
The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer

Abstract
:1. Introduction
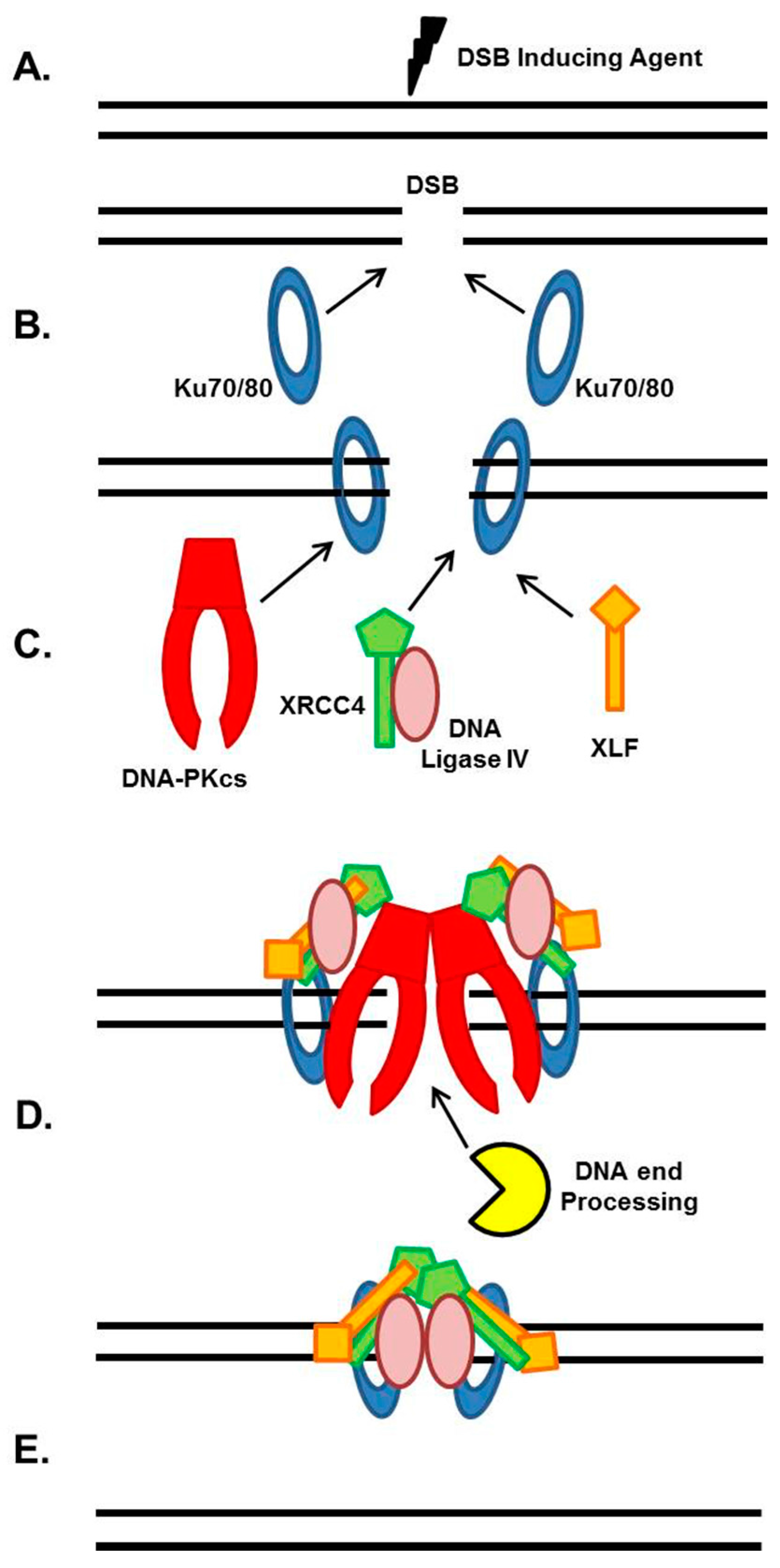
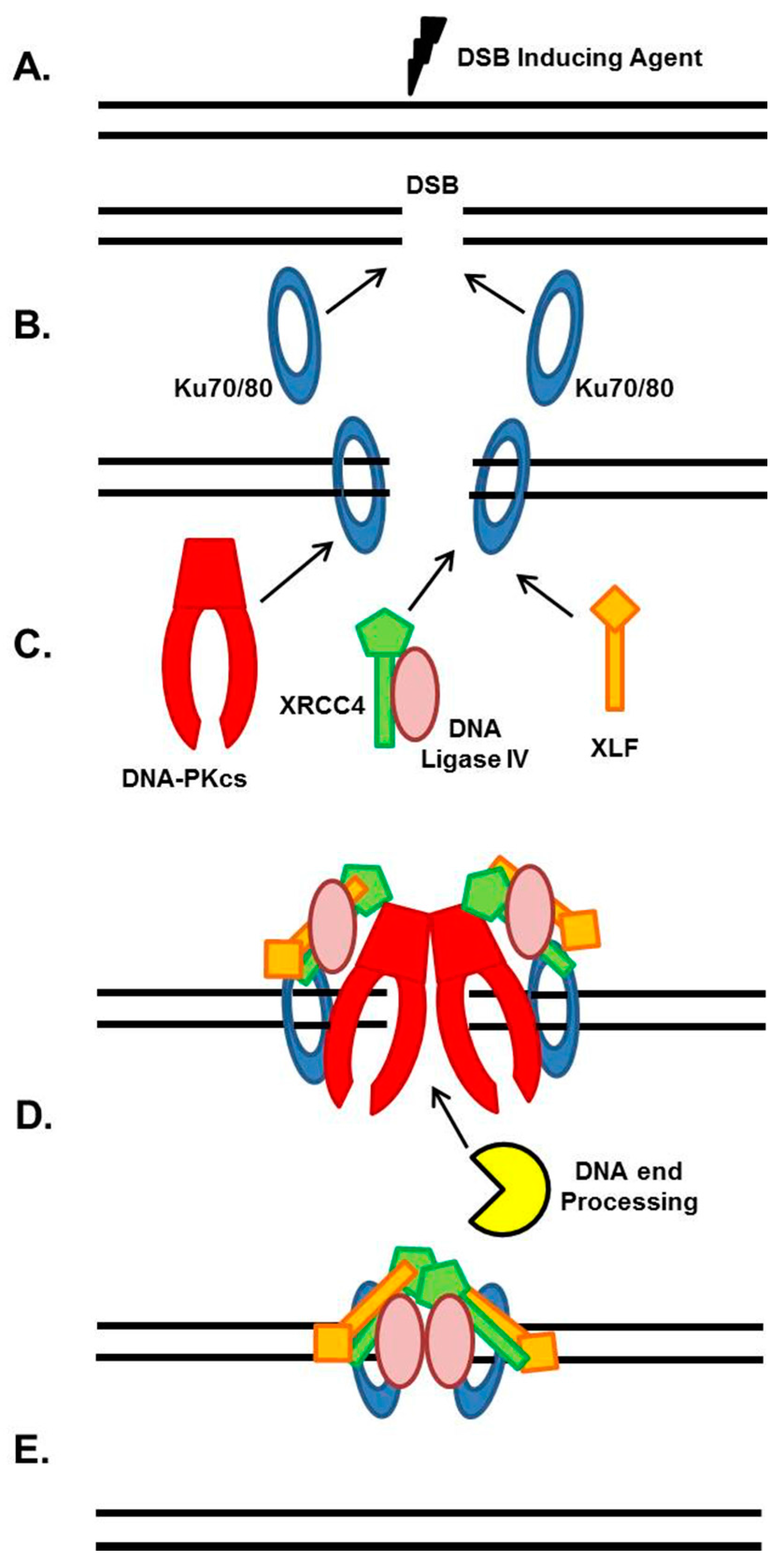
2. NHEJ General Mechanism
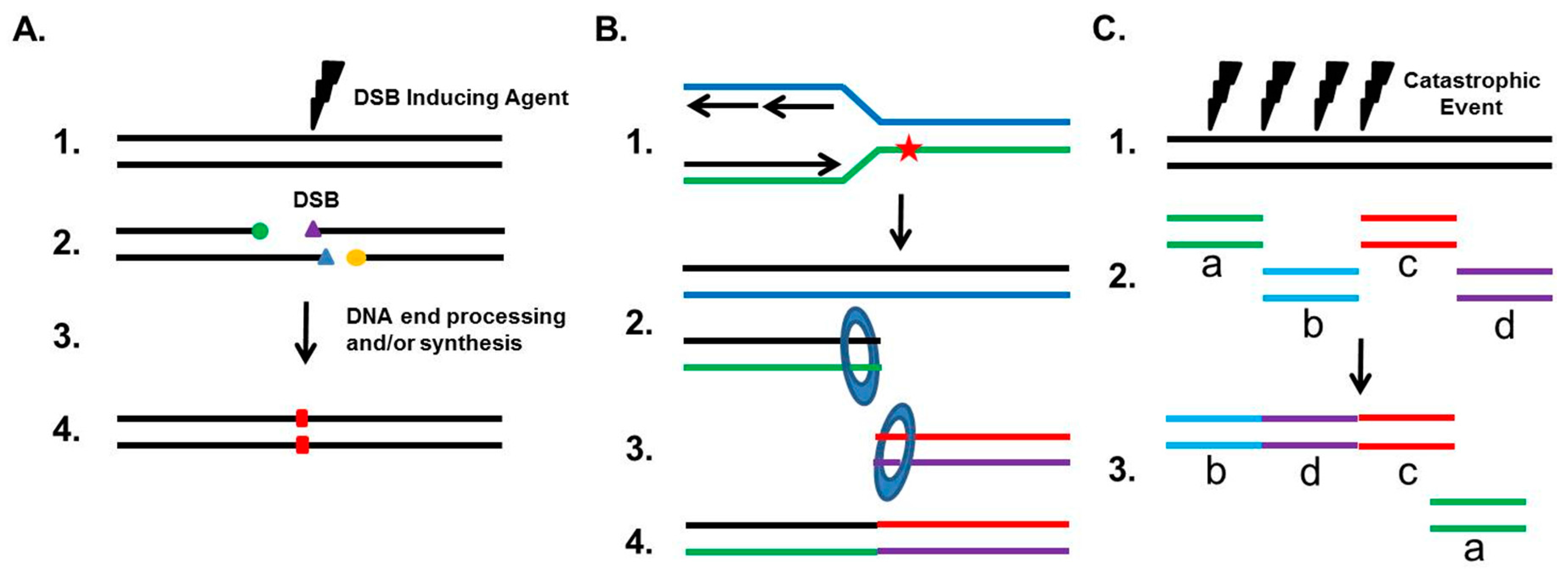
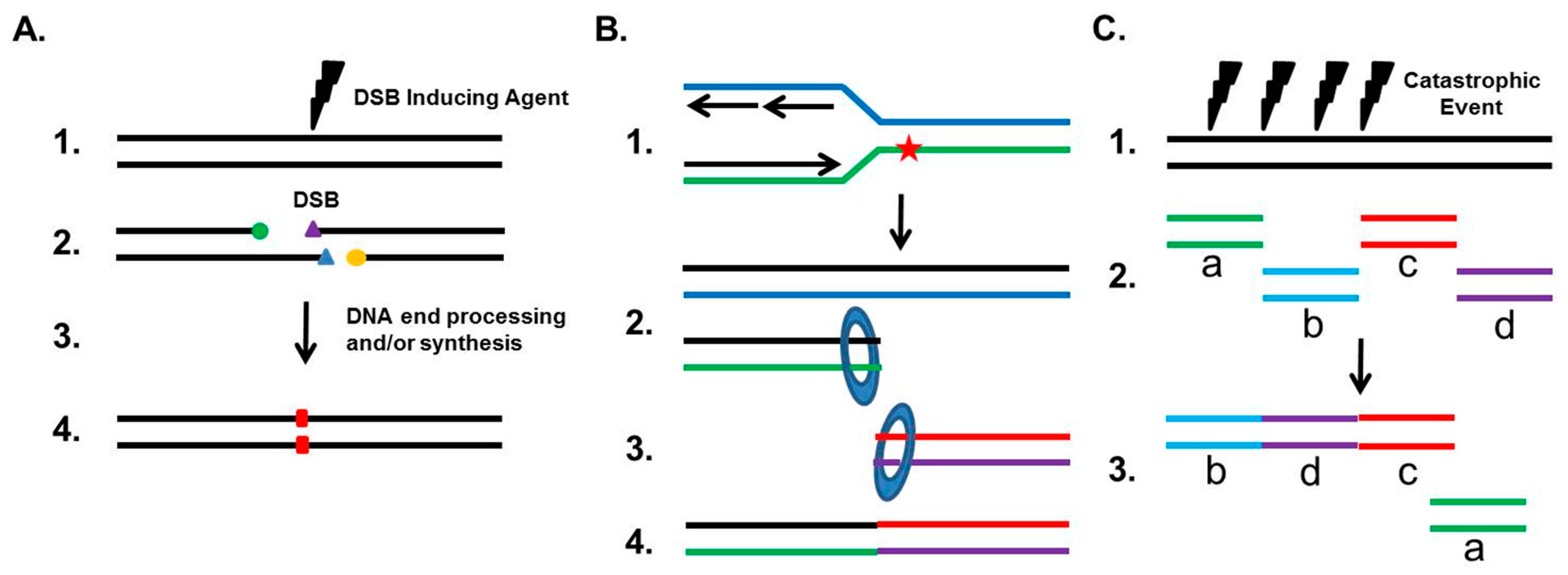
3. NHEJ’s General Role in Genome Maintenance and Instability
4. Core NHEJ Factors in Carcinogenesis and Cancer
4.1. Rodent Models and Human Patients with Defects in the Core NHEJ Factors
4.2. Differential Expression of the Core NHEJ Factors and Carcinogenesis
4.3. Single Nucleotide Polymorphisms in Core NHEJ Factors and Carcinogenesis
4.4. Differential Expression of Core NHEJ Factors in Cancer Progression and Survival
4.5. Differential Expression of the Core NHEJ Factors and Its Influence on Therapy Responsiveness
5. Manipulating NHEJ as an Anti-Cancer Therapy
5.1. DNA-PKcs
5.2. Ku70/80
5.3. LIG4
6. Perspective on the NHEJ Conundrum and Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Hiom, K. Coping with DNA double strand breaks. DNA Repair 2010, 9, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Malu, S.; Malshetty, V.; Francis, D.; Cortes, P. Role of non-homologous end joining in V(D)J recombination. Immunol. Res. 2012, 54, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, B.P.; Chen, D.J. DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 2014, 17, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Jette, N.; Lees-Miller, S.P. Non-homologous end joining: Emerging themes and unanswered questions. DNA Repair 2014, 17, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Ellenberger, T.; Tomkinson, A.E. Eukaryotic DNA ligases: Structural and functional insights. Annu. Rev. Biochem. 2008, 77, 313–338. [Google Scholar] [CrossRef] [PubMed]
- Bryans, M.; Valenzano, M.C.; Stamato, T.D. Absence of DNA ligase IV protein in XR-1 cells: Evidence for stabilization by XRCC4. Mutat. Res. 1999, 433, 53–58. [Google Scholar] [CrossRef]
- Mahaney, B.L.; Hammel, M.; Meek, K.; Tainer, J.A.; Lees-Miller, S.P. XRCC4 and XLF form long helical protein filaments suitable for DNA end protection and alignment to facilitate DNA double strand break repair. Biochem. Cell Biol. 2013, 91, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Riballo, E.; Woodbine, L.; Stiff, T.; Walker, S.A.; Goodarzi, A.A.; Jeggo, P.A. XLF-Cernunnos promotes DNA ligase IV-XRCC4 re-adenylation following ligation. Nucleic Acids Res. 2009, 37, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.M.; Cooper, D.N.; Ferec, C.; Kehrer-Sawatzki, H.; Patrinos, G.P. Genomic rearrangements in inherited disease and cancer. Semin. Cancer Biol. 2010, 20, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Kasparek, T.R.; Humphrey, T.C. DNA double-strand break repair pathways, chromosomal rearrangements and cancer. Semin. Cell Dev. Biol. 2011, 22, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Karanjawala, Z.E.; Grawunder, U.; Hsieh, C.L.; Lieber, M.R. The nonhomologous DNA end joining pathway is important for chromosome stability in primary fibroblasts. Curr. Biol. 1999, 9, 1501–1504. [Google Scholar] [CrossRef]
- Rothkamm, K.; Kuhne, M.; Jeggo, P.A.; Lobrich, M. Radiation-induced genomic rearrangements formed by nonhomologous end-joining of DNA double-strand breaks. Cancer Res. 2001, 61, 3886–3893. [Google Scholar] [PubMed]
- Jeggo, P.A.; Lobrich, M. How cancer cells hijack DNA double-strand break repair pathways to gain genomic instability. Biochem. J. 2015, 471, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Betermier, M.; Bertrand, P.; Lopez, B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [PubMed]
- Waters, C.A.; Strande, N.T.; Pryor, J.M.; Strom, C.N.; Mieczkowski, P.; Burkhalter, M.D.; Oh, S.; Qaqish, B.F.; Moore, D.T.; Hendrickson, E.A.; et al. The fidelity of the ligation step determines how ends are resolved during nonhomologous end joining. Nat. Commun. 2014, 5, 4286. [Google Scholar] [CrossRef] [PubMed]
- Sakata, K.; Someya, M.; Matsumoto, Y.; Hareyama, M. Ability to repair DNA double-strand breaks related to cancer susceptibility and radiosensitivity. Radiat. Med. 2007, 25, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Brady, N.; Gaymes, T.J.; Cheung, M.; Mufti, G.J.; Rassool, F.V. Increased error-prone NHEJ activity in myeloid leukemias is associated with DNA damage at sites that recruit key nonhomologous end-joining proteins. Cancer Res. 2003, 63, 1798–1805. [Google Scholar] [PubMed]
- Gaymes, T.J.; Mufti, G.J.; Rassool, F.V. Myeloid leukemias have increased activity of the nonhomologous end-joining pathway and concomitant DNA misrepair that is dependent on the Ku70/86 heterodimer. Cancer Res. 2002, 62, 2791–2797. [Google Scholar] [PubMed]
- Bau, D.T.; Mau, Y.C.; Ding, S.L.; Wu, P.E.; Shen, C.Y. DNA double-strand break repair capacity and risk of breast cancer. Carcinogenesis 2007, 28, 1726–1730. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, A.; Mao, Z.; Tian, X.; Spencer, B.; Seluanov, A.; Gorbunova, V. Knock-in reporter mice demonstrate that DNA repair by non-homologous end joining declines with age. PLoS Genet. 2014, 10, e1004511. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, D.M.; Richardson, C.A.; Elliott, B.; Jasin, M. Modeling oncogenic translocations: Distinct roles for double-strand break repair pathways in translocation formation in mammalian cells. DNA Repair 2006, 5, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R.; Gu, J.; Lu, H.; Shimazaki, N.; Tsai, A.G. Nonhomologous DNA end joining (NHEJ) and chromosomal translocations in humans. Subcell. Biochem. 2010, 50, 279–296. [Google Scholar] [CrossRef] [PubMed]
- Ghezraoui, H.; Piganeau, M.; Renouf, B.; Renaud, J.B.; Sallmyr, A.; Ruis, B.; Oh, S.; Tomkinson, A.E.; Hendrickson, E.A.; Giovannangeli, C.; et al. Chromosomal translocations in human cells are generated by canonical nonhomologous end-joining. Mol. Cell 2014, 55, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Simsek, D.; Jasin, M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat. Struct. Mol. Biol. 2010, 17, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yang, L.; Tanasa, B.; Hutt, K.; Ju, B.G.; Ohgi, K.; Zhang, J.; Rose, D.W.; Fu, X.D.; Glass, C.K.; et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009, 139, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. Mechanisms of human lymphoid chromosomal translocations. Nat. Rev. Cancer 2016, 16, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Daser, A.; Dear, P.; Wood, H.; Rabbitts, P.; Rabbitts, T. Nonreciprocal chromosomal translocations in renal cancer involve multiple DSBs and NHEJ associated with breakpoint inversion but not necessarily with transcription. Genes Chromosom. Cancer 2013, 52, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Collis, S.J.; Adelman, C.A.; Silva, N.; Horejsi, Z.; Ward, J.D.; Martinez-Perez, E.; Boulton, S.J.; La Volpe, A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol. Cell 2010, 39, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Pace, P.; Mosedale, G.; Hodskinson, M.R.; Rosado, I.V.; Sivasubramaniam, M.; Patel, K.J. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010, 329, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Romick-Rosendale, L.E.; Hoskins, E.E.; Privette Vinnedge, L.M.; Foglesong, G.D.; Brusadelli, M.G.; Potter, S.S.; Komurov, K.; Brugmann, S.A.; Lambert, P.F.; Kimple, R.J.; et al. Defects in the Fanconi Anemia Pathway in Head and Neck Cancer Cells Stimulate Tumor Cell Invasion through DNA-PK and Rac1 Signaling. Clin. Cancer Res. 2016, 22, 2062–2073. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, M.L.; Zhang, C.Z.; Pellman, D. Chromothripsis: A New Mechanism for Rapid Karyotype Evolution. Annu. Rev. Genet. 2015, 49, 183–211. [Google Scholar] [CrossRef] [PubMed]
- Rode, A.; Maass, K.K.; Willmund, K.V.; Lichter, P.; Ernst, A. Chromothripsis in cancer cells: An update. Int. J. Cancer 2016, 138, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Qi, X.; Guo, H.; Cai, M.; Li, C.; Zhu, J.; Chen, F.; Guo, H.; Li, J.; Zhao, Y.; et al. Novel role for non-homologous end joining in the formation of double minutes in methotrexate-resistant colon cancer cells. J. Med. Genet. 2015, 52, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Jin, S.; Gao, Y.; Weaver, D.T.; Alt, F.W. Ku70-deficient embryonic stem cells have increased ionizing radiosensitivity, defective DNA end-binding activity, and inability to support V(D)J recombination. Proc. Natl. Acad. Sci. USA 1997, 94, 8076–8081. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Inamdar, K.V.; Pfeiffer, P.; Feldmann, E.; Hannah, M.F.; Yu, Y.; Lee, J.W.; Zhou, T.; Lees-Miller, S.P.; Povirk, L.F. Accurate in vitro end joining of a DNA double strand break with partially cohesive 3′-overhangs and 3′-phosphoglycolate termini: Effect of Ku on repair fidelity. J. Biol. Chem. 2001, 276, 24323–24330. [Google Scholar] [CrossRef] [PubMed]
