Abstract
Due to the unique structures, photoelectric properties, good catalytic activity, and broad potential applications, gold nanoclusters (Au) received extensive attention in catalysis, bioengineering, environmental engineering, and so on. In the present work, the structures and properties of Au adsorbed on the MgO(001) and TiO(101) surfaces were investigated by density functional theory. The results showed that the catalytic properties of Au will be enhanced when Au is adsorbed on certain supports. Because the difference of the outer electronic structure of metals in supports, the direction of the charge transfer was different, thus inducing the different charge distribution on Au. When Au was adsorbed on MgO(001) [TiO(101)] surface, Au will have negative [positive] charges and thus higher catalytic activity in oxidation [reduction] reaction. The variation of surface charges caused by the support makes Au possess different catalytic activity in different systems. Moreover, the electronic structure of the support will make an obvious influence on the s and d density of states of Au, which should be the intrinsic reason that induces the variations of its structure and properties. These results should be an important theoretical reference for designing Au as the photocatalyst applied to the different oxidation and reduction reactions.
1. Introduction
The research on gold nanoclusters (Au) started to attract extensive attention when Haruta et al. [1] found that Au on TiO have a good catalytic activity in CO low-temperature oxidation reaction. Further deep explorations of Au uncovered that Au not only exhibited a variety of physical and chemical features [2,3,4], but also had broad applications in many fields, such as catalysis, biological engineering, nanotechnology, and so on [5,6,7,8,9,10]. More importantly, the study on the Au is still the research focus even now.
Among the factors of determining the Au physical and chemical features, the structures of Au are found to play a crucial role. In the theoretical aspect, Häkkinen et al. [11] and Min et al. [12] found that the neutral Au will prefer the two-dimensional structures when the atom number n is less than 13. Research of the Au (2 ≤ n ≤9) structures carried out by Mao et al. [13] indicates that with the increasing of n the average binding energy per atom will increase gradually, and an odd-even oscillation will appear in the Fermi energy, the electron affinity energy, and the ionization potential energy. Moreover, Bulusu et al. [14] investigated the Au with n = 15∼19 and revealed that the Au structure will be transformed to the hollow structure at n = 17 and the pyramid structure at n = 19. Also, Fa et al. [15] found that Au will be the two-dimensional planar structures in the case of n ≤ 12 and the transition from the planar to the three-dimensional structures will take place at n = 13∼15. Zhao et al. [16] found that the ground-state geometry of Au (n = 19∼22) is tetrahedron, and Au is a tube-like structure. All the researches indicate that the Au structures are closely dependent on the Au size.
On the other hand, great attention has also been paid to the study of the Au adsorbed on metal oxide surface, especially MgO and TiO. Yoon et al. [17] found that Au on the MgO surface will show a good catalytic activity in the CO oxidation reaction because the charges are transferred from MgO surface to Au. Liu et al. [18] reported that the structure of Au will be a three-dimensional structure at n = 7 in the study of Au (n = 1∼8) vertically adsorbed on the surface of MgO(001). Roldan et al. [19] studied the adsorption activity of the O activation on Au/MgO, and Stamatakis et al. [20] investigated the CO oxidation on Au/MgO. Both these two works demonstrate that the charge transfer occurring between the clusters and the O promotes the adsorption of oxygen. Meanwhile, many researches on Au/TiO have also begun. Kim et al. [21] found that the charge transfer between support materials and the Au will increase the catalytic activity of the clusters. The study of the stability of Au on anatase TiO(110) surface carried out by Pabisiak et al. [22] showed that the finite linear Au clusters will be formed on the highly defected surface. Asakura [23] found that Au anion is very important for the stability of Au adsorbed on TiO(110) surface. At the same time, Li et al. [24] explored the influence of Au/TiO on CO oxidation and showed that the reaction rate is dependent on the size and shape of the Au. Recently, Cao et al. [25] found that the charge transfer between Au and TiO(110) depends on the number of oxygen free radicals and the size of Au.
All the researches indicate that the chemical and physical properties of Au are closely related to its structure, its size, and the surfaces of the supports. Although many researches of Au adsorbed on metal oxide surfaces have been done, the intrinsic mechanism and the meso-scale control mechanism combining the macroscopic properties with the microscopic electronic structures are still not quite clear. So a further systematic research of Au on MgO(001) and anatase TiO(101) surfaces is required. In this paper, Au, Au/MgO(001), and Au/TiO(101) will be studied by using the density functional theory (DFT) [26,27]. The transition of the geometric structure, the variations of the average bond lengths and the partial density of states (PDOS), and the charge transfer will be discussed. Furthermore, the reasons why these variations appear and the possible intrinsic mechanisms will be analyzed.
