
DephosNet: A Novel Transfer Learning Approach for Dephosphorylation Site Prediction


Abstract
:1. Introduction
2. Methods
2.1. Data and Data Processing
2.1.1. Dephosphorylation Dataset
2.1.2. Phosphorylation Dataset
2.1.3. Data Preprocessing
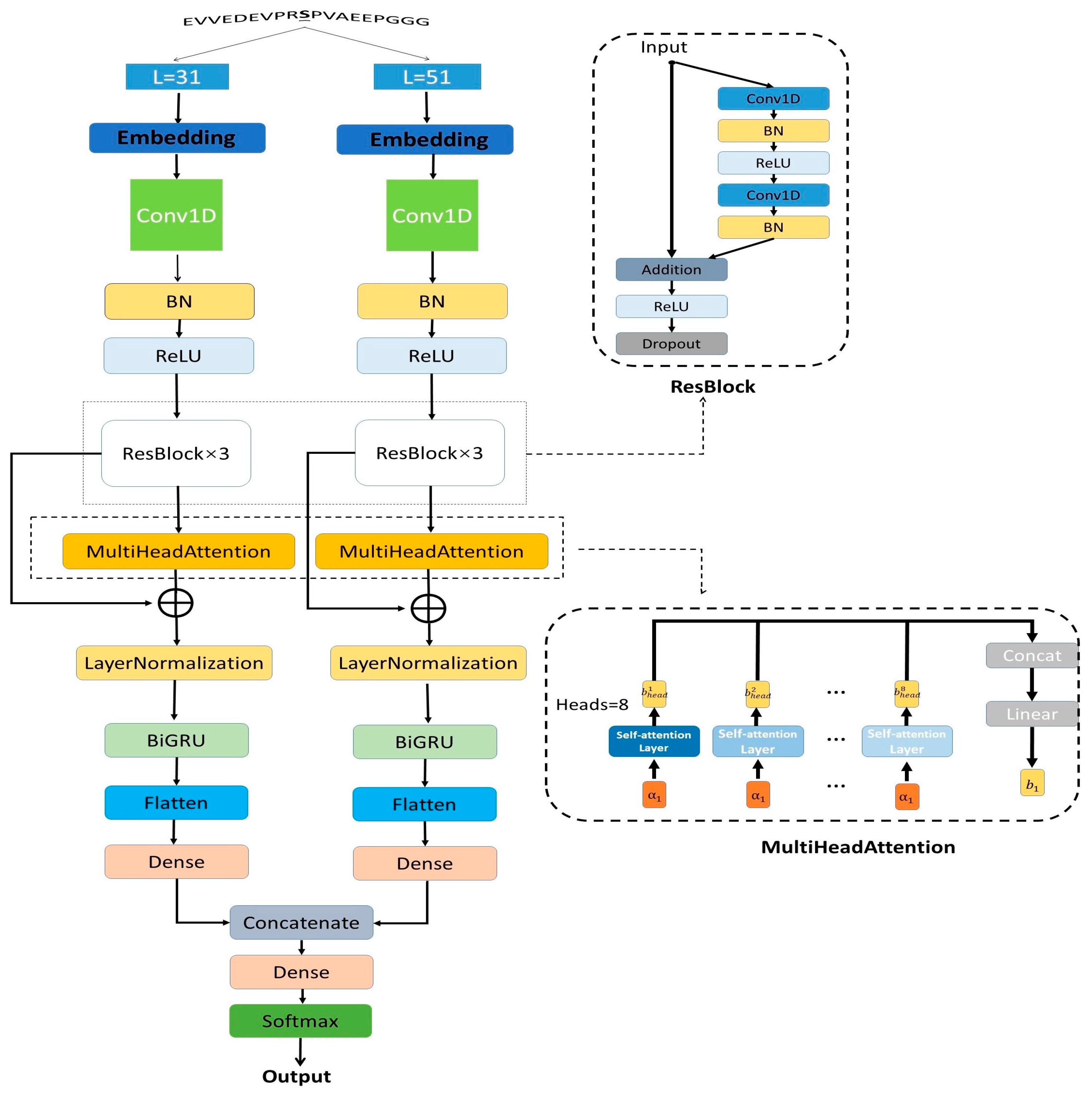
2.2. DephosNet Architecture
2.3. Transfer Learning
2.4. Performance Evaluation
3. Results
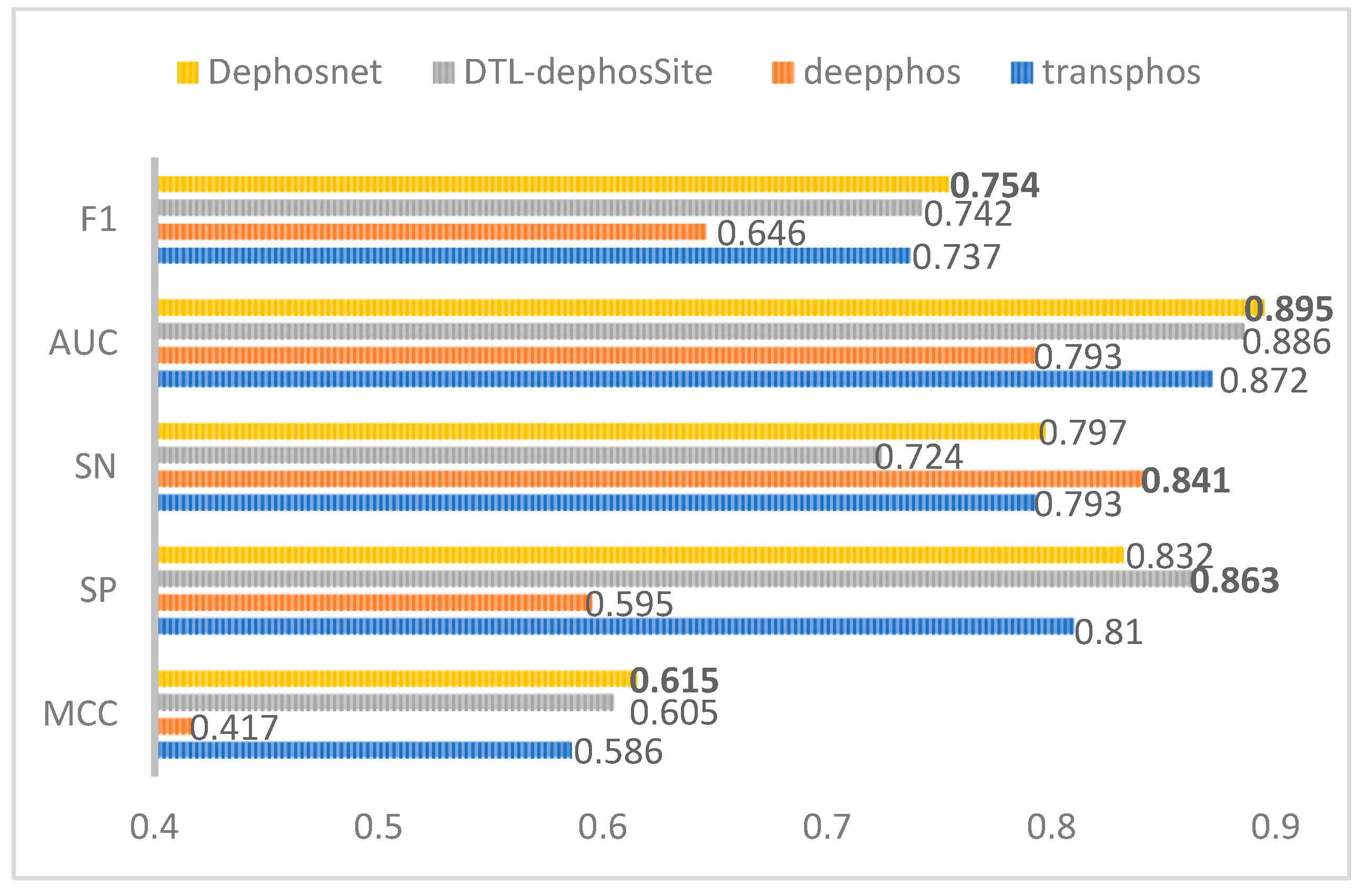
3.1. Comparison with Other State-of-the-Art Models on Dephosphorylation Site Datasets
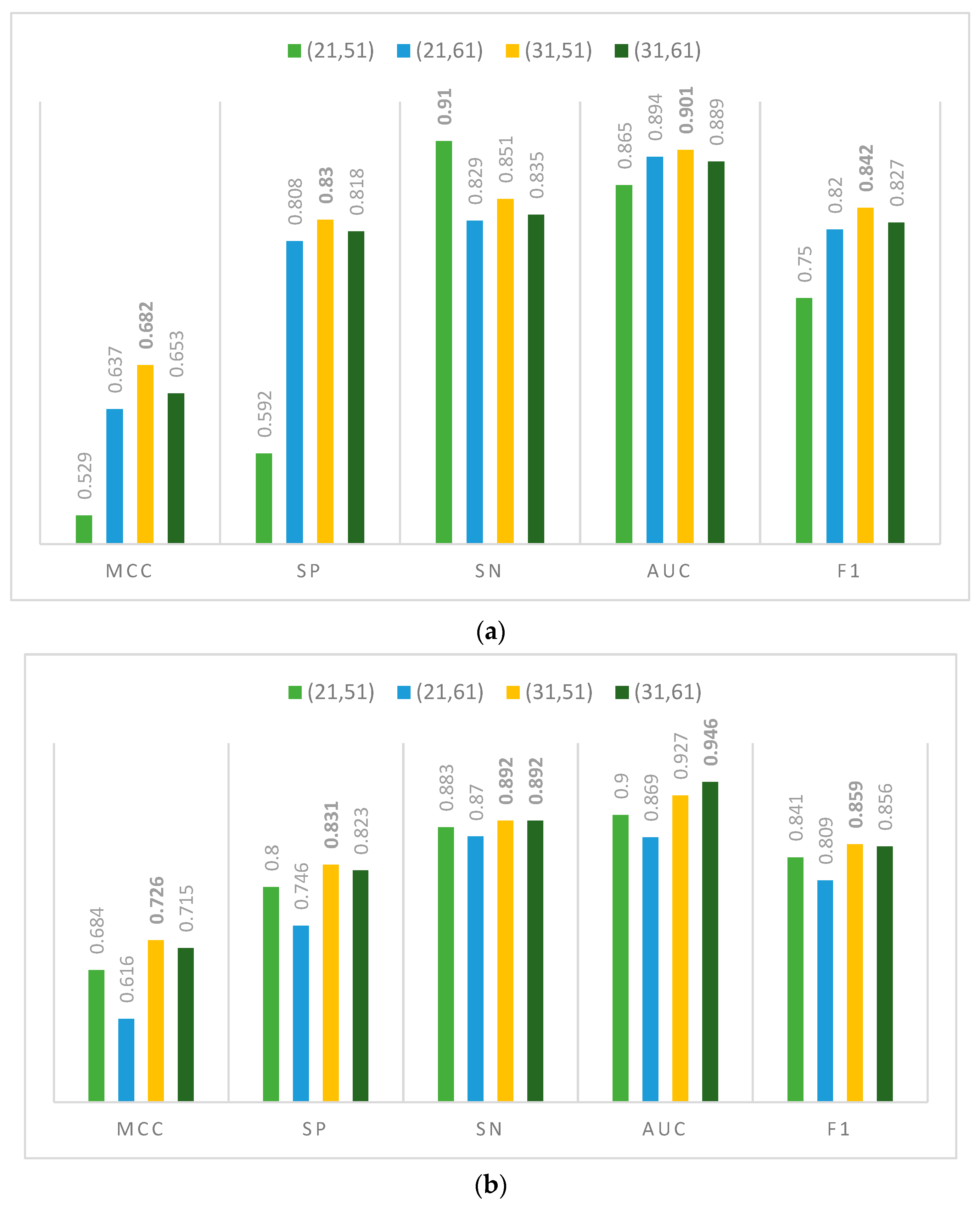
3.2. Ablation Experiment
3.3. Comparison with Other State-of-the-Art Models Based on Transfer Learning
3.4. Comparison with Other State-of-the-Art Models on Phosphorylation Site Datasets
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chaudhari, M. Machine Learning Based Strategies to Predict Sites of Arginine Methylation, Dephosphorylation and Redox-Sensitive ERK2 Substrates. Ph.D. Thesis, North Carolina Agricultural and Technical State University, Greensboro, NC, USA, 2021. [Google Scholar]
- Cohen, P. The origins of protein phosphorylation. Nat. Cell Biol. 2002, 4, E127–E130. [Google Scholar] [CrossRef] [PubMed]
- Ubersax, J.A.; Ferrell, J.E., Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Krebs, E.G.; Beavo, J.A. Phosphorylation-Dephosphorylation of Enzymes. Annu. Rev. Biochem. 1979, 48, 923–959. [Google Scholar] [CrossRef]
- Tomar, V.S.; Baral, T.K.; Nagavelu, K.; Somasundaram, K. Serine/threonine/tyrosine-interacting-like protein 1 (STYXL1), a pseudo phosphatase, promotes oncogenesis in glioma. Biochem. Biophys. Res. Commun. 2019, 515, 241–247. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro-Oncology 2014, 16, iv1–iv63. [Google Scholar] [CrossRef]
