
Chimeric Antigen Receptor (CAR) T Cell Therapy for Malignant Pleural Mesothelioma (MPM)

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Adoptive Cell Therapy (ACT)
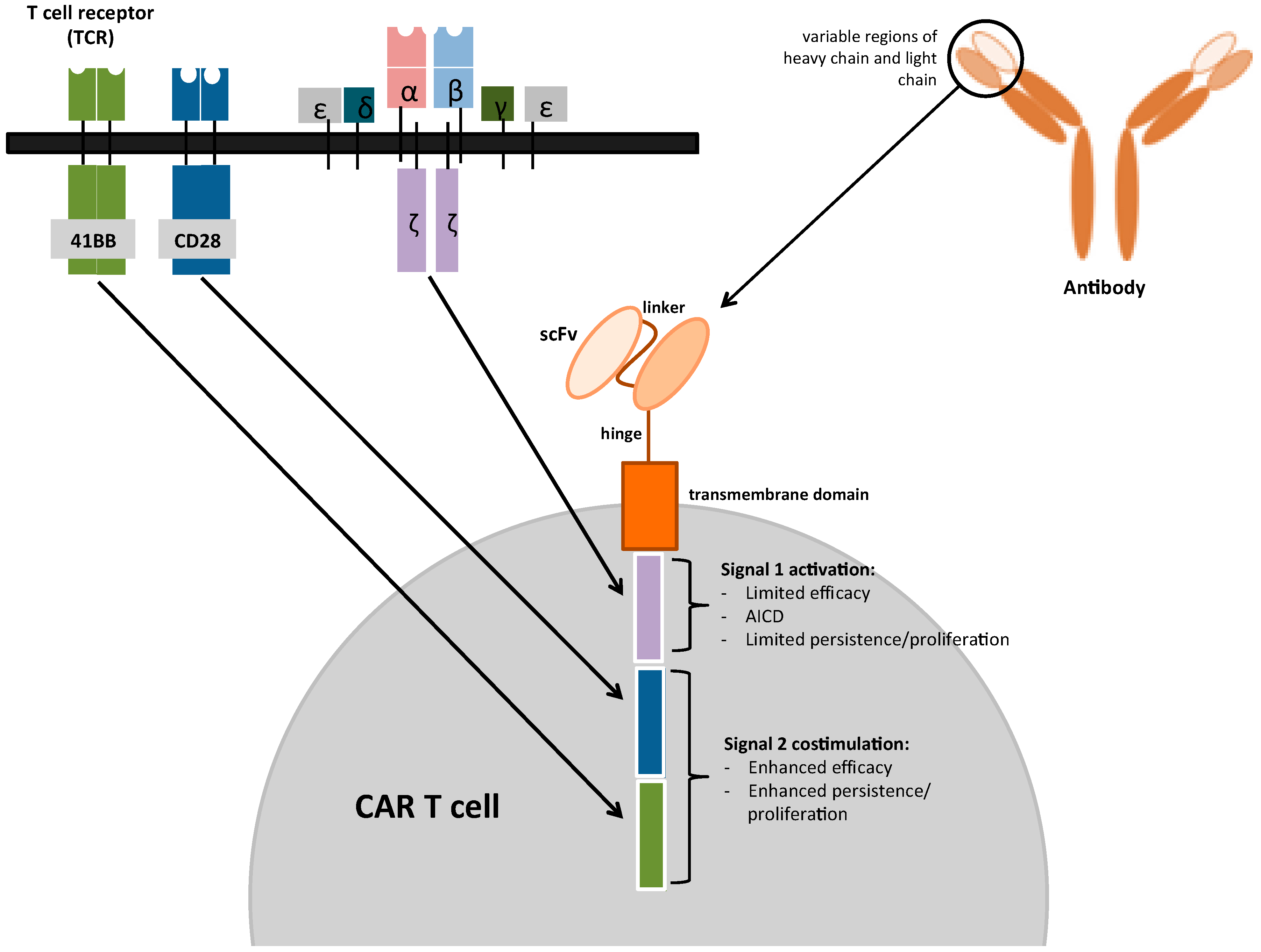
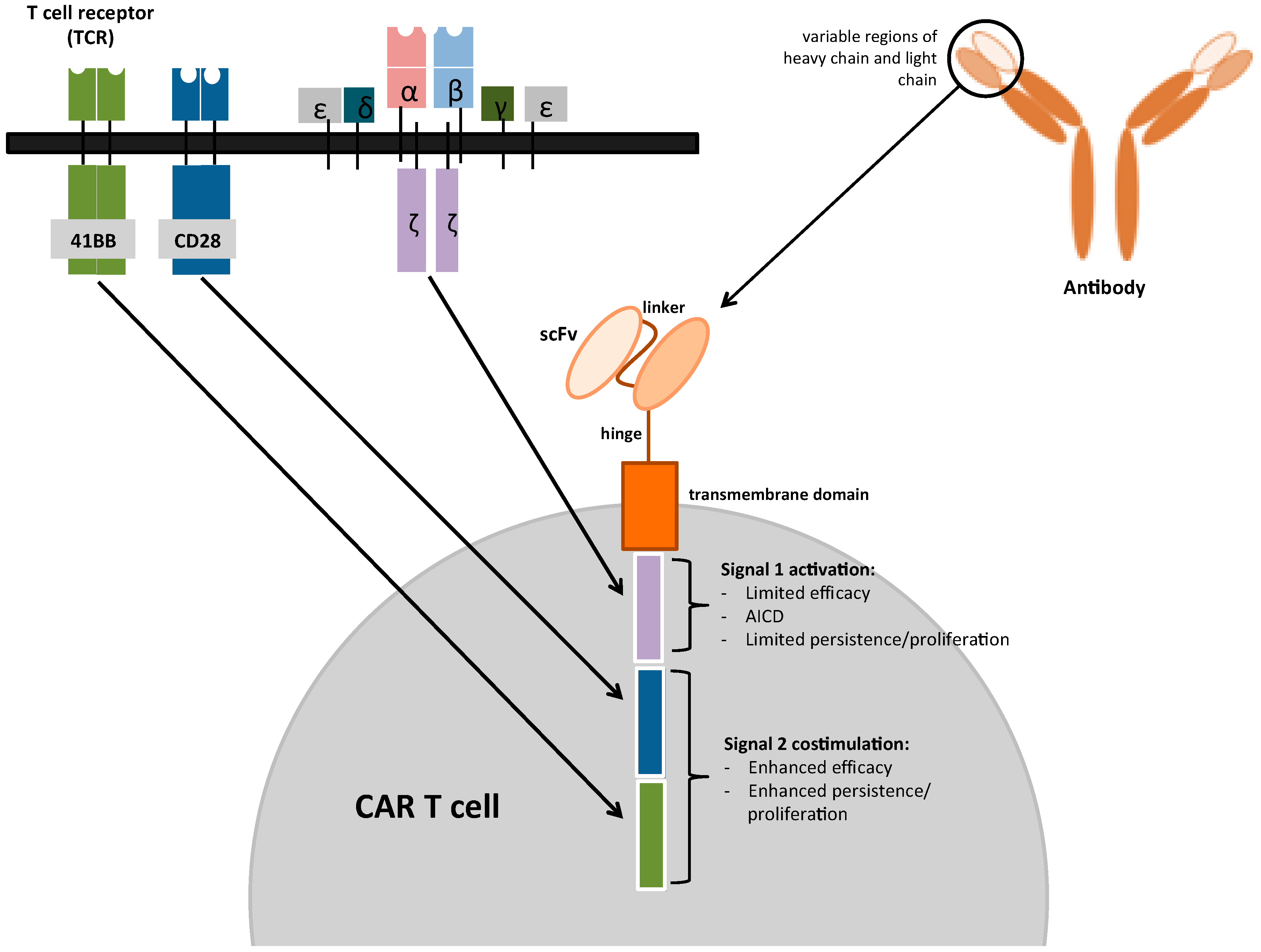
3. Chimeric Antigen Receptors (CARs)
4. Mesothelin CARs
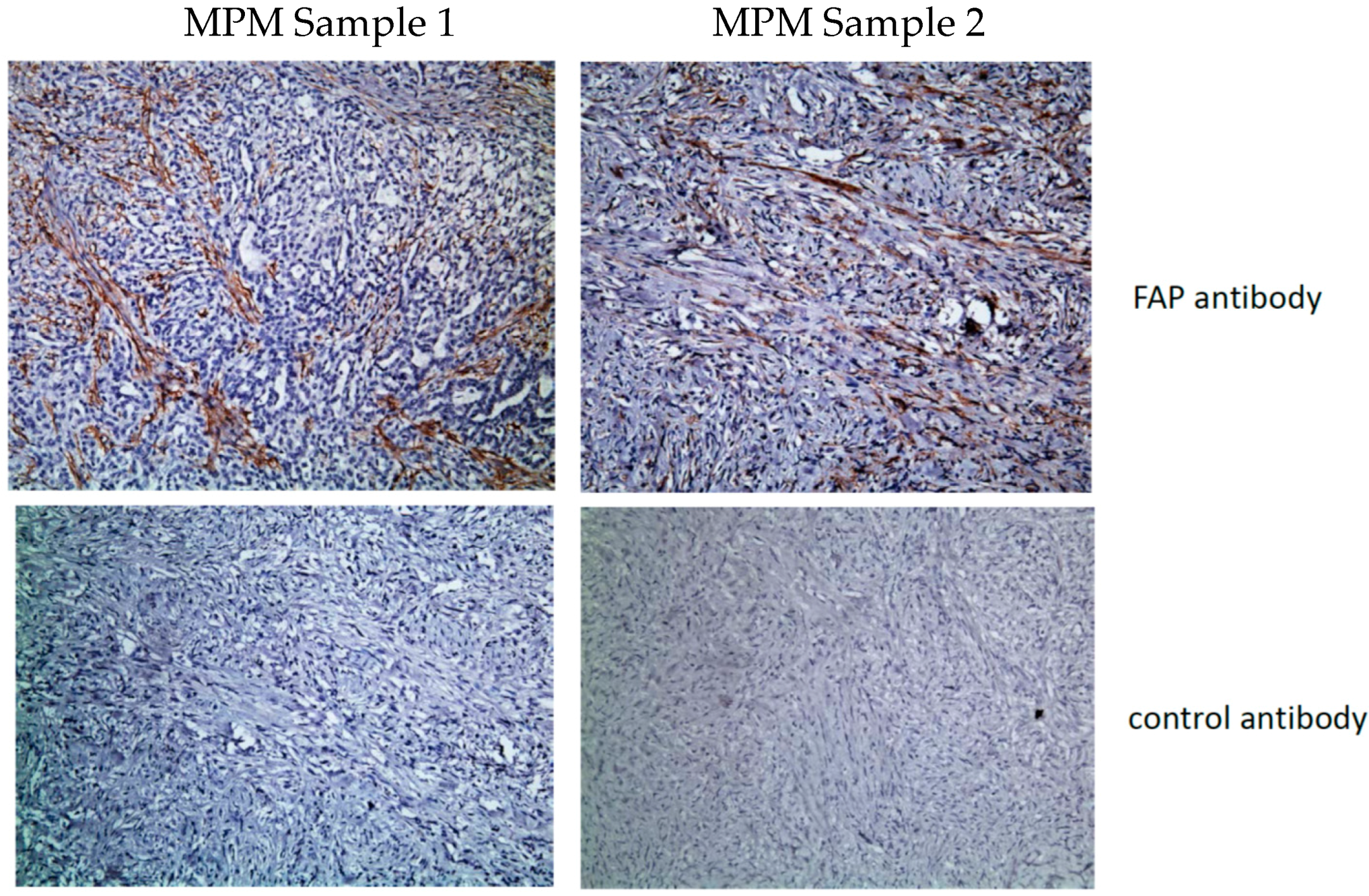
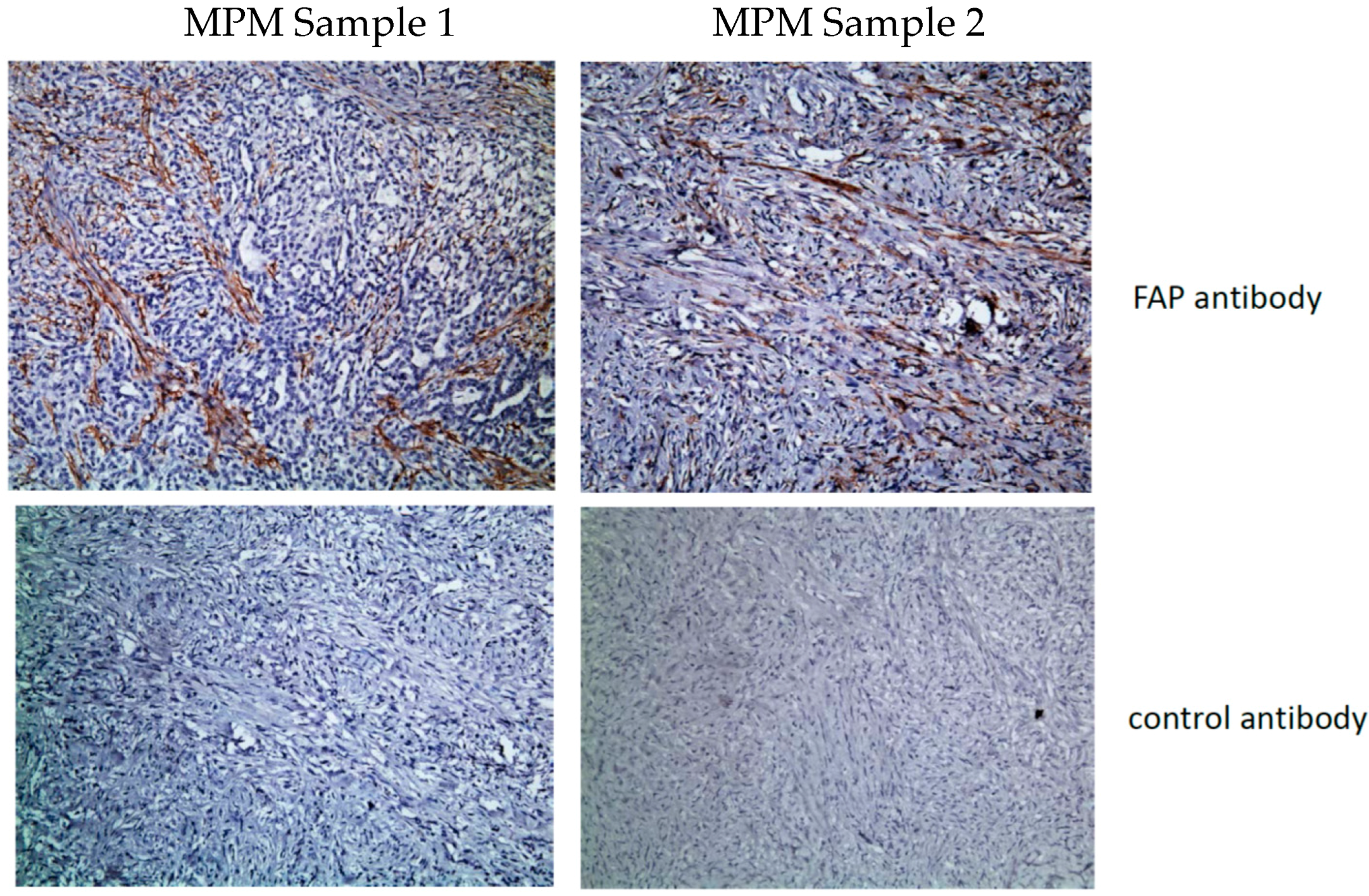
5. FAP CARs
6. Other CARs Targeting Potential Targets for MPM
6.1. Pan-ErbB “T4” CAR
6.2. 5T4 CAR
6.3. Chondroitin Sulfate Proteoglycan CARs
6.4. The Future: CAR Augmentation Strategies
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Stumphius, J.; Meyer, P.B. Asbestos bodies and mesothelioma. Ann. Occup. Hyg. 1968, 11, 283–293. [Google Scholar] [PubMed]
- Wagner, J.C.; Sleggs, C.A.; Marchand, P. Diffuse pleural mesothelioma and asbestos exposure in the North Western Cape Province. Br. J. Ind. Med. 1960, 17, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, C.; Bianchi, T. Malignant mesothelioma: Global incidence and relationship with asbestos. Ind. Health 2007, 45, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, J.T.; McElvenny, D.M.; Darnton, A.J.; Price, M.J.; Peto, J. The expected burden of mesothelioma mortality in Great Britain from 2002 to 2050. Br. J. Cancer 2005, 92, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.M. Malignant pleural mesothelioma: An epidemiological perspective. Ann. Cardiothorac. Surg. 2012, 1, 491–496. [Google Scholar] [PubMed]