2. Results and Discussion
2.1. Geometric Structures of Au
To find the steadiest adsorption site of Au atom on the supports, four types of initial structures (Figure 1) will be considered including O atom top, Mg atom top, Mg-O bridge, and Mg-O hole. After optimization, Au atoms initially adsorbed on Mg-O bridge or Mg-O hole will be moved to the site of O atom top. Also, the adsorption energy of the Au atom on the top site of O atoms is found to be less than that on Mg atoms. Therefore, we can draw a conclusion that the steadiest adsorption site should be the top site of O atom, which is in accordance with the results reported in the literature [28,29,30]. For each Au with a certain atom number n, its steadiest configuration on the supports will be determined by comparing lots of optimized structures obtained from different initial configurations.

Figure 1.
Typical initial structures of Au/MgO.
Figure 2, Figure 3 and Figure 4 show the optimized structures of Au in vacuum, Au on MgO(001) surface, and Au on TiO(101) surface, respectively. By comparing the corresponding stable structures of Au, Au/MgO(001) and Au/TiO(101), we can see that Au structures are distorted obviously when they are adsorbed on the MgO(001) and TiO(101) surfaces. In the case of Au/MgO(001), the bond length of Au on MgO(001) almost does not change and Au is askew adsorbed on the surface, as shown in Figure 3. Au on MgO(001), retaining a planar structure similar to that in vacuum, is adsorbed with the plane perpendicular to MgO(001) surface. In the corresponding side view, the middle Au atom is far away from the surface of MgO(001), but still on the top of oxygen atoms, which is in accordance with the references [28,29]. The structures of Au (n = 4∼13) on MgO(001) surface show partial distortion. In the top view, they seem to maintain the same basic structures as in vacuum, while in the side view obvious distortions can be observed. Au atoms located on the sites of the top and bridge of O atom are moved far away from the MgO(001) surface, but Au atoms on other sites are moved close to the MgO(001) surface, which makes the flat structures distort. Au shows a three dimensional flat cage structure in vacuum, but distorts seriously when it is adsorbed on MgO(001) with the atoms near MgO surface almost in one plane. For another three-dimensional flat cage structure, Au will change little when it is adsorbed on MgO(001). On TiO(101) surface, Au structure does not distort obviously, and the bond length is only slightly stretched. For other Au, roughly speaking, they show similar distortion with its bond lengths longer than those in vacuum, and with these variations less than those on MgO(001) surface. The changes of the structures of Au on MgO(001) and TiO(101) surfaces show that the adsorption properties of Au (n ≲ 4) are different from Au (n ≳ 5). Moreover, these results show that when Au are adsorbed on metal oxide surface, some atoms in the Au plane will move up and others will move down relative to the original Au plane, making the bonds stretched and the flat structure become a quasi-flat structure, which will be beneficial for improving the catalytic activity of Au.
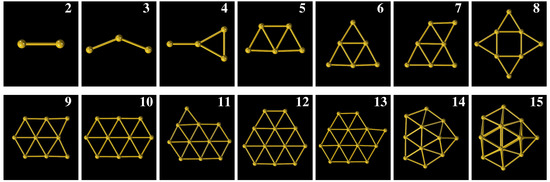
Figure 2.
Structures of Au with n = 2, 3, , 15 in vacuum.
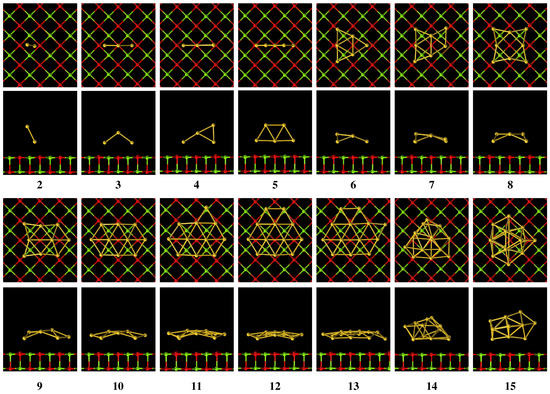
Figure 3.