- Nussenzweig, A.; Sokol, K.; Burgman, P.; Li, L.; Li, G.C. Hypersensitivity of Ku80-deficient cell lines and mice to DNA damage: The effects of ionizing radiation on growth, survival, and development. Proc. Natl. Acad. Sci. USA 1997, 94, 13588–13593. [Google Scholar] [CrossRef] [PubMed]
- Difilippantonio, M.J.; Zhu, J.; Chen, H.T.; Meffre, E.; Nussenzweig, M.C.; Max, E.E.; Ried, T.; Nussenzweig, A. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 2000, 404, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, D.O.; Sekiguchi, J.M.; Chang, S.; Frank, K.M.; Gao, Y.; DePinho, R.A.; Alt, F.W. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc. Natl. Acad. Sci. USA 2000, 97, 6630–6633. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Saha, J.; Sun, J.; Fattah, K.R.; Wang, S.C.; Jakob, B.; Chi, L.; Wang, S.Y.; Taucher-Scholz, G.; Davis, A.J.; et al. Phosphorylation of Ku dictates DNA double-strand break (DSB) repair pathway choice in S phase. Nucleic Acids Res. 2016, 44, 1732–1745. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Seidl, K.J.; Rathbun, G.A.; Zhu, C.; Manis, J.P.; van der Stoep, N.; Davidson, L.; Cheng, H.L.; Sekiguchi, J.M.; Frank, K.; et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity 1997, 7, 653–665. [Google Scholar] [CrossRef]
- Li, G.C.; Ouyang, H.; Li, X.; Nagasawa, H.; Little, J.B.; Chen, D.J.; Ling, C.C.; Fuks, Z.; Cordon-Cardo, C. Ku70: A candidate tumor suppressor gene for murine T cell lymphoma. Mol. Cell 1998, 2, 1–8. [Google Scholar] [CrossRef]
- Nussenzweig, A.; Chen, C.; da Costa Soares, V.; Sanchez, M.; Sokol, K.; Nussenzweig, M.C.; Li, G.C. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature 1996, 382, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Bogue, M.A.; Lim, D.S.; Hasty, P.; Roth, D.B. Ku86-deficient mice exhibit severe combined immunodeficiency and defective processing of V(D)J recombination intermediates. Cell 1996, 86, 379–389. [Google Scholar] [CrossRef]
- Tong, W.M.; Cortes, U.; Hande, M.P.; Ohgaki, H.; Cavalli, L.R.; Lansdorp, P.M.; Haddad, B.R.; Wang, Z.Q. Synergistic role of Ku80 and poly(ADP-ribose) polymerase in suppressing chromosomal aberrations and liver cancer formation. Cancer Res. 2002, 62, 6990–6996. [Google Scholar] [PubMed]
- Bogue, M.A.; Jhappan, C.; Roth, D.B. Analysis of variable (diversity) joining recombination in DNAdependent protein kinase (DNA-PK)-deficient mice reveals DNA-PK-independent pathways for both signal and coding joint formation. Proc. Natl. Acad. Sci. USA 1998, 95, 15559–15564. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; Maser, R.S.; Sahin, E.; Bailey, S.T.; Xia, H.; Ji, H.; McNamara, K.; Naylor, M.; Bronson, R.T.; Ghosh, S.; et al. Diminished lifespan and acute stress-induced death in DNA-PKcs-deficient mice with limiting telomeres. Oncogene 2007, 26, 2815–2821. [Google Scholar] [CrossRef] [PubMed]
- Espejel, S.; Martin, M.; Klatt, P.; Martin-Caballero, J.; Flores, J.M.; Blasco, M.A. Shorter telomeres, accelerated ageing and increased lymphoma in DNA-PKcs-deficient mice. EMBO Rep. 2004, 5, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Blunt, T.; Gell, D.; Fox, M.; Taccioli, G.E.; Lehmann, A.R.; Jackson, S.P.; Jeggo, P.A. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc. Natl. Acad. Sci. USA 1996, 93, 10285–10290. [Google Scholar] [CrossRef] [PubMed]
- Danska, J.S.; Holland, D.P.; Mariathasan, S.; Williams, K.M.; Guidos, C.J. Biochemical and genetic defects in the DNA-dependent protein kinase in murine scid lymphocytes. Mol. Cell Biol. 1996, 16, 5507–5517. [Google Scholar] [CrossRef] [PubMed]
- Kurimasa, A.; Ouyang, H.; Dong, L.J.; Wang, S.; Li, X.; Cordon-Cardo, C.; Chen, D.J.; Li, G.C. Catalytic subunit of DNA-dependent protein kinase: Impact on lymphocyte development and tumorigenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 1403–1408. [Google Scholar] [CrossRef] [PubMed]
- Guidos, C.J.; Williams, C.J.; Grandal, I.; Knowles, G.; Huang, M.T.; Danska, J.S. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes Dev. 1996, 10, 2038–2054. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, R.; Suetomi, K.; Yu, Y.; Silver, A.; Bedford, J.S.; Cox, R.; Ullrich, R.L. A deficiency in DNA repair and DNA-PKcs expression in the radiosensitive BALB/c mouse. Cancer Res. 2000, 60, 4342–4345. [Google Scholar] [PubMed]
- Yu, Y.; Okayasu, R.; Weil, M.M.; Silver, A.; McCarthy, M.; Zabriskie, R.; Long, S.; Cox, R.; Ullrich, R.L. Elevated breast cancer risk in irradiated BALB/c mice associates with unique functional polymorphism of the Prkdc (DNA-dependent protein kinase catalytic subunit) gene. Cancer Res. 2001, 61, 1820–1824. [Google Scholar] [PubMed]
- Zhang, S.; Matsunaga, S.; Lin, Y.F.; Sishc, B.; Shang, Z.; Sui, J.; Shih, H.Y.; Zhao, Y.; Foreman, O.; Story, M.D.; et al. Spontaneous tumor development in bone marrow-rescued DNA-PKcs(3A/3A) mice due to dysfunction of telomere leading strand deprotection. Oncogene 2016, 35, 3909–3918. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yajima, H.; Huynh, H.; Zheng, J.; Callen, E.; Chen, H.T.; Wong, N.; Bunting, S.; Lin, Y.F.; Li, M.; et al. Congenital bone marrow failure in DNA-PKcs mutant mice associated with deficiencies in DNA repair. J. Cell Biol. 2011, 193, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Barnes, D.E.; Stamp, G.; Rosewell, I.; Denzel, A.; Lindahl, T. Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr. Biol. 1998, 8, 1395–1398. [Google Scholar] [CrossRef]
- Lee, Y.; McKinnon, P.J. DNA ligase IV suppresses medulloblastoma formation. Cancer Res. 2002, 62, 6395–6399. [Google Scholar] [PubMed]
- Frank, K.M.; Sharpless, N.E.; Gao, Y.; Sekiguchi, J.M.; Ferguson, D.O.; Zhu, C.; Manis, J.P.; Horner, J.; DePinho, R.A.; Alt, F.W. DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol. Cell 2000, 5, 993–1002. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Ferguson, D.O.; O’Hagan, R.C.; Castrillon, D.H.; Lee, C.; Farazi, P.A.; Alson, S.; Fleming, J.; Morton, C.C.; Frank, K.; et al. Impaired nonhomologous end-joining provokes soft tissue sarcomas harboring chromosomal translocations, amplifications, and deletions. Mol. Cell 2001, 8, 1187–1196. [Google Scholar] [CrossRef]
- Giaccia, A.J.; Denko, N.; MacLaren, R.; Mirman, D.; Waldren, C.; Hart, I.; Stamato, T.D. Human chromosome 5 complements the DNA double-strand break-repair deficiency and gamma-ray sensitivity of the XR-1 hamster variant. Am. J. Hum. Genet. 1990, 47, 459–469. [Google Scholar] [PubMed]
- Gao, Y.; Ferguson, D.O.; Xie, W.; Manis, J.P.; Sekiguchi, J.; Frank, K.M.; Chaudhuri, J.; Horner, J.; DePinho, R.A.; Alt, F.W. Interplay of p53 and DNA-repair protein XRCC4 in tumorigenesis, genomic stability and development. Nature 2000, 404, 897–900. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.T.; Kaushal, D.; Murphy, M.; Zhang, Y.; Datta, A.; Chen, C.; Monroe, B.; Mostoslavsky, G.; Coakley, K.; Gao, Y.; et al. XRCC4 suppresses medulloblastomas with recurrent translocations in p53-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7378–7383. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Alt, F.W.; Cheng, H.L.; Brush, J.W.; Goff, P.H.; Murphy, M.M.; Franco, S.; Zhang, Y.; Zha, S. Lymphocyte-specific compensation for XLF/cernunnos end-joining functions in V(D)J recombination. Mol. Cell 2008, 31, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Fattah, F.J.; Lichter, N.F.; Fattah, K.R.; Oh, S.; Hendrickson, E.A. Ku70, an essential gene, modulates the frequency of rAAV-mediated gene targeting in human somatic cells. Proc. Natl. Acad. Sci. USA 2008, 105, 8703–8708. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ghosh, G.; Hendrickson, E.A. Ku86 represses lethal telomere deletion events in human somatic cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12430–12435. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, M.; Ijspeert, H.; Verkaik, N.S.; Turul, T.; Wiegant, W.W.; Morotomi-Yano, K.; Mari, P.O.; Tezcan, I.; Chen, D.J.; Zdzienicka, M.Z.; et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J. Clin. Investig. 2009, 119, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Woodbine, L.; Neal, J.A.; Sasi, N.K.; Shimada, M.; Deem, K.; Coleman, H.; Dobyns, W.B.; Ogi, T.; Meek, K.; Davies, E.G.; et al. PRKDC mutations in a SCID patient with profound neurological abnormalities. J. Clin. Investig. 2013, 123, 2969–2980. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, A.L.; Verronese, E.; Rice, G.I.; Fouyssac, F.; Bertrand, Y.; Picard, C.; Chansel, M.; Walter, J.E.; Notarangelo, L.D.; Butte, M.J.; et al. PRKDC mutations associated with immunodeficiency, granuloma, and autoimmune regulator-dependent autoimmunity. J. Allergy Clin. Immunol. 2015, 135, 1578–1588. [Google Scholar] [CrossRef] [PubMed]
