 15 November 2019).
- Jacobs, G.; Das, T.K.; Zhang, Y.; Li, J.; Racoillet, G.; Davis, B.H. Fischer-Tropsch synthesis: Support, loading and promoter effects on the reducibility of cobalt catalysts. Appl. Catal. A Gen. 2002, 233, 263–281. [Google Scholar] [CrossRef]
- Jacobs, G.; Ji, Y.; Davis, B.H.; Cronauer, D.C.; Kropf, A.J.; Marshall, C.L. Fischer-Tropsch synthesis: Temperature programmed EXAFS/XANES investigation of the influence of support type, cobalt loading, and noble metal promoter addition to the reduction behavior of cobalt oxide particles. Appl. Catal. A Gen. 2007, 333, 177–191. [Google Scholar] [CrossRef]
- Jacobs, G.; Chaney, J.A.; Patterson, P.M.; Das, T.K.; Maillot, J.C.; Davis, B.H. Fischer-Tropsch synthesis: Study of the promotion of Pt on the reduction property of Co/Al2O3 catalysts by in situ EXAFS of Co K and Pt LIII edges and XPS. J. Synchrotron Radiat. 2004, 11, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.M.; Perez, H.D.; Bartholomew, C.H.; Hecker, W.C. Effect of promoter deposition order on platinum-, ruthenium-, or rhenium-promoted cobalt Fischer-Tropsch catalysts. Appl. Catal. A Gen. 2014, 482, 275–286. [Google Scholar] [CrossRef]
- Cook, K.M.; Hecker, W.C. Reducibility of alumina-supported cobalt Fischer-Tropsch catalysts: Effects of noble metal type, distribution, retention, chemical state, bonding, and influence on cobalt crystallite size. Appl. Catal. A Gen. 2012, 449, 69–80. [Google Scholar] [CrossRef]
- Ma, W.; Jacobs, G.; Ji, Y.; Bhatelia, T.; Bukur, D.B.; Khalid, S.; Davis, B.H. Fischer-Tropsch synthesis: Influence of CO conversion on selectivities, H2/CO usage ratios, and catalyst stability for a Ru promoted Co/Al2O3 catalyst using a slurry phase reactor. Top. Catal. 2011, 54, 757–767. [Google Scholar] [CrossRef]
- Iglesia, E.; Soled, S.L.; Fiato, R.A.; Via, G.H. Bimetallic synergy in cobalt-ruthenium Fischer-Tropsch synthesis catalysts. J. Catal. 1993, 143, 345–368. [Google Scholar] [CrossRef]
- Hilmen, A.M.; Schanke, D.; Holmen, A. TPR study of the mechanism of rhenium promotion of alumina-supported cobalt Fischer-Tropsch catalysts. Catal. Lett. 1996, 38, 143–147. [Google Scholar] [CrossRef]
- Vada, S.; Hoff, A.; Ådnanes, E.; Schanke, D.; Holmen, A. Fischer-Tropsch synthesis on supported cobalt catalysts promoted by platinum and rhenium. Top. Catal. 1995, 2, 155–162. [Google Scholar] [CrossRef]
- Ronning, M.; Nicholson, D.G.; Holmen, A. In situ EXAFS study of the bimetallic interaction in a rhenium-promoted alumina-supported cobalt Fischer-Tropsch catalyst. Catal. Lett. 2001, 72, 141–146. [Google Scholar] [CrossRef]
- Jacobs, G.; Chaney, J.A.; Patterson, P.M.; Das, T.K.; Davis, B.H. Fischer-Tropsch synthesis: Study of the promotion of Re on the reduction property of Co/Al2O3 catalysts by in situ EXAFS/XANES of Co K and Re LIII edges and XPS. Appl. Catal. A 2004, 264, 203–212. [Google Scholar] [CrossRef]
- Jacobs, G.; Ribeiro, M.C.; Ma, W.; Ji, Y.; Khalid, S.; Sumodjo, P.T.A.; Davis, B.H. Group 11 (Cu, Ag, Au) promotion of 15%Co/Al2O3 Fischer-Tropsch catalysts. Appl. Catal. A Gen. 2009, 361, 137–151. [Google Scholar] [CrossRef]