Top and side view of the Au/MgO(001) structures.
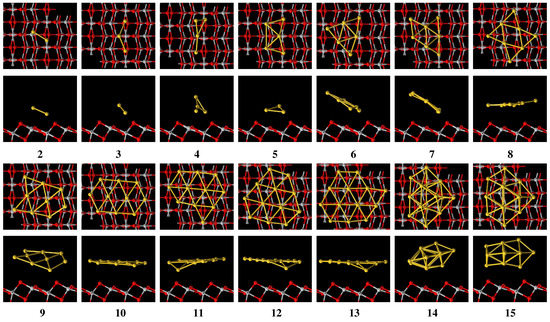
Figure 4.
Top and side view of the Au/TiO(101) structures.
Furthermore, we analyze the adsorption energy E, the binding energy E, the internal binding energy E and the average bond length for Au on MgO(001) and TiO(101) with different n in Figure 5. In Figure 5a, it can be found that the adsorption energy of some Au is very large, such as 1.51 eV (0.78 eV) for Au (Au) on the MgO(001), and 0.62 eV for Au on the TiO(101), possibly implying that the adsorption between Au (n ≲ 4) and supports should be the chemisorption, while other Au (n ≳ 5) may be adsorption. Meanwhile, the average adsorption energy of Au tends to decrease with the increase of the number n of Au on MgO(001) surface, and approaches zero. This means that Au in enough large size is difficult to be adsorbed on metal oxide surface. Except Au/TiO(101), all the adsorption energy values of other Au on TiO(101) are smaller than the counterparts of Au on MgO(001). This demonstrates that Au on Mg(001) surface may possess stronger stability. For Au on TiO(101) surface, Au is almost the steadiest adsorption structure except the chemisorption of Au. Figure 5b gives the variations of E of Au adsorbed on MgO(001) and TiO(101). Either on MgO(001) or TiO(101) surface, the E increases with the increase of Au atom number n. The E of Au on MgO(001) becomes nearly invariant when n ≳ 6, while on TiO surface, it will become invariant when n ≳ 7. This indicates that Au of larger size is steadier than the smaller one, and the smaller Au cluster may incline to merge to a larger one. In Figure 5c, the E of Au also increases with the atom number n of Au. The corresponding E will be reduced when Au is adsorbed on metal oxide surface, especially on TiO(101) surface. The interaction between TiO and Au is more beneficial for polymerizing. By comparing the values of E with E for Au on the same metal support in the same size in Figure 5a,c, we can see that the E within cluster (n ≥ 5) is always larger than the E. This demonstrates that the interaction of Au-Au in clusters is stronger than that of Au-O and Au-Mg, likewise for Au-O and Au-Ti, further indicating that either adsorbed on TiO(101) or MgO(001), Au atoms prefer to exist in the form of clusters. The bond length of Au corresponding to different n is given in Figure 5d. For clarity, the corresponding structure data before and after Au adsorption are also shown in Table 1. It can be found that all the adsorption-induced variations of the structure data for Au-Au, Mg-O, and Ti-O are in the range −0.081∽0.081 Å. For Au of the same atom number, except Au, all the average Au-Au bond lengths of Au adsorbed on metal oxide is longer than that in vacuum, indicating the activity will be improved. In addition, the variation of Au-Au bond length in Au/MgO(001) is larger than that in Au/TiO(101), meaning that the activity of Au on MgO(001) is higher, which agrees about the analysis of adsorption energy. As presented in references [31,32,33], the bond length variation in cluster is closely dependent on the competition between the cluster-support interaction and the intrinsic interaction within the cluster. Here, since E is greater than E for Au (n ≳ 5), the increase of the average Au-Au bond length is very tiny.
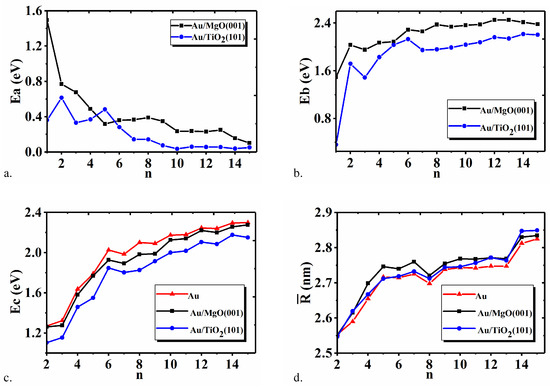
Figure 5.