- Ranaldi, L.; Fallucchi, F.; Zanzotto, F.M. Dis-cover ai minds to preserve human knowledge. Future Internet 2021, 14, 10. [Google Scholar] [CrossRef]
- Tyanova, S.; Cox, J. Perseus: A bioinformatics platform for integrative analysis of proteomics data in cancer research. Cancer Syst. Biol. Methods Protoc. 2018, 1711, 133–148. [Google Scholar]
- Blueggel, M.; Chamrad, D.; Meyer, H.E. Bioinformatics in proteomics. Curr. Pharm. Biotechnol. 2004, 5, 79–88. [Google Scholar] [CrossRef]
- Kimmel, J.C.; Kelley, D.R. Semisupervised adversarial neural networks for single-cell classification. Genome Res. 2021, 31, 1781–1793. [Google Scholar] [CrossRef]
- Alquicira-Hernandez, J.; Sathe, A.; Ji, H.P.; Nguyen, Q.; Powell, J.E. scPred: Accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome Biol. 2019, 20, 264. [Google Scholar] [CrossRef] [PubMed]
- Ciecholewski, M.; Kassjański, M. Computational methods for liver vessel segmentation in medical imaging: A review. Sensors 2021, 21, 2027. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, X.; Li, Y.; Song, R.; Wang, X. Crescent: A GPU-Based Targeted Nanopore Sequence Selector. In Proceedings of the 2022 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Las Vegas, NV, USA, 6–8 December 2022; pp. 2357–2365. [Google Scholar]
- Wang, X.; Song, R.; Xiao, J.; Li, T.; Li, X. Accelerating k-Shape Time Series Clustering Algorithm Using GPU. IEEE Trans. Parallel Distrib. Syst. 2023, 34, 2718–2734. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Kornhauser, J.M.; Latham, V.; Murray, B.; Nandhikonda, V.; Nord, A.; Skrzypek, E.; Wheeler, T.; Zhang, B.; Gnad, F. 15 years of PhosphoSitePlus®: Integrating post-translationally modified sites, disease variants and isoforms. Nucleic Acids Res. 2018, 47, D433–D441. [Google Scholar] [CrossRef] [PubMed]