- Abbaszadeh, F.; Clingen, P.H.; Arlett, C.F.; Plowman, P.N.; Bourton, E.C.; Themis, M.; Makarov, E.M.; Newbold, R.F.; Green, M.H.; Parris, C.N. A novel splice variant of the DNA-PKcs gene is associated with clinical and cellular radiosensitivity in a patient with xeroderma pigmentosum. J. Med. Genet. 2010, 47, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Virsik-Kopp, P.; Rave-Frank, M.; Hofman-Huther, H.; Schmidberger, H. Role of DNA-PK in the process of aberration formation as studied in irradiated human glioblastoma cell lines M059K and M059J. Int. J. Radiat. Biol. 2003, 79, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Lees-Miller, S.P.; Godbout, R.; Chan, D.W.; Weinfeld, M.; Day, R.S., 3rd; Barron, G.M.; Allalunis-Turner, J. Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science 1995, 267, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Buck, D.; Moshous, D.; de Chasseval, R.; Ma, Y.; le Deist, F.; Cavazzana-Calvo, M.; Fischer, A.; Casanova, J.L.; Lieber, M.R.; de Villartay, J.P. Severe combined immunodeficiency and microcephaly in siblings with hypomorphic mutations in DNA ligase IV. Eur. J. Immunol. 2006, 36, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Enders, A.; Fisch, P.; Schwarz, K.; Duffner, U.; Pannicke, U.; Nikolopoulos, E.; Peters, A.; Orlowska-Volk, M.; Schindler, D.; Friedrich, W.; et al. A severe form of human combined immunodeficiency due to mutations in DNA ligase IV. J. Immunol. 2006, 176, 5060–5068. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Lu, H.; Lan, S.; Liu, J.; Stein, M.N.; Haffty, B.G.; Shen, Z. Identification of the DNA repair defects in a case of Dubowitz syndrome. PLoS ONE 2013, 8, e54389. [Google Scholar] [CrossRef] [PubMed]
- Riballo, E.; Critchlow, S.E.; Teo, S.H.; Doherty, A.J.; Priestley, A.; Broughton, B.; Kysela, B.; Beamish, H.; Plowman, N.; Arlett, C.F.; et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Curr. Biol. 1999, 9, 699–702. [Google Scholar] [CrossRef]
- Guo, C.; Nakazawa, Y.; Woodbine, L.; Bjorkman, A.; Shimada, M.; Fawcett, H.; Jia, N.; Ohyama, K.; Li, T.S.; Nagayama, Y.; et al. XRCC4 deficiency in human subjects causes a marked neurological phenotype but no overt immunodeficiency. J. Allergy Clin. Immunol. 2015, 136, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Buck, D.; Malivert, L.; de Chasseval, R.; Barraud, A.; Fondaneche, M.C.; Sanal, O.; Plebani, A.; Stephan, J.L.; Hufnagel, M.; le Deist, F.; et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell 2006, 124, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.E.; Bicknell, L.S.; Yigit, G.; Duker, A.L.; van Kogelenberg, M.; Haghayegh, S.; Wieczorek, D.; Kayserili, H.; Albert, M.H.; Wise, C.A.; et al. Extreme growth failure is a common presentation of ligase IV deficiency. Hum. Mutat. 2014, 35, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Rigas, B.; Borgo, S.; Elhosseiny, A.; Balatsos, V.; Manika, Z.; Shinya, H.; Kurihara, N.; Go, M.; Lipkin, M. Decreased expression of DNA-dependent protein kinase, a DNA repair protein, during human colon carcinogenesis. Cancer Res. 2001, 61, 8381–8384. [Google Scholar] [PubMed]
- Beggs, A.D.; Domingo, E.; McGregor, M.; Presz, M.; Johnstone, E.; Midgley, R.; Kerr, D.; Oukrif, D.; Novelli, M.; Abulafi, M.; et al. Loss of expression of the double strand break repair protein ATM is associated with worse prognosis in colorectal cancer and loss of Ku70 expression is associated with CIN. Oncotarget 2012, 3, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- Lomnytska, M.I.; Becker, S.; Gemoll, T.; Lundgren, C.; Habermann, J.; Olsson, A.; Bodin, I.; Engstrom, U.; Hellman, U.; Hellman, K.; et al. Impact of genomic stability on protein expression in endometrioid endometrial cancer. Br. J. Cancer 2012, 106, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Perrot, A.; Pionneau, C.; Azar, N.; Baillou, C.; Lemoine, F.M.; Leblond, V.; Merle-Beral, H.; Bene, M.C.; Herbrecht, R.; Bahram, S.; et al. Waldenstrom’s macroglobulinemia harbors a unique proteome where Ku70 is severely underexpressed as compared with other B-lymphoproliferative disorders. Blood Cancer J. 2012, 2, e88. [Google Scholar] [CrossRef] [PubMed]
- Pozniak, Y.; Balint-Lahat, N.; Rudolph, J.D.; Lindskog, C.; Katzir, R.; Avivi, C.; Ponten, F.; Ruppin, E.; Barshack, I.; Geiger, T. System-wide Clinical Proteomics of Breast Cancer Reveals Global Remodeling of Tissue Homeostasis. Cell Syst. 2016, 2, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Someya, M.; Sakata, K.; Matsumoto, Y.; Yamamoto, H.; Monobe, M.; Ikeda, H.; Ando, K.; Hosoi, Y.; Suzuki, N.; Hareyama, M. The association of DNA-dependent protein kinase activity with chromosomal instability and risk of cancer. Carcinogenesis 2006, 27, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Auckley, D.H.; Crowell, R.E.; Heaphy, E.R.; Stidley, C.A.; Lechner, J.F.; Gilliland, F.D.; Belinsky, S.A. Reduced DNA-dependent protein kinase activity is associated with lung cancer. Carcinogenesis 2001, 22, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Mazzarelli, P.; Parrella, P.; Seripa, D.; Signori, E.; Perrone, G.; Rabitti, C.; Borzomati, D.; Gabbrielli, A.; Matera, M.G.; Gravina, C.; et al. DNA end binding activity and Ku70/80 heterodimer expression in human colorectal tumor. World J. Gastroenterol. 2005, 11, 6694–6700. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Zhang, Y.; Zou, M.; Yang, S.; Liang, X.Q. Expression of TRF1, TRF2, TIN2, TERT, KU70, and BRCA1 proteins is associated with telomere shortening and may contribute to multistage carcinogenesis of gastric cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, T.L.; Brum, I.S.; Biolchi, V.; Garicochea, B.; Fillmann, L.S.; Corleta, O.C. Is there any association between TACSTD2, KIAA1253, Ku70 and mutant KRAS gene expression and clinical-pathological features of colorectal cancer? Exp. Oncol. 2011, 33, 28–32. [Google Scholar] [PubMed]
- Zhang, X.; Wang, Y.; Ning, Y. Down-regulation of protein kinase, DNA-activated, catalytic polypeptide attenuates tumor progression and is an independent prognostic predictor of survival in prostate cancer. Urol. Oncol. 2017, 35, 111.e15–111.e23. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Cheng, S.; Zhu, Y.; Zhang, P.; Liu, N.; Xu, T.; Sun, C.; Lv, Y. Identification of PRKDC (Protein Kinase, DNA-Activated, Catalytic Polypeptide) as an essential gene for colorectal cancer (CRCs) cells. Gene 2016, 584, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, Y.; Watanabe, T.; Nakagawa, K.; Matsumoto, Y.; Enomoto, A.; Morita, A.; Nagawa, H.; Suzuki, N. Up-regulation of DNA-dependent protein kinase activity and Sp1 in colorectal cancer. Int. J. Oncol. 2004, 25, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Wu, X.; Vaporciyan, A.A.; Spitz, M.R.; Gu, J. Prognostic significance of ataxia-telangiectasia mutated, DNA-dependent protein kinase catalytic subunit, and Ku heterodimeric regulatory complex 86-kD subunit expression in patients with nonsmall cell lung cancer. Cancer 2008, 112, 2756–2764. [Google Scholar] [CrossRef] [PubMed]
- Evert, M.; Frau, M.; Tomasi, M.L.; Latte, G.; Simile, M.M.; Seddaiu, M.A.; Zimmermann, A.; Ladu, S.; Staniscia, T.; Brozzetti, S.; et al. Deregulation of DNA-dependent protein kinase catalytic subunit contributes to human hepatocarcinogenesis development and has a putative prognostic value. Br. J. Cancer 2013, 109, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Cornell, L.; Munck, J.M.; Alsinet, C.; Villanueva, A.; Ogle, L.; Willoughby, C.E.; Televantou, D.; Thomas, H.D.; Jackson, J.; Burt, A.D.; et al. DNA-PK-A candidate driver of hepatocarcinogenesis and tissue biomarker that predicts response to treatment and survival. Clin. Cancer Res. 2015, 21, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Tonotsuka, N.; Hosoi, Y.; Miyazaki, S.; Miyata, G.; Sugawara, K.; Mori, T.; Ouchi, N.; Satomi, S.; Matsumoto, Y.; Nakagawa, K.; et al. Heterogeneous expression of DNA-dependent protein kinase in esophageal cancer and normal epithelium. Int. J. Mol. Med. 2006, 18, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xie, C.; Yang, Z.; Chen, J.; Lu, N.H. Abnormal DNA-PKcs and Ku 70/80 expression may promote malignant pathological processes in gastric carcinoma. World J. Gastroenterol. 2013, 19, 6894–6901. [Google Scholar] [CrossRef] [PubMed]