Average adsorption energy Ea (a), average binding energy Eb (b), internal binding energy Ec (c), and average Au-Au bond length (d) of Au, Au/MgO(001), and Au/TiO(101) versus the atom number n.

Table 1.
Structure data of Au, Au/MgO(001), and Au/TiO(101).
2.2. PDOS
To study the effect of the adsorption-induced variation of the internal electronic structures of Au on the catalytic activity, we present all the related PDOS in Figure 6. First, we show the PDOS of O and Mg atoms in the surface of pure MgO(001) in Figure 6a. Clearly, it is the PDOS of p state of O atom that plays the dominant contribution. Therefore, we further only show the PDOS of p state of the O atom in the surface layer of the pure MgO(001), MgO(001) adsorbed with Au, and Mg(001) adsorbed with Au in Figure 6b. Obviously, a whole shift of the PDOS toward the low energy direction will appear due to the adsorption of Au or Au. This will cause the decrease of the corresponding PDOS value at Fermi energy, which also indicates that the adsorption of Au or Au will lead to the variation of the electron structure of the O atom in MgO(001) surface. In Figure 6c, we plot the PDOS of Au, Au/MgO(001), and Au/TiO(101). It can be found that in Au the PDOS of s and d states of Au will play the dominant contribution. Also, the peaks become broader in width and lower in height, and eventually three peaks incline to merge into a wider peak. Furthermore, we can see that the adsorption will push the three peaks below the Fermi energy to the low energy direction. In addition, the two peaks above the Fermi energy disappear for Au/TiO(101), and the two peaks will approach each other for Au/MgO(001). These imply that the different supports are able to induce the different variations of the PDOS of the s and d states in Au. Thus, the electronic structures of Au on MgO(001) and TiO(101) surface will be changed due to adsorption. Eventually, this should be beneficial to improving the Au catalytic activity.
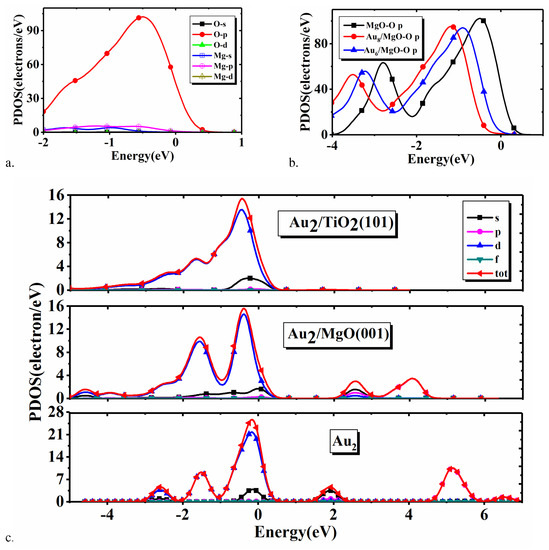
Figure 6.
(a) PDOS of O and Mg atoms in MgO(001), (b) PDOS of p state of O atom in the surface layer of pure MgO(001), MgO(001) adsorbed with Au, and Au clusters, respectively, and (c) PDOS of Au, Au/MgO(001) and Au/TiO(101).
2.3. Charge Transfer
As demonstrated in many works [34,35,36], charge transfer is a key factor in improving the catalytic activity of Au. Therefore, we intend to investigate the charge transfer occuring in Au/MgO(001) and Au/TiO(101). As examples, the electron density differences of Au/MgO(001) and Au/TiO(101) are shown in Figure 7a,b, respectively. Also, in Figure 8 we show the Mülliken charge distribution of Au, TiO, and Au/TiO(101). Meanwhile, for clarity, the corresponding charge transfer data are listed in Table 2. In Figure 7a, the negative charges accumulate around atoms of Au, while the negative charges near the O and Mg atoms connected with Au atoms decrease. This demonstrates that the electrons transferring from Mg-O to Au atoms, makes Au have negative charges. On the other hand, from the Mülliken charge distribution of Au/MgO(001) (see Table 2), the same conclusion can be drawn. Obviously, the whole charges of each Au and the charges of every Au atom in Au are all negative when Au is adsorbed on MgO(001). This indicates that the negative charges transfer from MgO surface to Au, which is consistent with the studies of Roldan [19] and Stamatakis [20]. Different from the case of MgO(001) surface, charges will accumulate near O atom of the TiO(101) which is close to Au atoms when Au is adsorbed on TiO(101), as shown in Figure 7b and Figure 8, and Table 2. The charges of the two Au atoms directly connected to TiO surface change from −0.06 to 0.06, and the charges of the central Au atom change from 0.09 to 0.03. Consequently, the whole charges of Au change from 0.00 to 0.23. These phenomena suggest that the negative charges will transfer from Au atoms to TiO surface, inducing that Au has positive charges. Therefore we conclude that the charge transfer direction in Au/MgO(001) is different from that in Au/TiO(101).