- Mazurek, J.M.; Syamlal, G.; Wood, J.M.; Hendricks, S.A.; Weston, A. Malignant mesothelioma mortality—United States, 1999–2015. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Tsao, A.S. Local and systemic therapies for malignant pleural mesothelioma. Curr. Treat. Options Oncol. 2014, 15, 683–699. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.P.; Klabatsa, A. Chemotherapy options and new advances in malignant pleural mesothelioma. Ann. Oncol. 2005, 16, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.; Emi, M.; Tanabe, K. Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Oizumi, S.; Kikuchi, E.; Shinagawa, N.; Konishi-Sakakibara, J.; Ishimine, A.; Aoe, K.; Gemba, K.; Kishimoto, T.; Torigoe, T.; et al. CD8+ tumor-infiltrating lymphocytes predict favorable prognosis in malignant pleural mesothelioma after resection. Cancer Immunol. Immunother. 2010, 59, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Forde, P.M.; Hammers, H.J.; Emens, L.A.; Taube, J.M.; Topalian, S.L. Antagonists of PD-1 and PD-L1 in Cancer Treatment. Semin. Oncol. 2015, 42, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Hegmans, J.P.; Hemmes, A.; Hammad, H.; Boon, L.; Hoogsteden, H.C.; Lambrecht, B.N. Mesothelioma environment comprises cytokines and T-regulatory cells that suppress immune responses. Eur. Respir. J. 2006, 27, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; O’Brien, S.; Sun, J.; Kapoor, V.; Maceyko, S.; Lo, A.; Pure, E.; Moon, E.; Albelda, S.M. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer Immunol. Res. 2016, 4, 541–551. [Google Scholar] [CrossRef] [PubMed]
- DeLong, P.; Tanaka, T.; Kruklitis, R.; Henry, A.C.; Kapoor, V.; Kaiser, L.R.; Sterman, D.H.; Albelda, S.M. Use of cyclooxygenase-2 inhibition to enhance the efficacy of immunotherapy. Cancer Res. 2003, 63, 7845–7852. [Google Scholar] [PubMed]