- Dinkel, H.; Chica, C.; Via, A.; Gould, C.M.; Jensen, L.J.; Gibson, T.J.; Diella, F. Phospho.ELM: A database of phosphorylation sites—Update 2011. Nucleic Acids Res. 2010, 39, D261–D267. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Z.; Zhang, C.; Meng, X.; Shi, X.; Qu, P. TransPhos: A Deep-Learning Model for General Phosphorylation Site Prediction Based on Transformer-Encoder Architecture. Int. J. Mol. Sci. 2022, 23, 4263. [Google Scholar] [CrossRef]
- Luo, F.; Wang, M.; Liu, Y.; Zhao, X.-M.; Li, A. DeepPhos: Prediction of protein phosphorylation sites with deep learning. Bioinformatics 2019, 35, 2766–2773. [Google Scholar] [CrossRef]
- Yang, H.; Wang, M.; Liu, X.; Zhao, X.-M.; Li, A. PhosIDN: An integrated deep neural network for improving protein phosphorylation site prediction by combining sequence and protein–protein interaction information. Bioinformatics 2021, 37, 4668–4676. [Google Scholar] [CrossRef]
- Gao, J.; Thelen, J.J.; Dunker, A.K.; Xu, D. Musite, a tool for global prediction of general and kinase-specific phosphorylation sites. Mol. Cell. Proteom. 2010, 9, 2586–2600. [Google Scholar] [CrossRef]
- Chaudhari, M.; Thapa, N.; Ismail, H.; Chopade, S.; Caragea, D.; Köhn, M.; Newman, R.H.; Kc, D.B. DTL-DephosSite: Deep Transfer Learning Based Approach to Predict Dephosphorylation Sites. Front. Cell Dev. Biol. 2021, 9, 662983. [Google Scholar] [CrossRef]