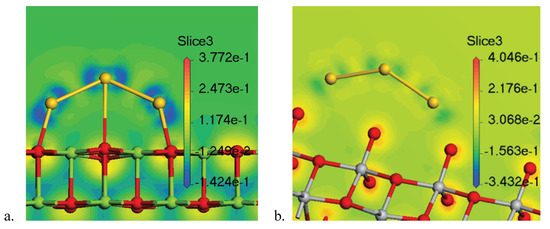
Figure 7.
Electron density differences of Au/MgO(001) (a) and Au/TiO(101) (b).
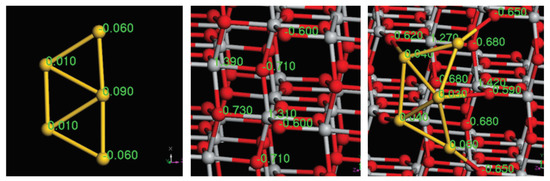
Figure 8.
Mülliken charge distribution of Au, TiO(101), and Au/TiO(101).
Table 2.
The Mulliken charge distribution of Au in different systems.
To understand the direction of the electron transfer between Au and supports, we should resort to the outer electron structure of the metal as reported in reference [33]. The outer electron structure of Au, Mg, and Ti are 5d6s, 3s, and 3d4s, respectively. Obviously, Au has an almost filled 5d orbit and half-filled s orbit, and Mg has no d orbit, while Ti has unfilled 3d orbits, inducing that the charge can flow from Mg-O to Au or from Au to Ti-O. Fundamentally speaking, the different direction of charge flow is determined by the difference of the outer electron structure of the metal d orbit. Therefore, the negative charges flow from Mg-O in MgO(001) to Au and finally Au has negative charges, which makes the Au possess high catalytic activity in oxidation reaction. However, the negative charges flow from Au to Ti-O in TiO(101) and finally Au has positive charges, which makes the Au possess high catalytic activity in reduction reaction. Therefore, catalytic activity of Au can be controlled by choosing suitable metal oxide supports.
3. Materials and Methods
All calculations are performed by the plane-wave ultrasoft pseudo potential method based on DFT with the CASTEP [37] program of Material Studio package. The GGA-PBE [38,39] version of exchange-correlation energy function is applied, which was proved to be reasonable [40]. The cutoff energy for the plane wave basis set is 340 eV and the total energy convergence criterion for the self-consistent field (SCF) is 1.0 × 10 eV/atom. Based on the literature [41,42,43,44,45,46] and our experience, the MgO(001) and TiO(101) should be the absorption surfaces. Calculations of the two systems are performed for a two layer slab [41] with 6 × 6 surface periodicity and 3 Å [47,48] with 3 × 3 surface periodicity respectively, and both separated from their replicas by vacuum region of 15 Å width.
First of all, the stablest structures can be determined by optimizing Au structures in vacuum with the atom number ranging from 1 to 15. Then the chosen structures are adsorbed on MgO(001) and TiO(101) surfaces, respectively, which will be optimized further. Finally, the steadiest structures of the two systems can be determined according to the adsorption energy, the binding energy, and the average bond length. The adsorption energy per Au atom is calculated by the formula
Here E stands for the total energy of the Au/sub system while E and E represent the energies of the isolated oxide substrate and the Au clusters, respectively. Therefore, a larger adsorption energy will indicate a stronger adsorption between cluster and support. The binding energy (per Au atom) of Au atom and oxide substrate is calculated via the formula
where E is the energy of an isolate Au atom calculated in a large cell. In addition, the internal binding energy E and the average bond length of Au are calculated according to the following formulae:
Here R denotes the distance between Au atoms i and j in Au and m represents the number of bonds in Au.