- Sterman, D.H.; Alley, E.; Stevenson, J.P.; Friedberg, J.; Metzger, S.; Recio, A.; Moon, E.K.; Haas, A.R.; Vachani, A.; Katz, S.I.; et al. Pilot and feasibility trial evaluating immuno-gene therapy of malignant mesothelioma using intrapleural delivery of adenovirus-IFNalpha combined with chemotherapy. Clin. Cancer Res. 2016, 22, 3791–3800. [Google Scholar] [CrossRef] [PubMed]
- Krug, L.M.; Dao, T.; Brown, A.B.; Maslak, P.; Travis, W.; Bekele, S.; Korontsvit, T.; Zakhaleva, V.; Wolchok, J.; Yuan, J.; et al. WT1 peptide vaccinations induce CD4 and CD8 T cell immune responses in patients with mesothelioma and non-small cell lung cancer. Cancer Immunol. Immunother. 2010, 59, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, R.; Hegmans, J.P.; Maat, A.P.; Kaijen-Lambers, M.E.; Bezemer, K.; Hendriks, R.W.; Hoogsteden, H.C.; Aerts, J.G. Extended tumor control after dendritic cell vaccination with low-dose cyclophosphamide as adjuvant treatment in patients with malignant pleural mesothelioma. Am. J. Respir. Crit. Care Med. 2016, 193, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Alley, E.W.; Lopez, J.; Santoro, A.; Morosky, A.; Saraf, S.; Piperdi, B.; van Brummelen, E. Clinical safety and activity of pembrolizumab in patients with malignant pleural mesothelioma (KEYNOTE-028): Preliminary results from a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2017, 18, 623–630. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Sadelain, M.; Riviere, I.; Brentjens, R. Targeting tumours with genetically enhanced T lymphocytes. Nat. Rev. Cancer 2003, 3, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Morello, A.; Sadelain, M.; Adusumilli, P.S. Mesothelin-targeted CARs: Driving T cells to solid tumors. Cancer Discov. 2016, 6, 133–146. [Google Scholar] [CrossRef] [PubMed]
- van der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Riviere, I. Clinical manufacturing of CAR T cells: Foundation of a promising therapy. Mol. Ther. Oncol. 2016, 3, 16015. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Haas, A.R.; Maus, M.V.; Torigian, D.A.; Soulen, M.C.; Plesa, G.; Chew, A.; Zhao, Y.; Levine, B.L.; Albelda, S.M.; et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2014, 2, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224–225. [Google Scholar] [CrossRef] [PubMed]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Rosenberg, S.A. Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 2013, 10, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.C.; Riddell, S.R. Design and implementation of adoptive therapy with chimeric antigen receptor-modified T cells. Immunol. Rev. 2014, 257, 127–144. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Thomas, A.; Alewine, C.; Le, D.T.; Jaffee, E.M.; Pastan, I. Mesothelin Immunotherapy for Cancer: Ready for Prime Time? J. Clin. Oncol. 2016, 34, 4171–4179. [Google Scholar] [CrossRef] [PubMed]
- Servais, E.L.; Colovos, C.; Rodriguez, L.; Bograd, A.J.; Nitadori, J.; Sima, C.; Rusch, V.W.; Sadelain, M.; Adusumilli, P.S. Mesothelin overexpression promotes mesothelioma cell invasion and MMP-9 secretion in an orthotopic mouse model and in epithelioid pleural mesothelioma patients. Clin. Cancer Res. 2012, 18, 2478–2489. [Google Scholar] [CrossRef] [PubMed]