- Jermwongratanachai, T.; Jacobs, G.; Ma, W.; Shafer, W.D.; Gnanamani, M.K.; Gao, P.; Kitiyanan, B.; Davis, B.H.; Klettlinger, J.L.S.; Yen, C.H.; et al. Fischer-Tropsch synthesis: Comparisons between Pt and Ag promoted Co/Al2O3 catalysts for reducibility, local atomic structure, catalytic activity, and oxidation-reduction (OR) cycles. Appl. Catal. A 2013, 464–465, 165–180. [Google Scholar] [CrossRef]
- Shafer, W.D.; Gnanamani, M.K.; Graham, U.M.; Yang, J.; Masuku, C.M.; Jacobs, G.; Davis, B.H. Fischer-Tropsch: Product Selectivity—The Fingerprint of Synthetic Fuels. Catalysts 2019, 9, 259. [Google Scholar] [CrossRef]
- van Steen, E.; Claeys, M.; Dry, M.E.; van de Loosdrecht, J.; Viljoen, E.L.; Visagie, J.L. Stability of nanocrystals: Thermodynamic analysis of oxidation and re-reduction of cobalt in water/hydrogen mixtures. J. Phys. Chem. B 2005, 109, 3575–3577. [Google Scholar] [CrossRef]
- Jacobs, G.; Das, T.K.; Patterson, P.M.; Luo, M.; Conner, W.A.; Davis, B.H. Fischer-Tropsch synthesis: Effect of water on Co/Al2O3 catalysts and XAFS characterization of reoxidation phenomena. Appl. Catal. A 2004, 270, 65–76. [Google Scholar] [CrossRef]
- Logdberg, S.; Boutonnet, M.; Walmsley, J.C.; Jaras, S.; Holmen, A.; Blekkan, E.A. Effect of water on the space-time yield of different supported cobalt catalysts during Fischer-Tropsch synthesis. Appl. Catal. A 2011, 393, 109–121. [Google Scholar] [CrossRef]
- Hughes, N.A.; Gloriot, V.; Smiley, D.D.; Jacobs, G.; Pendyala, V.R.R.; Graham, U.M.; Ma, W.; Gnanamani, M.K.; Shafer, W.D.; Maclennan, A.; et al. Fischer-Tropsch synthesis: comparisons of Al2O3 and TiO2 supported Co catalysts prepared by aqueous impregnation and CVD methods. In Fischer-Tropsch Synthesis, Catalysts and Catalysis: Advances and Applications; Davis, B.H., Occelli, M.L., Eds.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2016; Ch. 6; pp. 85–106. [Google Scholar]
- Singh, A.K.; Xu, Q. Synergistic catalysis over bimetallic alloy nanoparticles. ChemCatChem 2013, 5, 652–676. [Google Scholar] [CrossRef]
- Nam, K.M.; Shim, J.H.; Han, D.W.; Kwon, H.S.; Kang, Y.M.; Li, Y.; Song, H.; Seo, W.S.; Park, J.T. Syntheses and characterization of wurtzite CoO, rocksalt CoO, and spinel Co3O4 nanocrystals: Their interconversion and tuning of phase and morphology. Chem. Mater. 2010, 22, 4446–4454. [Google Scholar] [CrossRef]
- Morelos-Santos, O.; de la Torre, A.R.; Schacht-Hernández, P.; Portales-Martínez, B.; Soto-Escalante, I.; Mendoza-Martínez, A.M.; Mendoza-Cruz, R.; Velázquez-Salazar, J.J.; José-Yacamán, M. NiFe2O4 Nanocatalyst for Heavy Crude Oil Upgrading in Low Hydrogen/Feedstock Ratio. Catal. Today 2019. [Google Scholar] [CrossRef]
- Hu, L.; Wu, L.; Liao, M.; Hu, X.; Fang, X. Electrical transport properties of large, individual NiCo2O4 nanoplates. Adv. Funct. Mater. 2012, 22, 998–1004. [Google Scholar] [CrossRef]
- Taylor, A.; Floyd, R.W. Precision measurements of lattice parameters of non-cubic crystals. Acta Crystallogr. 1950, 3, 285–289. [Google Scholar] [CrossRef]