Aiming at uncovering the intrinsic mechanism of the adsorption-induced influences on the properties of Au, our research was carried out in vacuum at 0 K and 1 atm.
4. Conclusions
The structures and properties of Au adsorbed on different metal oxide supports were studied by DFT. The following results are obtained: (1) The catalytic activity of Au becomes higher when they are adsorbed on metal oxide surface; (2) With the increase of Au atom number n, the binding energy increases while the adsorption energy between clusters and metal oxide decreases; (3) For the same size, the Au of Au/MgO(001) has higher catalytic activity than that of Au/TiO(101); (4) The direction of charge flow is determined by the outer electron structure of Au atom and metal atom of the oxide surface when Au is adsorbed on the oxide surface. In the system of Au/MgO(001), charges flow from Mg-O to Au, indicating that Au has negative charges, which leads to high catalytic activity in oxidation reaction, while in the system of Au/TiO(101), the direction of charge flow is reversed and Au has positive charges, leading to high catalytic activity in reduction reaction. This research may provide a theoretical reference for the design of the structures of Au catalyst, as well as the further study of its stability and catalytic properties.
Author Contributions
Conceptualization, H.W. and Z.J.; Data analysis, J.G.; Writing—original draft preparation, Q.H.; Writing—review and editing, Y.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Key Research and Development Program of China (No. 2017YFC0210304), the National Natural Science Foundation of China (Nos. 11774029, 11504374), and the CAS Informatization Program of the Thirteenth Five-Year Plan (No. XXH1350303-103).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 ∘C. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Chang, C.M.; Chou, M.Y. Alternative Low-Symmetry Structure for 13 Atom Metal Clusters. Phys. Rev. Lett. 2004, 93, 133401. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, G.; Zhao, J. Density-functional study of Aun (n = 2–20) clusters: Lowest-energy structures and electronic properties. Phys. Rev. B 2002, 66, 035418. [Google Scholar] [CrossRef]
- Fernandez, E.M.; Soler, J.M.; Balbás, L.C. Planar and cagelike structures of gold clusters: Density-functional pseudopotential calculations. Phys. Rev. B 2006, 73, 235433. [Google Scholar] [CrossRef]
- Yang, Q.; Liu, J.; Chen, H.; Wang, X.; Huang, Q.; Shan, Z. Preparation of noble metallic nanoclusters and its application in biological detection. Prog. Chem. 2011, 23, 880–892. [Google Scholar]
- Yan, F.; Liu, X.; Zhao, D.; Bao, W.; Xi, F. Application of Fluorescent Gold Nanoclusters for the Determination of Small Molecules. Prog. Chem. 2013, 25, 799–808. [Google Scholar]
- Shang, L.; Dong, S.; Nienhaus, G.U. Ultra-small fluorescent metal nanoclusters: Synthesis and biological applications. Nano Today 2011, 6, 401–418. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Y.; House, S.D.; Zhao, S.; Wahab, Z.; Yang, J.C.; Jin, R. Interface Engineering of Gold Nanoclusters for CO Oxidation Catalysis. ACS Appl. Mater. Interfaces 2018, 10, 29425–29434. [Google Scholar] [CrossRef]
- Moon, Y.K.; Jeong, S.Y.; Kang, Y.C.; Lee, J.H. Metal Oxide Gas Sensors with Au Nanocluster Catalytic Overlayer: Toward Tuning Gas Selectivity and Response Using a Novel Bilayer Sensor Design. ACS Appl. Mater. Interfaces 2019, 11, 32169–32177. [Google Scholar] [CrossRef]