- Kachala, S.S.; Bograd, A.J.; Villena-Vargas, J.; Suzuki, K.; Servais, E.L.; Kadota, K.; Chou, J.; Sima, C.S.; Vertes, E.; Rusch, V.W.; et al. Mesothelin overexpression is a marker of tumor aggressiveness and is associated with reduced recurrence-free and overall survival in early-stage lung adenocarcinoma. Clin. Cancer Res. 2014, 20, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Moon, E.; Carpenito, C.; Paulos, C.M.; Liu, X.; Brennan, A.L.; Chew, A.; Carroll, R.G.; Scholler, J.; Levine, B.L.; et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010, 70, 9053–9061. [Google Scholar] [CrossRef] [PubMed]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, P.S.; Cherkassky, L.; Villena-Vargas, J.; Colovos, C.; Servais, E.; Plotkin, J.; Jones, D.R.; Sadelain, M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Schuberth, P.C.; Hagedorn, C.; Jensen, S.M.; Gulati, P.; van den Broek, M.; Mischo, A.; Soltermann, A.; Jungel, A.; Marroquin Belaunzaran, O.; Stahel, R.; et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J. Transl. Med. 2013, 11, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrausch, U.; Schuberth, P.C.; Hagedorn, C.; Soltermann, A.; Tomaszek, S.; Stahel, R.; Weder, W.; Renner, C. Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive malignant pleural mesothelioma (FAPME-1). BMC Cancer 2012, 12, 615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klampatsa, A.; Achkova, D.Y.; Davies, D.M.; Parente-Pereira, A.C.; Woodman, N.; Rosekilly, J.; Osborne, G.; Thayaparan, T.; Bille, A.; Sheaf, M.; et