- Grupp, K.; Roettger, L.; Kluth, M.; Hube-Magg, C.; Simon, R.; Lebok, P.; Minner, S.; Tsourlakis, M.C.; Koop, C.; Graefen, M.; et al. Expression of DNA ligase IV is linked to poor prognosis and characterizes a subset of prostate cancers harboring TMPRSS2:ERG fusion and PTEN deletion. Oncol. Rep. 2015, 34, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Kuhmann, C.; Li, C.; Kloor, M.; Salou, M.; Weigel, C.; Schmidt, C.R.; Ng, L.W.; Tsui, W.W.; Leung, S.Y.; Yuen, S.T.; et al. Altered regulation of DNA ligase IV activity by aberrant promoter DNA methylation and gene amplification in colorectal cancer. Hum. Mol. Genet. 2014, 23, 2043–2054. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Rabii, R.; Liang, G.; Song, C.; Chen, W.; Guo, M.; Wei, X.; Messadi, D.; Hu, S. The Expression Levels of XLF and Mutant P53 Are Inversely Correlated in Head and Neck Cancer Cells. J. Cancer 2016, 7, 1374–1382. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.F.; Tseng, H.C.; Chiu, C.F.; Liang, S.Y.; Tsai, C.W.; Tsai, M.H.; Bau, D.T. Association between DNA double strand break gene Ku80 polymorphisms and oral cancer susceptibility. Oral Oncol. 2009, 45, 789–793. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Chiu, C.F.; Liang, S.Y.; Wu, H.C.; Chang, C.L.; Tsai, C.W.; Wang, H.C.; Lee, H.Z.; Bau, D.T. Significant association of Ku80 single nucleotide polymorphisms with bladder cancer susceptibility in Taiwan. Anticancer Res. 2009, 29, 1275–1279. [Google Scholar] [PubMed]
- Li, J.Q.; Chen, J.; Liu, N.N.; Yang, L.; Zeng, Y.; Wang, B.; Wang, X.R. Ku80 gene G-1401T promoter polymorphism and risk of gastric cancer. World J. Gastroenterol. 2011, 17, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Liu, C.S.; Chiu, C.F.; Chiang, S.Y.; Wang, C.H.; Wang, R.F.; Lin, C.C.; Tsai, R.Y.; Bau, D.T. Significant association of DNA repair gene Ku80 genotypes with breast cancer susceptibility in Taiwan. Anticancer Res. 2009, 29, 5251–5254. [Google Scholar] [PubMed]
- Yang, M.D.; Hsu, Y.M.; Kuo, Y.S.; Chen, H.S.; Chang, C.L.; Wu, C.N.; Chang, C.H.; Liao, Y.M.; Wang, H.C.; Wang, M.F.; et al. Significant association of Ku80 single nucleotide polymorphisms with colorectal cancer susceptibility in Central Taiwan. Anticancer Res. 2009, 29, 2239–2242. [Google Scholar] [PubMed]
- Fu, Y.P.; Yu, J.C.; Cheng, T.C.; Lou, M.A.; Hsu, G.C.; Wu, C.Y.; Chen, S.T.; Wu, H.S.; Wu, P.E.; Shen, C.Y. Breast cancer risk associated with genotypic polymorphism of the nonhomologous end-joining genes: a multigenic study on cancer susceptibility. Cancer Res. 2003, 63, 2440–2446. [Google Scholar] [PubMed]
- Xu, H.; Zou, P.; Chen, P.; Zhao, L.; Zhao, P.; Lu, A. Association between the XRCC6 Promoter rs2267437 polymorphism and cancer risk: Evidence based on the current literature. Genet. Test. Mol. Biomark. 2013, 17, 607–614. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Luo, S.; Huang, T.; Ren, J.; Wu, X.; Shao, J.; Zhu, Q. The Ku70 -1310C/G promoter polymorphism is associated with breast cancer susceptibility in Chinese Han population. Mol. Biol. Rep. 2012, 39, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Willems, P.; De Ruyck, K.; Van den Broecke, R.; Makar, A.; Perletti, G.; Thierens, H.; Vral, A. A polymorphism in the promoter region of Ku70/XRCC6, associated with breast cancer risk and oestrogen exposure. J. Cancer Res. Clin. Oncol. 2009, 135, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.M.; Yang, M.D.; Chang, W.S.; Jeng, L.B.; Lee, M.H.; Lu, M.C.; Chang, S.C.; Tsai, C.W.; Tsai, Y.; Tsai, F.J.; et al. The contribution of XRCC6/Ku70 to hepatocellular carcinoma in Taiwan. Anticancer Res. 2013, 33, 529–535. [Google Scholar] [PubMed]
- Hsia, T.C.; Liu, C.J.; Chu, C.C.; Hang, L.W.; Chang, W.S.; Tsai, C.W.; Wu, C.I.; Lien, C.S.; Liao, W.L.; Ho, C.Y.; et al. Association of DNA double-strand break gene XRCC6 genotypes and lung cancer in Taiwan. Anticancer Res. 2012, 32, 1015–1020. [Google Scholar] [PubMed]
- Tseng, R.C.; Hsieh, F.J.; Shih, C.M.; Hsu, H.S.; Chen, C.Y.; Wang, Y.C. Lung cancer susceptibility and prognosis associated with polymorphisms in the nonhomologous end-joining pathway genes: A multiple genotype-phenotype study. Cancer 2009, 115, 2939–2948. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yang, Y.; An, Y.; Zhou, Y.; Liu, Y.; Yu, Q.; Lu, D.; Wang, H.; Jin, L.; Zhou, W.; et al. Genetic polymorphisms in DNA double-strand break repair genes XRCC5, XRCC6 and susceptibility to hepatocellular carcinoma. Carcinogenesis 2011, 32, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Henriquez-Hernandez, L.A.; Valenciano, A.; Foro-Arnalot, P.; Alvarez-Cubero, M.J.; Cozar, J.M.; Suarez-Novo, J.F.; Castells-Esteve, M.; Fernandez-Gonzalo, P.; De-Paula-Carranza, B.; Ferrer, M.; et al. Association between single-nucleotide polymorphisms in DNA double-strand break repair genes and prostate cancer aggressiveness in the Spanish population. Prostate Cancer Prostatic Dis. 2016, 19, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.D.; Wang, H.C.; Chang, W.S.; Tsai, C.W.; Bau, D.T. Genetic polymorphisms of DNA double strand break gene Ku70 and gastric cancer in Taiwan. BMC Cancer 2011, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Tsai, C.W.; Hsu, C.M.; Shih, L.C.; Chang, W.S.; Shui, H.A.; Bau, D.T. The role of XRCC6/Ku70 in nasopharyngeal carcinoma. Int. J. Oral Maxillofac Surg. 2015, 44, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Bau, D.T.; Tseng, H.C.; Wang, C.H.; Chiu, C.F.; Hua, C.H.; Wu, C.N.; Liang, S.Y.; Wang, C.L.; Tsai, C.W.; Tsai, M.H. Oral cancer and genetic polymorphism of DNA double strand break gene Ku70 in Taiwan. Oral Oncol. 2008, 44, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.S.; Ke, H.L.; Tsai, C.W.; Lien, C.S.; Liao, W.L.; Lin, H.H.; Lee, M.H.; Wu, H.C.; Chang, C.H.; Chen, C.C.; et al. The role of XRCC6 T-991C functional polymorphism in renal cell carcinoma. Anticancer Res. 2012, 32, 3855–3860. [Google Scholar] [PubMed]
- Wang, W.; Pan, X.; Huo, X.; Yan, F.; Wang, M.; Wang, D.; Gao, Y.; Cao, Q.; Luo, D.; Qin, C.; et al. A functional polymorphism C-1310G in the promoter region of Ku70/XRCC6 is associated with risk of renal cell carcinoma. Mol. Carcinog. 2012, 51 (Suppl. 1), E183–E190. [Google Scholar] [CrossRef] [PubMed]
- Sobczuk, A.; Smolarz, B.; Romanowicz, H.; Zadrozny, M.; Baszczynski, J.; Westfal, B.; Pertynski, T. Analysis of the polymorphisms in non-homologous DNA end joining (NHEJ) gene Ku70 and Ligase IV in sporadic breast cancer in women. Pol. J. Pathol. 2010, 61, 27–31. [Google Scholar] [PubMed]
- Liu, Y.; Zhang, H.; Zhou, K.; Chen, L.; Xu, Z.; Zhong, Y.; Liu, H.; Li, R.; Shugart, Y.Y.; Wei, Q.; et al. Tagging SNPs in non-homologous end-joining pathway genes and risk of glioma. Carcinogenesis 2007, 28, 1906–1913. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Ren, J.; Yan, D.; Xiao, L.; Sun, R. Association between the XRCC6 polymorphisms and cancer risks: A systematic review and meta-analysis. Medicine 2015, 94, e283. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Peng, L.; Li, C.P.; Li, A.P.; Zhou, J.W.; Zhang, Z.D.; Liu, Q.Z. Genetic variants of the XRCC7 gene involved in DNA repair and risk of human bladder cancer. Int. J. Urol. 2008, 15, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.E.; Bondy, M.L.; Shen, H.; El-Zein, R.; Aldape, K.; Cao, Y.; Pudavalli, V.; Levin, V.A.; Yung, W.K.; Wei, Q. Polymorphisms of DNA repair genes and risk of glioma. Cancer Res. 2004, 64, 5560–5563. [Google Scholar] [CrossRef] [PubMed]
- Zhi, Y.; Yu, J.; Liu, Y.; Wei, Q.; Yuan, F.; Zhou, X.; Song, B.; Chen, Z.; Yang, J. Interaction between polymorphisms of DNA repair genes significantly modulated bladder cancer risk. Int. J. Med. Sci. 2012, 9, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Mandal, R.K.; Kapoor, R.; Mittal, R.D. Polymorphic variants of DNA repair gene XRCC3 and XRCC7 and risk of prostate cancer: A study from North Indian population. DNA Cell Biol. 2010, 29, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Schildkraut, J.M.; Iversen, E.S.; Wilson, M.A.; Clyde, M.A.; Moorman, P.G.; Palmieri, R.T.; Whitaker, R.; Bentley, R.C.; Marks, J.R.; Berchuck, A. Association between DNA damage response and repair genes and risk of invasive serous ovarian cancer. PLoS ONE 2010, 5, e10061. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Qi, S.; Dou, C.; Shuang, L.; Yan, H. Association of LIG4 and XRCC4 gene polymorphisms with the risk of human glioma in a Chinese population. Int. J. Clin. Exp. Pathol. 2015, 8, 2057–2062. [Google Scholar] [PubMed]