- Burgos, J.C.; Mejia, S.M.; Metha, G.F. Effect of Charge and Phosphine Ligands on the Electronic Structure of the Au8 Cluster. ACS Omega 2019, 4, 9169–9180. [Google Scholar] [CrossRef]
- Haekkinen, H.; Yoon, B.; Landman, U.; Li, X.; Zhai, H.J.; Wang, L.S. On the Electronic and Atomic Structures of Small Aun(n = 4–14) Clusters: A Photoelectron Spectroscopy and Density Functional Study. Chem. Inf. 2003, 107, 6168–6175. [Google Scholar] [CrossRef]
- Min, B.J.; Shin, W.C.; Park, J.I. Plane-wave Density Functional Theory Study of the Electronic and Structural Properties of Ionized and Neutral Small Gold Clusters. New Phys. Sae Mulli 2017, 67, 480–484. [Google Scholar] [CrossRef]
- Mao, H.P.; Wang, H.Y.; Ni, Y.; Xu, G.L.; Ma, M.Z.; Zhu, Z.H.; Tang, Y.J. Geometry and electronic properties of Aun(n = 2–9) clusters. Acta Phys Sin. 2004, 53, 1766–1771. [Google Scholar]
- Bulusu, S.; Zeng, X.C. Structures and relative stability of neutral gold clusters: Aun (n = 15–19). J. Chem. Phys. 2006, 125, 154303. [Google Scholar] [CrossRef]
- Fa, W.; Luo, C.; Dong, J. Bulk-fragment and tube-like structures of Aun (n = 2–26). Phys. Rev. B 2005, 72, 3182–3184. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Ning, H.; Wang, J.; Su, X.J.; Guo, X.G.; Liu, Y. Structural evolution of Aun (n = 20–32) clusters: Lowest-lying structures and relativistic effects. Phys. Lett. A 2010, 374, 1033–1038. [Google Scholar] [CrossRef]
- Yoon, B. Charging Effects on Bonding and Catalyzed Oxidation of CO on Au8 Clusters on MgO. Science 2005, 307, 403–407. [Google Scholar] [CrossRef]
- Liu, J.; Shunfang, L.I.; Haisheng, L.I. Study on First-Pinciples Calculations of Adsorption of Gold Clusters Aun (n ≤ 8) on MgO(001) Surface. Mater. Rev. 2008, 2, 232–236. [Google Scholar]
- Roldan, A.; Ricart, J.M.; Illas, F.; Pacchioni, G. O2 Activation by Au5 Clusters Stabilized on Clean and Electron-Rich MgO Stepped Surfaces. J. Phys. Chem. C 2010, 114, 16973–16978. [Google Scholar] [CrossRef]
- Stamatakis, M.; Christiansen, M.A.; Vlachos, D.G.; Mpourmpakis, G. Multiscale Modeling Reveals Poisoning Mechanisms of MgO-Supported Au Clusters in CO Oxidation. Nano Lett. 2012, 12, 3621–3626. [Google Scholar] [CrossRef]
- Kim, Y.D.; Fischer, M.; Gantefor, G. Origin of unusual catalytic activities of Au-based catalysts. Chem. Phys. Lett. 2003, 377, 170–176. [Google Scholar] [CrossRef][Green Version]
- Pabisiak, T.; Kiejna, A. Stability of gold nanostructures on rutile TiO2(110) surface. Surf. Sci. 2011, 605, 668–674. [Google Scholar] [CrossRef]
- Asakura, K.; Takakusagi, S.; Ariga, H.; Chun, W.J.; Suzuki, S.; Koike, Y.; Uehara, H.; Miyazaki, K.; Iwasawa, Y. Preparation and structure of a single Au atom on the TiO2(110) surface: Control of the Au–metal oxide surface interaction. Faraday Discuss. 2013, 162, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gao, Y.; Li, H.; Zhao, Y.; Pei, Y.; Chen, Z.; Zeng, X.C. CO Oxidation on TiO2(110) Supported Subnanometer Gold Clusters: Size and Shape Effects. J. Am. Chem. Soc. 2013, 135, 19336–19346. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Cao, Y.; Hu, S.; Yu, M.; Wang, T.; Huang, S.; Yan, S. Manipulating the charge state of Au clusters on rutile TiO2(110) single crystal surfaces through molecular reactions probed by infrared spectroscopy. Phys. Chem. Chem. Phys. 2016, 18, 17660–17665. [Google Scholar]
- Kohn, W.; Sham, L.J. Quantum Density Oscillations in an Inhomogeneous Electron Gas. Phys. Rev. 1965, 137, 1697–1706. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Pacchioni, G.; Giordano, L.; Baistrocchi, M. Charging of metal atoms on ultrathin MgO/Mo(100) films. Phys. Rev. Lett. 2005, 94, 226104. [Google Scholar] [CrossRef]