- Duan, G.; Li, X.; Köhn, M. The human DEPhOsphorylation database DEPOD: A 2015 update. Nucleic Acids Res. 2014, 43, D531–D535. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zeng, S.; Xu, C.; Qiu, W.; Liang, Y.; Joshi, T.; Xu, D. MusiteDeep: A deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics 2017, 33, 3909–3916. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Huang, R.; Li, J.; Liao, Y.; Chen, Z.; He, G.; Yan, R.; Gryllias, K. A perspective survey on deep transfer learning for fault diagnosis in industrial scenarios: Theories, applications and challenges. Mech. Syst. Signal Process. 2022, 167, 108487. [Google Scholar] [CrossRef]
- Aslan, M.F.; Unlersen, M.F.; Sabanci, K.; Durdu, A. CNN-based transfer learning–BiLSTM network: A novel approach for COVID-19 infection detection. Appl. Soft Comput. 2021, 98, 106912. [Google Scholar] [CrossRef]
- Wang, X.; Gao, C.; Han, P.; Li, X.; Chen, W.; Rodríguez Patón, A.; Wang, S.; Zheng, P. PETrans: De Novo Drug Design with Protein-Specific Encoding Based on Transfer Learning. Int. J. Mol. Sci. 2023, 24, 1146. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Site | Train | Test |
|---|---|---|
| S/T | 1806 | 446 |
| Y | 201 | 50 |
| Site | Train | Test | ||
|---|---|---|---|---|
| Positive | Negative | Positive | Negative | |
| S/T | 34,401 | 64,126 | 2074 | 3912 |
| Y | 1883 | 4011 | 47 | 109 |
| PARAMETER SETTINGS | PARAMETER SETTINGS |
|---|---|
| EPOCHS_PRETRAINED EPOCHS | 100 200 |
| LEARNING_RATE_PRETRAINED LEARNING_RATE | 0.00001 0.000001 |
| DROPOUT | 0.2 |
| EMBEDDING DIM | 16 |
| DENSE | 256, 64, 2 |
| BATCH SIZE NUM_HEADS(MULTIHEADATTENTION) KEY_DIM(MULTIHEADATTENTION) VALIDATION_SPLIT | 16 8 64 0.2 |