- Jiao, K.; Qin, J.; Zhao, Y.; Zhang, H. Genetic effects of XRCC4 and ligase IV genes on human glioma. Neuroreport 2016, 27, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Sehl, M.E.; Langer, L.R.; Papp, J.C.; Kwan, L.; Seldon, J.L.; Arellano, G.; Reiss, J.; Reed, E.F.; Dandekar, S.; Korin, Y.; et al. Associations between single nucleotide polymorphisms in double-stranded DNA repair pathway genes and familial breast cancer. Clin. Cancer Res. 2009, 15, 2192–2203. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.Y.; Liu, S.Y.; Chen, C.H.; Tseng, H.F.; Chuang, L.Y.; Yang, C.H.; Lin, Y.C.; Wen, C.H.; Chiang, W.F.; Ho, C.H.; et al. Combinational polymorphisms of four DNA repair genes XRCC1, XRCC2, XRCC3, and XRCC4 and their association with oral cancer in Taiwan. J. Oral Pathol. Med. 2008, 37, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, L.N. Association between single nucleotide polymorphisms of X-ray repair cross-complementing protein 4 gene and development of pancreatic cancer. Genet. Mol. Res. 2015, 14, 9626–9632. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Tian, Y.; Li, K.; Jiang, Q.; Xue, H.; Yang, S. Association of single nucleotide polymorphisms of DNA repair gene and susceptibility to pancreatic cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 3180–3185. [Google Scholar] [PubMed]
- Tseng, H.C.; Tsai, M.H.; Chiu, C.F.; Wang, C.H.; Chang, N.W.; Huang, C.Y.; Tsai, C.W.; Liang, S.Y.; Wang, C.L.; Bau, D.T. Association of XRCC4 codon 247 polymorphism with oral cancer susceptibility in Taiwan. Anticancer Res. 2008, 28, 1687–1691. [Google Scholar] [PubMed]
- He, M.; Hu, X.; Chen, L.; Cao, A.Y.; Yu, K.D.; Shi, T.Y.; Kuang, X.Y.; Shi, W.B.; Ling, H.; Li, S.; et al. A recessive variant of XRCC4 predisposes to non- BRCA1/2 breast cancer in chinese women and impairs the DNA damage response via dysregulated nuclear localization. Oncotarget 2014, 5, 12218–12232. [Google Scholar] [CrossRef] [PubMed]
- Long, X.D.; Zhao, D.; Wang, C.; Huang, X.Y.; Yao, J.G.; Ma, Y.; Wei, Z.H.; Liu, M.; Zeng, L.X.; Mo, X.Q.; et al. Genetic polymorphisms in DNA repair genes XRCC4 and XRCC5 and aflatoxin B1-related hepatocellular carcinoma. Epidemiology 2013, 24, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.F.; Tsai, M.H.; Tseng, H.C.; Wang, C.L.; Wang, C.H.; Wu, C.N.; Lin, C.C.; Bau, D.T. A novel single nucleotide polymorphism in XRCC4 gene is associated with oral cancer susceptibility in Taiwanese patients. Oral Oncol. 2008, 44, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Jiang, W.Y.; Qiao, D.; Zhang, S.G.; Wu, Y.; Zhang, X.X.; Hua, L.X.; Ding, Y.; Feng, N.H. An updated meta-analysis of XRCC4 polymorphisms and cancer risk based on 31 case-control studies. Cancer Biomark. 2012, 12, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Margulis, V.; Lin, J.; Yang, H.; Wang, W.; Wood, C.G.; Wu, X. Genetic susceptibility to renal cell carcinoma: The role of DNA double-strand break repair pathway. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2366–2373. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, J.D.; Malats, N.; Rothman, N.; Real, F.X.; Silverman, D.; Kogevinas, M.; Chanock, S.; Yeager, M.; Welch, R.; Dosemeci, M.; et al. Evaluation of genetic variation in the double-strand break repair pathway and bladder cancer risk. Carcinogenesis 2007, 28, 1788–1793. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, N.; Matsui, S.; Narita, S.; Kamba, T.; Mitsuzuka, K.; Hatakeyama, S.; Horikawa, Y.; Inoue, T.; Saito, S.; Ohyama, C.; et al. Distinct cancer-specific survival in metastatic prostate cancer patients classified by a panel of single nucleotide polymorphisms of cancer-associated genes. Genes Cancer 2013, 4, 54–60. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Chang, S.C.; Wallar, G.M.; Zhang, Z.F.; Cai, L. Association of XRCC3 and XRCC4 gene polymorphisms, family history of cancer and tobacco smoking with non-small-cell lung cancer in a Chinese population: A case-control study. J. Hum. Genet. 2013, 58, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Ming-Zhong, S.; Hui-Xiang, J.; Zhong-Wei, Z.; Hao, J.; Rong, Z. Genetic variants of the DNA damage repair genes XRCC4 and RAD51 are associated with susceptibility to esophageal cancer. Clin. Res. Hepatol. Gastroenterol. 2015, 39, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Chang, C.L.; Tsai, C.W.; Wu, H.C.; Chiu, C.F.; Wang, R.F.; Liu, C.S.; Lin, C.C.; Bau, D.T. Significant association of an XRCC4 single nucleotide polymorphism with bladder cancer susceptibility in Taiwan. Anticancer Res. 2009, 29, 1777–1782. [Google Scholar] [PubMed]
- Chang, C.H.; Chiu, C.F.; Wu, H.C.; Tseng, H.C.; Wang, C.H.; Lin, C.C.; Tsai, C.W.; Liang, S.Y.; Wang, C.L.; Bau, D.T. Significant association of XRCC4 single nucleotide polymorphisms with prostate cancer susceptibility in Taiwanese males. Mol. Med. Rep. 2008, 1, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.F.; Wang, C.H.; Wang, C.L.; Lin, C.C.; Hsu, N.Y.; Weng, J.R.; Bau, D.T. A novel single nucleotide polymorphism in XRCC4 gene is associated with gastric cancer susceptibility in Taiwan. Ann. Surg. Oncol. 2008, 15, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Bau, D.T.; Yang, M.D.; Tsou, Y.A.; Lin, S.S.; Wu, C.N.; Hsieh, H.H.; Wang, R.F.; Tsai, C.W.; Chang, W.S.; Hsieh, H.M.; et al. Colorectal cancer and genetic polymorphism of DNA double-strand break repair gene XRCC4 in Taiwan. Anticancer Res. 2010, 30, 2727–2730. [Google Scholar] [PubMed]
- Roddam, P.L.; Rollinson, S.; O’Driscoll, M.; Jeggo, P.A.; Jack, A.; Morgan, G.J. Genetic variants of NHEJ DNA ligase IV can affect the risk of developing multiple myeloma, a tumour characterised by aberrant class switch recombination. J. Med. Genet. 2002, 39, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Andreae, J.; Varon, R.; Sperling, K.; Seeger, K. Polymorphisms in the DNA ligase IV gene might influence the risk of acute lymphoblastic leukemia in children. Leukemia 2007, 21, 2226–2227. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Shan, X.F.; Shang, K.; Xu, H.; He, J.; Cai, Z.G. Relevance of LIG4 gene polymorphisms with cancer susceptibility: Evidence from a meta-analysis. Sci. Rep. 2014, 4, 6630. [Google Scholar] [CrossRef] [PubMed]
- Werbrouck, J.; De Ruyck, K.; Duprez, F.; Van Eijkeren, M.; Rietzschel, E.; Bekaert, S.; Vral, A.; De Neve, W.; Thierens, H. Single-nucleotide polymorphisms in DNA double-strand break repair genes: Association with head and neck cancer and interaction with tobacco use and alcohol consumption. Mutat. Res. 2008, 656, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Goode, E.L.; Dunning, A.M.; Kuschel, B.; Healey, C.S.; Day, N.E.; Ponder, B.A.; Easton, D.F.; Pharoah, P.P. Effect of germ-line genetic variation on breast cancer survival in a population-based study. Cancer Res. 2002, 62, 3052–3057. [Google Scholar] [PubMed]
- Kuschel, B.; Auranen, A.; McBride, S.; Novik, K.L.; Antoniou, A.; Lipscombe, J.M.; Day, N.E.; Easton, D.F.; Ponder, B.A.; Pharoah, P.D.; et al. Variants in DNA double-strand break repair genes and breast cancer susceptibility. Hum. Mol. Genet. 2002, 11, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Andrew, A.S.; Gui, J.; Hu, T.; Wyszynski, A.; Marsit, C.J.; Kelsey, K.T.; Schned, A.R.; Tanyos, S.A.; Pendleton, E.M.; Ekstrom, R.M.; et al. Genetic polymorphisms modify bladder cancer recurrence and survival in a USA population-based prognostic study. BJU Int. 2015, 115, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Adel Fahmideh, M.; Lavebratt, C.; Schuz, J.; Roosli, M.; Tynes, T.; Grotzer, M.A.; Johansen, C.; Kuehni, C.E.; Lannering, B.; Prochazka, M.; et al. Common genetic variations in cell cycle and DNA repair pathways associated with pediatric brain tumor susceptibility. Oncotarget 2016, 7, 63640–63650. [Google Scholar] [CrossRef] [PubMed]