- Nishizawa, T.; Ishida, K. The Co-Ni (Cobalt-Nickel) System. Bull. Alloy Phase Diagr. 1983, 4, 390–395. [Google Scholar] [CrossRef]
- Downs, R.T.; Bartelmehs, K.; Gibbs, G.; Boisen, M. Interactive Software for Calculating and Displaying X-Ray or Neutron Powder Diffractometer Patterns of Crystalline Materials. Am. Mineral. 1993, 78, 1104–1107. [Google Scholar]
- Mendoza-Cruz, R.; Bazán-Diaz, L.; Velázquez-Salazar, J.J.; Samaniego-Benitez, J.E.; Ascencio-Aguirre, F.M.; Herrera-Becerra, R.; José-Yacamán, M.; Guisbiers, G. Order-disorder phase transitions in Au-Cu nanocubes: From nano-thermodynamics to synthesis. Nanoscale 2017, 9, 9267–9274. [Google Scholar] [CrossRef]
- Guisbiers, G.; Mendoza-Pérez, R.; Bazán-Díaz, L.; Mendoza-Cruz, R.; Velázquez-Salazar, J.J.; José-Yacamán, M. Size and shape effects on the phase diagrams of nickel-based bimetallic nanoalloys. J. Phys. Chem. C 2017, 121, 6930–6939. [Google Scholar] [CrossRef]
- Gallego, G.S.; Batiot-Dupeyrat, C.; Barrault, J.; Florez, E.; Mondragon, F. Dry Reforming of Methane over LaNi1−yByO3±δ (B = Mg, Co) Perovskites Used as Catalyst Precursor. Appl. Catal. A Gen. 2008, 334, 251–258. [Google Scholar] [CrossRef]
- Li, H.; Liao, J.; Du, Y.; You, T.; Liao, W.; Wen, L. Magnetic-Field-Induced Deposition to Fabricate Multifunctional Nanostructured Co, Ni, And Coni Alloy Films as Catalysts, Ferromagnetic and Superhydrophobic Materials. Chem. Commun. 2013, 49, 1768–1770. [Google Scholar] [CrossRef]
- Sun, S.; Zeng, H.; Robinson, D.B.; Raoux, S.; Rice, P.M.; Wang, S.X.; Li, G. Monodisperse mfe2o4 (m = fe, co, mn) nanoparticles. J. Am. Chem. Soc. 2004, 126, 273–279. [Google Scholar] [CrossRef]
- Mourdikoudis, S.; Liz-Marzan, L.M. Oleylamine in nanoparticle synthesis. Chem. Mater. 2013, 25, 1465–1476. [Google Scholar] [CrossRef]
- Tsakoumis, N.E.; Ronning, M.; Borg, O.; Rytter, E.; Holmen, A. Deactivation of cobalt based Fischer-Tropsch catalyst: A review. Catal. Today 2010, 154, 162–182. [Google Scholar] [CrossRef]
- Jahangiri, H.; Bennet, J.; Mahjoubi, P.; Wilson, K.; Gu, S. A review of advanced catalyst development for Fischer-Tropsch synthesis of hydrocarbon from biomass derived syn-gas. Catal. Sci. Technol. 2014, 4, 2210–2229. [Google Scholar] [CrossRef]
- Rytter, E.; Skagseth, T.H.; Eri, S.; Sjastad, A.O. Cobalt Fischer-Tropsch catalysts using nickel promoter as a rhenium substitute to suppress deactivation. Ind. Eng. Chem. Res. 2010, 49, 4140–4148. [Google Scholar] [CrossRef]
- Nikparsa, P.; Mirzaei, A.A.; Rauch, R. Modification of Co/Al2O3 Fischer-Tropsch nanocatalysts by adding Ni: A kinetic approach. Int. J. Chem. Kinet. 2016, 48, 131–143. [Google Scholar] [CrossRef]
- Shimura, K.; Miyazawa, T.; Hanaoka, T.; Hirata, S. Fischer-Tropsch synthesis over alumina supported bimetallic Co-Ni catalyst: Effect of impregnation sequence and solution. J. Mol. Catal. 2015, 407, 15–24. [Google Scholar] [CrossRef]
- Yu, H.; Zhao, A.; Zhang, H.; Ying, W.; Fang, D. Bimetallic catalyst of Co and Ni for Fischer-Tropsch synthesis supported on alumina. Energy Sources Part A Recovery Util. Environ. Eff. 2015, 37, 47–54. [Google Scholar] [CrossRef]