| Site | Methods | MCC | SP | SN | ROC_AUC |
|---|---|---|---|---|---|
| S/T | RF | 0.48 | 0.72 | 0.76 | 0.8 |
| DTL-DephosSite | 0.46 | 0.71 | 0.76 | 0.81 | |
| DephosNet | 0.49 | 0.67 | 0.81 | 0.82 | |
| Y | RF | 0.16 | 0.65 | 0.5 | 0.68 |
| DTL-DephosSite | 0.42 | 0.81 | 0.62 | 0.8 | |
| DephosNet | 0.52 | 0.73 | 0.79 | 0.83 |
| S/T | MCC | SP | SN | AUC | F1 |
| DephosNet | 0.713 | 0.853 | 0.86 | 0.906 | 0.857 |
| DTL-DephosSite | 0.639 | 0.777 | 0.86 | 0.896 | 0.825 |
| Transphos | 0.634 | 0.786 | 0.847 | 0.893 | 0.821 |
| Deepphos | 0.633 | 0.835 | 0.797 | 0.876 | 0.812 |
| Y | MCC | SP | SN | AUC | F1 |
| DephosNet | 0.726 | 0.808 | 0.917 | 0.952 | 0.863 |
| DTL-DephosSite | 0.599 | 0.808 | 0.792 | 0.852 | 0.792 |
| Deepphos | 0.655 | 0.731 | 0.917 | 0.883 | 0.83 |
| Transphos | 0.53 | 0.692 | 0.833 | 0.8 | 0.769 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Wang, X.; Zheng, P. DephosNet: A Novel Transfer Learning Approach for Dephosphorylation Site Prediction. Computers 2023, 12, 229. https://doi.org/10.3390/computers12110229
Yang Q, Wang X, Zheng P. DephosNet: A Novel Transfer Learning Approach for Dephosphorylation Site Prediction. Computers. 2023; 12(11):229. https://doi.org/10.3390/computers12110229
Chicago/Turabian StyleYang, Qing, Xun Wang, and Pan Zheng. 2023. "DephosNet: A Novel Transfer Learning Approach for Dephosphorylation Site Prediction" Computers 12, no. 11: 229. https://doi.org/10.3390/computers12110229
APA StyleYang, Q., Wang, X., & Zheng, P. (2023). DephosNet: A Novel Transfer Learning Approach for Dephosphorylation Site Prediction. Computers, 12(11), 229. https://doi.org/10.3390/computers12110229