- Parrella, P.; Mazzarelli, P.; Signori, E.; Perrone, G.; Marangi, G.F.; Rabitti, C.; Delfino, M.; Prencipe, M.; Gallo, A.P.; Rinaldi, M.; et al. Expression and heterodimer-binding activity of Ku70 and Ku80 in human non-melanoma skin cancer. J. Clin. Pathol. 2006, 59, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Abdelbaqi, K.; Di Paola, D.; Rampakakis, E.; Zannis-Hadjopoulos, M. Ku protein levels, localization and association to replication origins in different stages of breast tumor progression. J. Cancer 2013, 4, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Komuro, Y.; Watanabe, T.; Hosoi, Y.; Matsumoto, Y.; Nakagawa, K.; Tsuno, N.; Kazama, S.; Kitayama, J.; Suzuki, N.; Nagawa, H. The expression pattern of Ku correlates with tumor radiosensitivity and disease free survival in patients with rectal carcinoma. Cancer 2002, 95, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Saygili, U.; Gorkay, I.B.; Koyuncuoglu, M.; Gol, M.; Uslu, T.; Erten, O. The relationship between expression of Ku70 and survival in irradiated patients with endometrial carcinoma. Gynecol. Oncol. 2004, 95, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Robson, T.; Doig, T.; Brenn, T.; Mathers, M.; Brown, E.R.; Doherty, V.; Bartlett, J.M.; Anderson, N.; Melton, D.W. DNA repair and replication proteins as prognostic markers in melanoma. Histopathology 2013, 62, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Li, P.; Xu, M.; Yin, J.; Su, Z.; Li, W.; Zhang, J. Ku80 is highly expressed in lung adenocarcinoma and promotes cisplatin resistance. J. Exp. Clin. Cancer Res. 2012, 31, 99. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, P.; Liu, W.; Xia, Z.; Shi, F.; Zhong, M. Expression and significance of Ku80 and PDGFR-alpha in nasal NK/T-cell lymphoma. Pathol. Res. Pract. 2016, 212, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Alshareeda, A.T.; Negm, O.H.; Albarakati, N.; Green, A.R.; Nolan, C.; Sultana, R.; Madhusudan, S.; Benhasouna, A.; Tighe, P.; Ellis, I.O.; et al. Clinicopathological significance of KU70/KU80, a key DNA damage repair protein in breast cancer. Breast Cancer Res. Treat. 2013, 139, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Pavon, M.A.; Parreno, M.; Leon, X.; Sancho, F.J.; Cespedes, M.V.; Casanova, I.; Lopez-Pousa, A.; Mangues, M.A.; Quer, M.; Barnadas, A.; et al. Ku70 predicts response and primary tumor recurrence after therapy in locally advanced head and neck cancer. Int. J. Cancer 2008, 123, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Someya, M.; Sakata, K.; Matsumoto, Y.; Satoh, M.; Narimatsu, H.; Hareyama, M. Immunohistochemical analysis of Ku70/86 expression of breast cancer tissues. Oncol. Rep. 2007, 18, 1483–1487. [Google Scholar] [CrossRef] [PubMed]
- Korabiowska, M.; Quentin, T.; Schlott, T.; Bauer, H.; Kunze, E. Down-regulation of Ku 70 and Ku 80 mRNA expression in transitional cell carcinomas of the urinary bladder related to tumor progression. World J. Urol. 2004, 22, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Gao, J.; Lu, Y. Downregulated Ku70 and ATM associated to poor prognosis in colorectal cancer among Chinese patients. Onco Targets Ther. 2014, 7, 1955–1961. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.S.; Liu, L.; Liu, Z.G.; Zeng, M.S.; Song, L.B.; Xia, Y.F. [Expression and clinical significance of DNA-PKcs in nasopharyngeal carcinoma]. Ai Zheng 2008, 27, 979–983. [Google Scholar] [PubMed]
- Shin, K.; Kim, K.H.; Yoon, M.S.; Suh, D.S.; Lee, J.Y.; Kim, A.; Eo, W. Expression of Interactive Genes Associated with Apoptosis and Their Prognostic Value for Ovarian Serous Adenocarcinoma. Adv. Clin. Exp. Med. 2016, 25, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Bouchaert, P.; Guerif, S.; Debiais, C.; Irani, J.; Fromont, G. DNA-PKcs expression predicts response to radiotherapy in prostate cancer. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fatah, T.M.; Arora, A.; Moseley, P.; Coveney, C.; Perry, C.; Johnson, K.; Kent, C.; Ball, G.; Chan, S.; Madhusudan, S. ATM, ATR and DNA-PKcs expressions correlate to adverse clinical outcomes in epithelial ovarian cancers. BBA Clin. 2014, 2, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.F.; Kothari, V.; Drake, J.M.; Zhao, S.; Dylgjeri, E.; Dean, J.L.; Schiewer, M.J.; McNair, C.; Jones, J.K.; Aytes, A.; et al. DNA-PKcs-Mediated Transcriptional Regulation Drives Prostate Cancer Progression and Metastasis. Cancer Cell 2015, 28, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Kotula, E.; Berthault, N.; Agrario, C.; Lienafa, M.C.; Simon, A.; Dingli, F.; Loew, D.; Sibut, V.; Saule, S.; Dutreix, M. DNA-PKcs plays role in cancer metastasis through regulation of secreted proteins involved in migration and invasion. Cell Cycle 2015, 14, 1961–1972. [Google Scholar] [CrossRef] [PubMed]
- Um, J.H.; Kwon, J.K.; Kang, C.D.; Kim, M.J.; Ju, D.S.; Bae, J.H.; Kim, D.W.; Chung, B.S.; Kim, S.H. Relationship between antiapoptotic molecules and metastatic potency and the involvement of DNA-dependent protein kinase in the chemosensitization of metastatic human cancer cells by epidermal growth factor receptor blockade. J. Pharmacol. Exp. Ther. 2004, 311, 1062–1070. [Google Scholar] [CrossRef] [PubMed]
- Pyun, B.J.; Seo, H.R.; Lee, H.J.; Jin, Y.B.; Kim, E.J.; Kim, N.H.; Kim, H.S.; Nam, H.W.; Yook, J.I.; Lee, Y.S. Mutual regulation between DNA-PKcs and Snail1 leads to increased genomic instability and aggressive tumor characteristics. Cell Death Dis. 2013, 4, e517. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xu, X.; Hao, Y.; Chen, J.; Lu, H.; Qin, J.; Peng, L.; Chen, B. Expression of DNA-PKcs and BRCA1 as prognostic indicators in nasopharyngeal carcinoma following intensity-modulated radiation therapy. Oncol. Lett. 2013, 5, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Someya, M.; Sakata, K.I.; Matsumoto, Y.; Kamdar, R.P.; Kai, M.; Toyota, M.; Hareyama, M. The association of DNA-dependent protein kinase activity of peripheral blood lymphocytes with prognosis of cancer. Br. J. Cancer 2011, 104, 1724–1729. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Choe, G.; Park, K.U.; Park, D.J.; Yang, H.K.; Lee, B.L.; Kim, W.H. Altered expression of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) during gastric carcinogenesis and its clinical implications on gastric cancer. Int. J. Oncol. 2007, 31, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Yang, H.K.; Kim, W.H.; Choe, G. Loss of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) expression in gastric cancers. Cancer Res. Treat. 2005, 37, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.; Villalobos, V.M.; Gevaert, O.; Abramovitz, M.; Williams, C.; Sikic, B.I.; Leyland-Jones, B. Single Gene Prognostic Biomarkers in Ovarian Cancer: A Meta-Analysis. PLoS ONE 2016, 11, e0149183. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Xu, X.; Wang, X.; Kuang, H.; Osterman, M.; Feng, S.; Han, D.; Wu, Y.; Li, M.; Guo, H. Increasing sensitivity to DNA damage is a potential driver for human ovarian cancer. Oncotarget 2016, 7, 49710–49721. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.R.; Davidson, S.E.; Margison, G.P.; Jackson, S.P.; Hendry, J.H.; West, C.M. Expression of Ku70 correlates with survival in carcinoma of the cervix. Br. J. Cancer 2000, 83, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, J.; Sakata, K.I.; Someya, M.; Matsumoto, Y.; Satoh, M.; Nakata, K.; Hori, M.; Takagi, M.; Kondoh, A.; Himi, T.; et al. Analysis and results of Ku and XRCC4 expression in hypopharyngeal cancer tissues treated with chemoradiotherapy. Oncol. Lett. 2012, 4, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Al-Ubaidi, F.L.; Schultz, N.; Loseva, O.; Egevad, L.; Granfors, T.; Helleday, T. Castration therapy results in decreased Ku70 levels in prostate cancer. Clin. Cancer Res. 2013, 19, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Tarish, F.L.; Schultz, N.; Tanoglidi, A.; Hamberg, H.; Letocha, H.; Karaszi, K.; Hamdy, F.C.; Granfors, T.; Helleday, T. Castration radiosensitizes prostate cancer tissue by impairing DNA double-strand break repair. Sci. Transl. Med. 2015, 7, 312re311. [Google Scholar] [CrossRef] [PubMed]
- Harima, Y.; Sawada, S.; Miyazaki, Y.; Kin, K.; Ishihara, H.; Imamura, M.; Sougawa, M.; Shikata, N.; Ohnishi, T. Expression of Ku80 in cervical cancer correlates with response to radiotherapy and survival. Am. J. Clin. Oncol. 2003, 26, e80–e85. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Qu, Y.; Xu, X.; Xu, Q.; Geng, J.; Xu, J. Nuclear survivin and its relationship to DNA damage repair genes in non-small cell lung cancer investigated using tissue array. PLoS ONE 2013, 8, e74161. [Google Scholar] [CrossRef]
- Moeller, B.J.; Yordy, J.S.; Williams, M.D.; Giri, U.; Raju, U.; Molkentine, D.P.; Byers, L.A.; Heymach, J.V.; Story, M.D.; Lee, J.J.; et al. DNA repair biomarker profiling of head and neck cancer: Ku80 expression predicts locoregional failure and death following radiotherapy. Clin. Cancer Res. 2011, 17, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Beskow, C.; Skikuniene, J.; Holgersson, A.; Nilsson, B.; Lewensohn, R.; Kanter, L.; Viktorsson, K. Radioresistant cervical cancer shows upregulation of the NHEJ proteins DNA-PKcs, Ku70 and Ku86. Br. J. Cancer 2009, 101, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Zhang, H.M.; Li, H.H.; Zhou, R.; Mao, H.L.; Chen, Y.B.; Cui, M.H. A preliminary study on the radiation-resistance mechanism in ovarian cancer. J. Cancer Res. Ther. 2013, 9, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Sirzen, F.; Nilsson, A.; Zhivotovsky, B.; Lewensohn, R. DNA-dependent protein kinase content and activity in lung carcinoma cell lines: Correlation with intrinsic radiosensitivity. Eur. J. Cancer 1999, 35, 111–116. [Google Scholar] [CrossRef]