- Yulikov, M.; Sterrer, M.; Heyde, M.; Rust, H.P.; Risse, T.; Freund, H.J.; Pacchioni, G.; Scagnelli, A. Binding of Single Gold Atoms on Thin MgO(001) Films. Phys. Rev. Lett. 2006, 96, 146804. [Google Scholar] [CrossRef]
- Yulikov, M.; Sterrer, M.; Risse, T.; Freund, H.J. Gold atoms and clusters on MgO(100) films; an EPR and IRAS study. Surf. Sci. 2009, 603, 1622–1628. [Google Scholar] [CrossRef]
- Ankudinov, A.L.; Rehr, J.J.; Low, J.J.; Bare, S.R. Sensitivity of Pt X-ray absorption near edge structure to the morphology of small Pt clusters. J. Chem. Phys. 2002, 116, 1911–1919. [Google Scholar] [CrossRef]
- Boyanov, B.I.; Morrison, T.I. Support and Temperature Effects in Platinum Clusters. 1. Spatial Structure. J. Phys. Chem. 1996, 100, 16310–16317. [Google Scholar] [CrossRef]
- Wang, L.L.; Khare, S.V.; Chirita, V.; Johnson, D.D.; Rockett, A.A.; Frenkel, A.I.; Mack, N.H.; Nuzzo, R.G. Origin of Bulklike Structure and Bond Length Disorder of Pt37 and Pt6Ru31 Clusters on Carbon: Comparison of Theory and Experiment. J. Am. Chem. Soc. 2006, 128, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Costello, C.K.; Kung, M.C.; Oh, H.S.; Wang, Y.; Kung, H.H. Nature of the active site for CO oxidation on highly active Au/γ-Al2O3. Appl. Catal. A 2002, 232, 159–168. [Google Scholar] [CrossRef]
- Fu, Q.; Saltsburg, H.; Flytzani-Stephanopoulos, M. Active Nonmetallic Au and Pt Species on Ceria-Based Water-Gas Shift Catalysts. Science 2003, 301, 935–938. [Google Scholar] [CrossRef]
- Guzman, J. Structure and reactivity of a mononuclear gold complex catalyst supported on magnesium oxide. Angew. Chem. Int. Ed. Engl. 2003, 42, 690–693. [Google Scholar] [CrossRef]
- Matsuzawa, N.; Seto, J.; Dixon, D.A. Density Functional Theory Predictions of Second-Order Hyperpolarizabilities of Metallocenes. J. Phys. Chem. A 1997, 101, 9391–9398. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Aikens, C.M. Modelling small gold and silver nanoparticles with electronic structure methods. Mol. Simul. 2012, 38, 607–614. [Google Scholar] [CrossRef]
- Ferrando, R.; Barcaro, G.; Fortunelli, A. Structures of small Au clusters on MgO(001) studied by density-functional calculations. Phys. Rev. B 2011, 83, 045418. [Google Scholar] [CrossRef]
- Henry, C.R. Surface studies of supported model catalysts. Surf. Sci. Rep. 1998, 31, 231–325. [Google Scholar] [CrossRef]
- Henry, C.R. Morphology of supported nanoparticles. Prog. Surf. Sci. 2005, 80, 92–116. [Google Scholar] [CrossRef]
- You, H.; Liu, C.J.; Ge, Q. Interaction of Pt clusters with the anatase TiO2(101) surface: A first principles study. J. Phys. Chem. B 2006, 110, 7463–7472. [Google Scholar]
- Gong, X.Q.; Selloni, A.; Dulub, O.; Jacobson, P.; Diebold, U. Small Au and Pt Clusters at the Anatase TiO2(101) Surface: Behavior at Terraces, Steps, and Surface Oxygen Vacancies. J. Am. Chem. Soc. 2008, 130, 370–381. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, M.; Han, Y.; Li, W.; Meng, X.; Zong, B. Nucleation and Growth of Palladium Clusters on Anatase TiO2(101) Surface: A First Principle Study. J. Phys. Chem. C 2008, 112, 19506–19515. [Google Scholar] [CrossRef]
- Pabisiak, T.; Kiejna, A. Energetics of oxygen vacancies at rutile TiO2(110) surface. Solid State Commun. 2007, 144, 324–328. [Google Scholar] [CrossRef]
- Kiejna, A.; Pabisiak, T.; Gao, S.W. The energetics and structure of rutile TiO2(110). J. Phys. Condens. Matter 2006, 18, 4207. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).