- van Helden, P.; Prinsloo, F.; van den Berg, J.A.; Xaba, B.; Erasmus, W.; Claeys, M.; van de Loosdrecht, J. Cobalt-nickel bimetallic Fischer-Tropsch catalysts: A combined theoretical and experimental approach. Catal. Today 2020, 342, 88–98. [Google Scholar] [CrossRef]
- Jacoby, M. X-ray absorption spectroscopy. Chem. Eng. News 2001, 79, 33–38. [Google Scholar] [CrossRef]
- Ressler, T. WinXAS: A Program for X-ray Absorption Spectroscopy Data Analysis under MS-Windows. J. Synchrotron Radiat. 1998, 5, 118–122. [Google Scholar] [CrossRef]
- Ravel, B. ATOMS: Crystallography for the X-ray absorption spectroscopist. J. Synchrotron Radiat. 2001, 8, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Newville, M.; Ravel, B.; Haskel, D.; Rehr, J.J.; Stern, E.A.; Yacoby, Y. Analysis of multiple-scattering XAFS data using theoretical standards. Phys. B Condens. Matter 1995, 208–209, 154–156. [Google Scholar] [CrossRef]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | As (BET) [m2/g] | Vp [cm3/g] | Dp [nm] |
|---|---|---|---|
| Sample #4 | 95.5 | 0.243 | 9.3 |
| Sample #5 | 96.5 | 0.226 | 9.3 |
| μmol H2 Desorbed/gcat | Uncorr.% Disp. | Uncorr.Diam. (nm) | O2 Uptake (μmol/gcat) | * % Red. | ** % Red. | * Corr. % Disp. | ** Corr. % Disp. | * Corr. Diam. (nm) | ** Corr. Diam. (nm) |
|---|---|---|---|---|---|---|---|---|---|
| Sample #4—25%metal (90%Co-10%Ni)/Al2O3 | |||||||||
| 92.5 | 4.4 | 24 | 1495 | 54.6 | 40.5 | 8.0 | 10.8 | 12.9 | 9.6 |
| Sample #5—25%Co/Al2O3 | |||||||||
| 91.3 | 4.3 | 24 | 1324 | 46.8 | 29.1 | 9.2 | 14.8 | 11.2 | 7.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López-Tinoco, J.; Mendoza-Cruz, R.; Bazán-Díaz, L.; Karuturi, S.C.; Martinelli, M.; Cronauer, D.C.; Kropf, A.J.; Marshall, C.L.; Jacobs, G. The Preparation and Characterization of Co–Ni Nanoparticles and the Testing of a Heterogenized Co–Ni/Alumina Catalyst for CO Hydrogenation. Catalysts 2020, 10, 18. https://doi.org/10.3390/catal10010018
López-Tinoco J, Mendoza-Cruz R, Bazán-Díaz L, Karuturi SC, Martinelli M, Cronauer DC, Kropf AJ, Marshall CL, Jacobs G. The Preparation and Characterization of Co–Ni Nanoparticles and the Testing of a Heterogenized Co–Ni/Alumina Catalyst for CO Hydrogenation. Catalysts. 2020; 10(1):18. https://doi.org/10.3390/catal10010018
Chicago/Turabian StyleLópez-Tinoco, Julián, Rubén Mendoza-Cruz, Lourdes Bazán-Díaz, Sai Charan Karuturi, Michela Martinelli, Donald C. Cronauer, A. Jeremy Kropf, Christopher L. Marshall, and Gary Jacobs. 2020. "The Preparation and Characterization of Co–Ni Nanoparticles and the Testing of a Heterogenized Co–Ni/Alumina Catalyst for CO Hydrogenation" Catalysts 10, no. 1: 18. https://doi.org/10.3390/catal10010018
APA StyleLópez-Tinoco, J., Mendoza-Cruz, R., Bazán-Díaz, L., Karuturi, S. C., Martinelli, M., Cronauer, D. C., Kropf, A. J., Marshall, C. L., & Jacobs, G. (2020). The Preparation and Characterization of Co–Ni Nanoparticles and the Testing of a Heterogenized Co–Ni/Alumina Catalyst for CO Hydrogenation. Catalysts, 10(1), 18. https://doi.org/10.3390/catal10010018