al. Intracavitary T4 immunotherapy of malignant mesothelioma using pan-ErbB re-targeted CAR T-cells. Cancer Lett. 2017, 393, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Southall, P.J.; Boxer, G.M.; Bagshawe, K.D.; Hole, N.; Bromley, M.; Stern, P.L. Immunohistological distribution of 5T4 antigen in normal and malignant tissues. Br. J. Cancer 1990, 61, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Al-Taei, S.; Salimu, J.; Lester, J.F.; Linnane, S.; Goonewardena, M.; Harrop, R.; Mason, M.D.; Tabi, Z. Overexpression and potential targeting of the oncofoetal antigen 5T4 in malignant pleural mesothelioma. Lung Cancer 2012, 77, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zheng, H.; Luo, W.; Zhang, Q.; Liu, J.; Yao, K. 5T4-specific chimeric antigen receptor modification promotes the immune efficacy of cytokine-induced killer cells against nasopharyngeal carcinoma stem cell-like cells. Sci. Rep. 2017, 7, 4859. [Google Scholar] [CrossRef] [PubMed]
- Rivera, Z.; Ferrone, S.; Wang, X.; Jube, S.; Yang, H.; Pass, H.I.; Kanodia, S.; Gaudino, G.; Carbone, M. CSPG4 as a target of antibody-based immunotherapy for malignant mesothelioma. Clin. Cancer Res. 2012, 18, 5352–5363. [Google Scholar] [CrossRef] [PubMed]
- Beard, R.E.; Zheng, Z.; Lagisetty, K.H.; Burns, W.R.; Tran, E.; Hewitt, S.M.; Abate-Daga, D.; Rosati, S.F.; Fine, H.A.; Ferrone, S.; et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J. Immunother. Cancer 2014, 2, 25. [Google Scholar] [CrossRef] [PubMed]