- Shao, C.J.; Fu, J.; Shi, H.L.; Mu, Y.G.; Chen, Z.P. Activities of DNA-PK and Ku86, but not Ku70, may predict sensitivity to cisplatin in human gliomas. J. NeuroOncol. 2008, 89, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Willmore, E.; Elliott, S.L.; Mainou-Fowler, T.; Summerfield, G.P.; Jackson, G.H.; O’Neill, F.; Lowe, C.; Carter, A.; Harris, R.; Pettitt, A.R.; et al. DNA-dependent protein kinase is a therapeutic target and an indicator of poor prognosis in B-cell chronic lymphocytic leukemia. Clin. Cancer Res. 2008, 14, 3984–3992. [Google Scholar] [CrossRef] [PubMed]
- Shintani, S.; Mihara, M.; Li, C.; Nakahara, Y.; Hino, S.; Nakashiro, K.; Hamakawa, H. Up-regulation of DNA-dependent protein kinase correlates with radiation resistance in oral squamous cell carcinoma. Cancer Sci. 2003, 94, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Molina, S.; Guerif, S.; Garcia, A.; Debiais, C.; Irani, J.; Fromont, G. DNA-PKcs Expression Is a Predictor of Biochemical Recurrence After Permanent Iodine 125 Interstitial Brachytherapy for Prostate Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2016, 95, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Shibata, T.; Fumoto, S.; Uchida, Y.; Mueller, W.; Takeno, S. DNA-PKcs expression in esophageal cancer as a predictor for chemoradiation therapeutic sensitivity. Ann. Surg. Oncol. 2002, 9, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.; Jung, Y.S.; Suh, H.N.; Wang, W.; Kim, M.J.; Oh, Y.S.; Lien, E.M.; Shen, X.; Matsumoto, Y.; McCrea, P.D.; et al. LIG4 mediates Wnt signalling-induced radioresistance. Nat. Commun. 2016, 7, 10994. [Google Scholar] [CrossRef] [PubMed]
- Gemenetzidis, E.; Gammon, L.; Biddle, A.; Emich, H.; Mackenzie, I.C. Invasive oral cancer stem cells display resistance to ionising radiation. Oncotarget 2015, 6, 43964–43977. [Google Scholar] [CrossRef] [PubMed]
- Salles, B.; Calsou, P.; Frit, P.; Muller, C. The DNA repair complex DNA-PK, a pharmacological target in cancer chemotherapy and radiotherapy. Pathologie-Biologie 2006, 54, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol. 2003, 4, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A. Studies on mammalian mutants defective in rejoining double-strand breaks in DNA. Mutat. Res. 1990, 239, 1–16. [Google Scholar] [CrossRef]
- Smith, G.C.; Jackson, S.P. The DNA-dependent protein kinase. Genes Dev. 1999, 13, 916–934. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Calsou, P.; Salles, B. The activity of the DNA-dependent protein kinase (DNA-PK) complex is determinant in the cellular response to nitrogen mustards. Biochimie 2000, 82, 25–28. [Google Scholar] [CrossRef]
- Belenkov, A.I.; Paiement, J.P.; Panasci, L.C.; Monia, B.P.; Chow, T.Y. An antisense oligonucleotide targeted to human Ku86 messenger RNA sensitizes M059K malignant glioma cells to ionizing radiation, bleomycin, and etoposide but not DNA cross-linking agents. Cancer Res. 2002, 62, 5888–5896. [Google Scholar] [PubMed]
- Li, G.C.; He, F.; Shao, X.; Urano, M.; Shen, L.; Kim, D.; Borrelli, M.; Leibel, S.A.; Gutin, P.H.; Ling, C.C. Adenovirus-mediated heat-activated antisense Ku70 expression radiosensitizes tumor cells in vitro and in vivo. Cancer Res. 2003, 63, 3268–3274. [Google Scholar] [PubMed]
- Marangoni, E.; Le Romancer, M.; Foray, N.; Muller, C.; Douc-Rasy, S.; Vaganay, S.; Abdulkarim, B.; Barrois, M.; Calsou, P.; Bernier, J.; et al. Transfer of Ku86 RNA antisense decreases the radioresistance of human fibroblasts. Cancer Gene Ther. 2000, 7, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Omori, S.; Takiguchi, Y.; Suda, A.; Sugimoto, T.; Miyazawa, H.; Takiguchi, Y.; Tanabe, N.; Tatsumi, K.; Kimura, H.; Pardington, P.E.; et al. Suppression of a DNA double-strand break repair gene, Ku70, increases radio- and chemosensitivity in a human lung carcinoma cell line. DNA Repair 2002, 1, 299–310. [Google Scholar] [CrossRef]
- Peng, Y.; Zhang, Q.; Nagasawa, H.; Okayasu, R.; Liber, H.L.; Bedford, J.S. Silencing expression of the catalytic subunit of DNA-dependent protein kinase by small interfering RNA sensitizes human cells for radiation-induced chromosome damage, cell killing, and mutation. Cancer Res. 2002, 62, 6400–6404. [Google Scholar] [PubMed]
- Hollick, J.J.; Golding, B.T.; Hardcastle, I.R.; Martin, N.; Richardson, C.; Rigoreau, L.J.; Smith, G.C.; Griffin, R.J. 2,6-disubstituted pyran-4-one and thiopyran-4-one inhibitors of DNA-Dependent protein kinase (DNA-PK). Bioorgan. Med. Chem. Lett. 2003, 13, 3083–3086. [Google Scholar] [CrossRef]
- Frit, P.; Canitrot, Y.; Muller, C.; Foray, N.; Calsou, P.; Marangoni, E.; Bourhis, J.; Salles, B. Cross-resistance to ionizing radiation in a murine leukemic cell line resistant to cis-dichlorodiammineplatinum(II): Role of Ku autoantigen. Mol. Pharmacol. 1999, 56, 141–146. [Google Scholar] [PubMed]
- Shen, H.; Kauvar, L.; Tew, K.D. Importance of glutathione and associated enzymes in drug response. Oncol. Res. 1997, 9, 295–302. [Google Scholar] [PubMed]
- Xu, W.; Liu, L.; Smith, G.C.; Charles l, G. Nitric oxide upregulates expression of DNA-PKcs to protect cells from DNA-damaging anti-tumour agents. Nat. Cell Biol. 2000, 2, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.J.; Hosoi, Y.; Miyachi, H.; Ishii, K.; Yoshida, M.; Nemoto, K.; Takai, Y.; Yamada, S.; Suzuki, N.; Ono, T. DNA-dependent protein kinase activity correlates with Ku70 expression and radiation sensitivity in esophageal cancer cell lines. Clin. Cancer Res. 2000, 6, 1073–1078. [Google Scholar] [PubMed]
- Ader, I.; Muller, C.; Bonnet, J.; Favre, G.; Cohen-Jonathan, E.; Salles, B.; Toulas, C. The radioprotective effect of the 24 kDa FGF-2 isoform in HeLa cells is related to an increased expression and activity of the DNA dependent protein kinase (DNA-PK) catalytic subunit. Oncogene 2002, 21, 6471–6479. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Christodoulopoulos, G.; Salles, B.; Panasci, L. DNA-Dependent protein kinase activity correlates with clinical and in vitro sensitivity of chronic lymphocytic leukemia lymphocytes to nitrogen mustards. Blood 1998, 92, 2213–2219. [Google Scholar] [PubMed]
- Muller, C.; Salles, B. Regulation of DNA-dependent protein kinase activity in leukemic cells. Oncogene 1997, 15, 2343–2348. [Google Scholar] [CrossRef] [PubMed]
- Marangoni, E.; Foray, N.; O’Driscoll, M.; Douc-Rasy, S.; Bernier, J.; Bourhis, J.; Jeggo, P. A Ku80 fragment with dominant negative activity imparts a radiosensitive phenotype to CHO-K1 cells. Nucleic Acids Res. 2000, 28, 4778–4782. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Park, S.J.; Lee, S.H. A targeted inhibition of DNA-dependent protein kinase sensitizes breast cancer cells following ionizing radiation. J. Pharmacol. Exp. Ther. 2002, 303, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.S.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res. 1999, 59, 4375–4382. [Google Scholar] [PubMed]
- Izzard, R.A.; Jackson, S.P.; Smith, G.C. Competitive and noncompetitive inhibition of the DNA-dependent protein kinase. Cancer Res. 1999, 59, 2581–2586. [Google Scholar] [PubMed]
- Boulton, S.; Kyle, S.; Yalcintepe, L.; Durkacz, B.W. Wortmannin is a potent inhibitor of DNA double strand break but not single strand break repair in Chinese hamster ovary cells. Carcinogenesis 1996, 17, 2285–2290. [Google Scholar] [CrossRef] [PubMed]
- Chernikova, S.B.; Wells, R.L.; Elkind, M.M. Wortmannin sensitizes mammalian cells to radiation by inhibiting the DNA-dependent protein kinase-mediated rejoining of double-strand breaks. Radiat. Res. 1999, 151, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, K.E.; Youmell, M.B.; Palayoor, S.T.; Price, B.D. Radiosensitization of human tumor cells by the phosphatidylinositol3-kinase inhibitors wortmannin and LY294002 correlates with inhibition of DNA-dependent protein kinase and prolonged G2-M delay. Clin. Cancer Res. 1997, 3, 1149–1156. [Google Scholar] [PubMed]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar] [PubMed]
- Walker, E.H.; Pacold, M.E.; Perisic, O.; Stephens, L.; Hawkins, P.T.; Wymann, M.P.; Williams, R.L. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 2000, 6, 909–919. [Google Scholar] [CrossRef]
- Veuger, S.J.; Curtin, N.J.; Richardson, C.J.; Smith, G.C.; Durkacz, B.W. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res. 2003, 63, 6008–6015. [Google Scholar] [PubMed]
- Willmore, E.; de Caux, S.; Sunter, N.J.; Tilby, M.J.; Jackson, G.H.; Austin, C.A.; Durkacz, B.W. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 2004, 103, 4659–4665. [Google Scholar] [CrossRef] [PubMed]
- Leahy, J.J.; Golding, B.T.; Griffin, R.J.; Hardcastle, I.R.; Richardson, C.; Rigoreau, L.; Smith, G.C. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorgan. Med. Chem. Lett. 2004, 14, 6083–6087. [Google Scholar] [CrossRef] [PubMed]
- Ismail, I.H.; Martensson, S.; Moshinsky, D.; Rice, A.; Tang, C.; Howlett, A.; McMahon, G.; Hammarsten, O. SU11752 inhibits the DNA-dependent protein kinase and DNA double-strand break repair resulting in ionizing radiation sensitization. Oncogene 2004, 23, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D.L.; Franz, H.R.; Ueno, A.M.; Smith, C.J.; Doolittle, D.J.; Waldren, C.A. Vanillin (3-methoxy-4-hydroxybenzaldehyde) inhibits mutation induced by hydrogen peroxide, N-methyl-N-nitrosoguanidine and mitomycin C but not (137)Cs gamma-radiation at the CD59 locus in human-hamster hybrid A(L) cells. Mutagenesis 2000, 15, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Keshava, C.; Keshava, N.; Ong, T.M.; Nath, J. Protective effect of vanillin on radiation-induced micronuclei and chromosomal aberrations in V79 cells. Mutation Res. 1998, 397, 149–159. [Google Scholar] [CrossRef]
- Take, Y.; Kumano, M.; Hamano, Y.; Fukatsu, H.; Teraoka, H.; Nishimura, S.; Okuyama, A. OK-1035, a selective inhibitor of DNA-dependent protein kinase. Biochem. Biophys. Res. Commun. 1995, 215, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Stockley, M.; Clegg, W.; Fontana, G.; Golding, B.T.; Martin, N.; Rigoreau, L.J.; Smith, G.C.; Griffin, R.J. Synthesis, crystal structure determination, and biological properties of the DNA-dependent protein kinase (DNA-PK) inhibitor 3-cyano-6-hydrazonomethyl-5-(4-pyridyl)pyrid-[1H]-2-one (OK-1035). Bioorgan. Med. Chem. Lett. 2001, 11, 2837–2841. [Google Scholar] [CrossRef]
- Douglas, P.; Moorhead, G.B.; Ye, R.; Lees-Miller, S.P. Protein phosphatases regulate DNA-dependent protein kinase activity. J. Biol. Chem. 2001, 276, 18992–18998. [Google Scholar] [CrossRef] [PubMed]
- Loong, S.L.; Korzh, S.; Price, A. Reduced DNA-dependent protein kinase activity in two cell lines derived from adult cancer patients with late radionecrosis. Oncogene 2004, 23, 5562–5566. [Google Scholar] [CrossRef] [PubMed]
- Riabinska, A.; Daheim, M.; Herter-Sprie, G.S.; Winkler, J.; Fritz, C.; Hallek, M.; Thomas, R.K.; Kreuzer, K.A.; Frenzel, L.P.; Monfared, P.; et al. Therapeutic targeting of a robust non-oncogene addiction to PRKDC in ATM-defective tumors. Sci. Transl. Med. 2013, 5, 189ra178. [Google Scholar] [CrossRef] [PubMed]
- Winkler, J.; Hofmann, K.; Chen, S. Novel targets for ATM-deficient malignancies. Mol. Cell. Oncol. 2014, 1, e29905. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, D.; Han, J.S.; Jeong, C.S.; Chung, B.S.; Kang, C.D.; Li, G.C. Ku autoantigen affects the susceptibility to anticancer drugs. Cancer Res. 1999, 59, 4012–4017. [Google Scholar] [PubMed]
- Nimura, Y.; Kawata, T.; Uzawa, K.; Okamura, J.; Liu, C.; Saito, M.; Shimada, H.; Seki, N.; Nakagawara, A.; Ito, H.; et al. Silencing Ku80 using small interfering RNA enhanced radiation sensitivity in vitro and in vivo. Int. J. Oncol. 2007, 30, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Jekimovs, C.; Bolderson, E.; Suraweera, A.; Adams, M.; O’Byrne, K.J.; Richard, D.J. Chemotherapeutic compounds targeting the DNA double-strand break repair pathways: The good, the bad, and the promising. Front. Oncol. 2014, 4, 86. [Google Scholar] [CrossRef] [PubMed]
- Weterings, E.; Gallegos, A.C.; Dominick, L.N.; Cooke, L.S.; Bartels, T.N.; Vagner, J.; Matsunaga, T.O.; Mahadevan, D. A novel small molecule inhibitor of the DNA repair protein Ku70/80. DNA Repair 2016, 43, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.; Nambiar, M.; Sharma, S.; Karki, S.S.; Goldsmith, G.; Hegde, M.; Kumar, S.; Pandey, M.; Singh, R.K.; Ray, P.; et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 2012, 151, 1474–1487. [Google Scholar] [CrossRef] [PubMed]
- Greco, G.E.; Matsumoto, Y.; Brooks, R.C.; Lu, Z.; Lieber, M.R.; Tomkinson, A.E. SCR7 is neither a selective nor a potent inhibitor of human DNA ligase IV. DNA Repair 2016, 43, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Menchon, G.; Bombarde, O.; Trivedi, M.; Negrel, A.; Inard, C.; Giudetti, B.; Baltas, M.; Milon, A.; Modesti, M.; Czaplicki, G.; et al. Structure-Based Virtual Ligand Screening on the XRCC4/DNA Ligase IV Interface. Sci. Rep. 2016, 6, 22878. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhong, S.; Zhu, X.; Dziegielewska, B.; Ellenberger, T.; Wilson, G.M.; MacKerell, A.D., Jr.; Tomkinson, A.E. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008, 68, 3169–3177. [Google Scholar] [CrossRef] [PubMed]
- Sarkaria, J.N.; Tibbetts, R.S.; Busby, E.C.; Kennedy, A.P.; Hill, D.E.; Abraham, R.T. Inhibition of phosphoinositide 3-kinase related kinases by the radiosensitizing agent wortmannin. Cancer Res. 1998, 58, 4375–4382. [Google Scholar] [PubMed]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef] [PubMed]
- Rainey, M.D.; Charlton, M.E.; Stanton, R.V.; Kastan, M.B. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008, 68, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, I.R.; Cockcroft, X.; Curtin, N.J.; El-Murr, M.D.; Leahy, J.J.; Stockley, M.; Golding, B.T.; Rigoreau, L.; Richardson, C.; Smith, G.C.; et al. Discovery of potent chromen-4-one inhibitors of the DNA-dependent protein kinase (DNA-PK) using a small-molecule library approach. J. Med. Chem. 2005, 48, 7829–7846. [Google Scholar] [CrossRef] [PubMed]
- Munck, J.M.; Batey, M.A.; Zhao, Y.; Jenkins, H.; Richardson, C.J.; Cano, C.; Tavecchio, M.; Barbeau, J.; Bardos, J.; Cornell, L.; et al. Chemosensitization of cancer cells by KU-0060648, a dual inhibitor of DNA-PK and PI-3K. Mol. Cancer Ther. 2012, 11, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, R.; Ter Burg, J.; Garrick, B.; van Bochove, G.G.; Brown, J.R.; Fernandes, S.M.; Rodriguez, M.S.; Michot, J.M.; Hallek, M.; Eichhorst, B.; et al. Dual TORK/DNA-PK inhibition blocks critical signaling pathways in chronic lymphocytic leukemia. Blood 2016, 128, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Anker, J.F.; Carneiro, B.A.; Chandra, S.; Kaplan, J.; Kalyan, A.; Santa-Maria, C.A.; Platanias, L.C.; Giles, F.J. Genomic landscape of DNA repair genes in cancer. Oncotarget 2016, 7, 23312–23321. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Hemann, M.T. From breaking bad to worse: Exploiting homologous DNA repair deficiency in cancer. Cancer Discov. 2014, 4, 516–518. [Google Scholar] [CrossRef] [PubMed]
- Michl, J.; Zimmer, J.; Tarsounas, M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016, 35, 909–923. [Google Scholar] [CrossRef] [PubMed]
- Middleton, F.K.; Patterson, M.J.; Elstob, C.J.; Fordham, S.; Herriott, A.; Wade, M.A.; McCormick, A.; Edmondson, R.; May, F.E.; Allan, J.M.; et al. Common cancer-associated imbalances in the DNA damage response confer sensitivity to single agent ATR inhibition. Oncotarget 2015, 6, 32396–32409. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Molecular Target | Compound Name | IC50 | References |
|---|---|---|---|
| DNA-PKcs | Caffeine | ATM: 1.2 mM | [224] |
| ATR: 1.1 mM | |||
| DNA-PKcs: 10 mM/L | |||
| Wortmannin | ATM: 150 nM | [252] | |
| ATR: 1.8 μM/L | |||
| DNA-PKcs: 16 nM/L | |||
| LY294002 | PI3K: 1.4 μM | [229] | |
| KU55933 | ATM: 13 nM | [253] | |
| ATR, DNA-PKcs: 16 nM/L | |||
| CP466722 | [254] | ||
| NU7026 | DNA-PKcs: 230 nM | [231] | |
| ATM, ATR: 13 μM | |||
| NU7441 | DNA-PKcs: 230 nM | [214,233,255] | |
| ATM, ATR: ≥100 μM | |||
| KU0060648 | DNA-PKcs: 0.019–0.17 μM | [256] | |
| MSC2490484A | NCT02316197 | ||
| NCT02516813 | |||
| VX-984 | NCT0264427 | ||
| CC-115 | NCT01353625 | ||
| [257] | |||
| Ku70/80 | STL127705 | [247] | |
| DNA Ligase IV | SCR-7 | [248,249] | |
| Compound #3101 | [250] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sishc, B.J.; Davis, A.J. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers 2017, 9, 81. https://doi.org/10.3390/cancers9070081
Sishc BJ, Davis AJ. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers. 2017; 9(7):81. https://doi.org/10.3390/cancers9070081
Chicago/Turabian StyleSishc, Brock J., and Anthony J. Davis. 2017. "The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer" Cancers 9, no. 7: 81. https://doi.org/10.3390/cancers9070081
APA StyleSishc, B. J., & Davis, A. J. (2017). The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers, 9(7), 81. https://doi.org/10.3390/cancers9070081