- Newick, K.; O’Brien, S.; Moon, E.; Albelda, S.M. CAR T cell therapy for solid tumors. Annu. Rev. Med. 2017, 68, 139–152. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, S.; Narala, N.; Huye, L.; Yagyu, S.; Savoldo, B.; Dotti, G.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Ramos, C.A. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 2015, 125, 3905–3916. [Google Scholar] [CrossRef] [PubMed]
- Bollard, C.M.; Rossig, C.; Calonge, M.J.; Huls, M.H.; Wagner, H.J.; Massague, J.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood 2002, 99, 3179–3187. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.; Kapoor, V.; Predina, J.; Powell, D.J., Jr.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Wang, L.C.; Tsai, C.Y.; Bhoj, V.; Gershenson, Z.; Moon, E.; Newick, K.; Sun, J.; Lo, A.; Baradet, T.; et al. Generation of potent T-cell immunotherapy for cancer using DAP12-based, multichain, chimeric immunoreceptors. Cancer Immunol. Res. 2015, 3, 815–826. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klampatsa, A.; Haas, A.R.; Moon, E.K.; Albelda, S.M. Chimeric Antigen Receptor (CAR) T Cell Therapy for Malignant Pleural Mesothelioma (MPM). Cancers 2017, 9, 115. https://doi.org/10.3390/cancers9090115
Klampatsa A, Haas AR, Moon EK, Albelda SM. Chimeric Antigen Receptor (CAR) T Cell Therapy for Malignant Pleural Mesothelioma (MPM). Cancers. 2017; 9(9):115. https://doi.org/10.3390/cancers9090115
Chicago/Turabian StyleKlampatsa, Astero, Andrew R. Haas, Edmund K. Moon, and Steven M. Albelda. 2017. "Chimeric Antigen Receptor (CAR) T Cell Therapy for Malignant Pleural Mesothelioma (MPM)" Cancers 9, no. 9: 115. https://doi.org/10.3390/cancers9090115