
Androgen Receptor-Dependent and -Independent Mechanisms Involved in Prostate Cancer Therapy Resistance



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
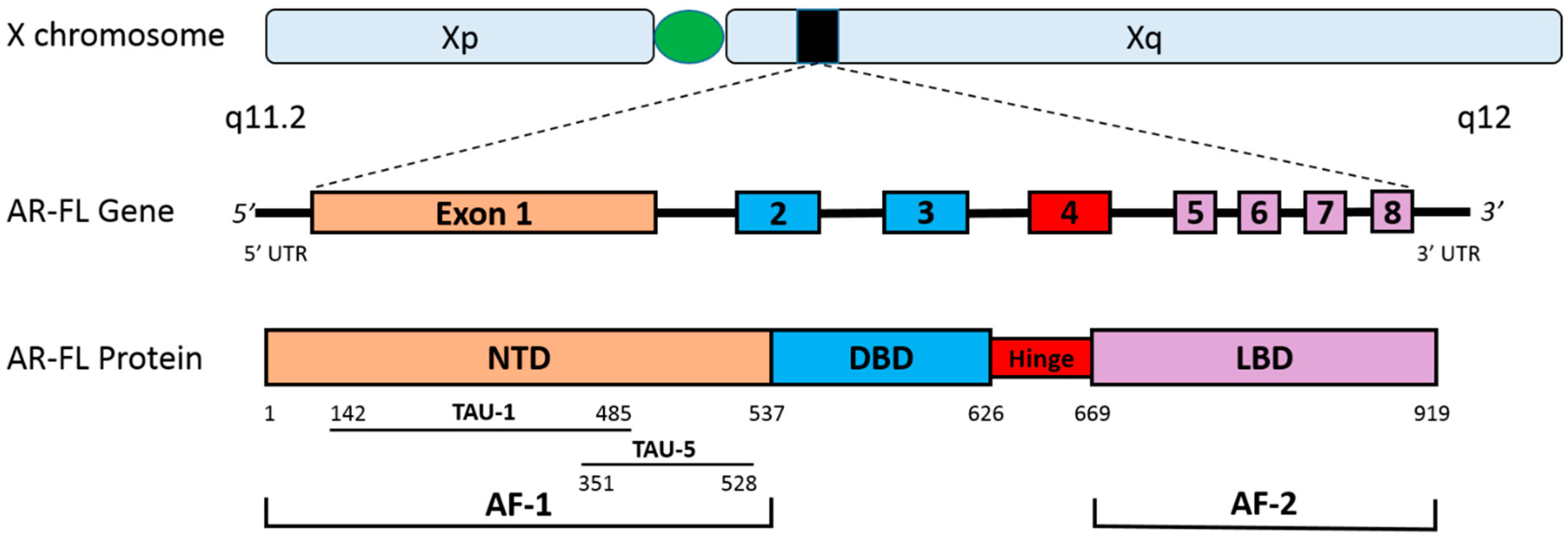
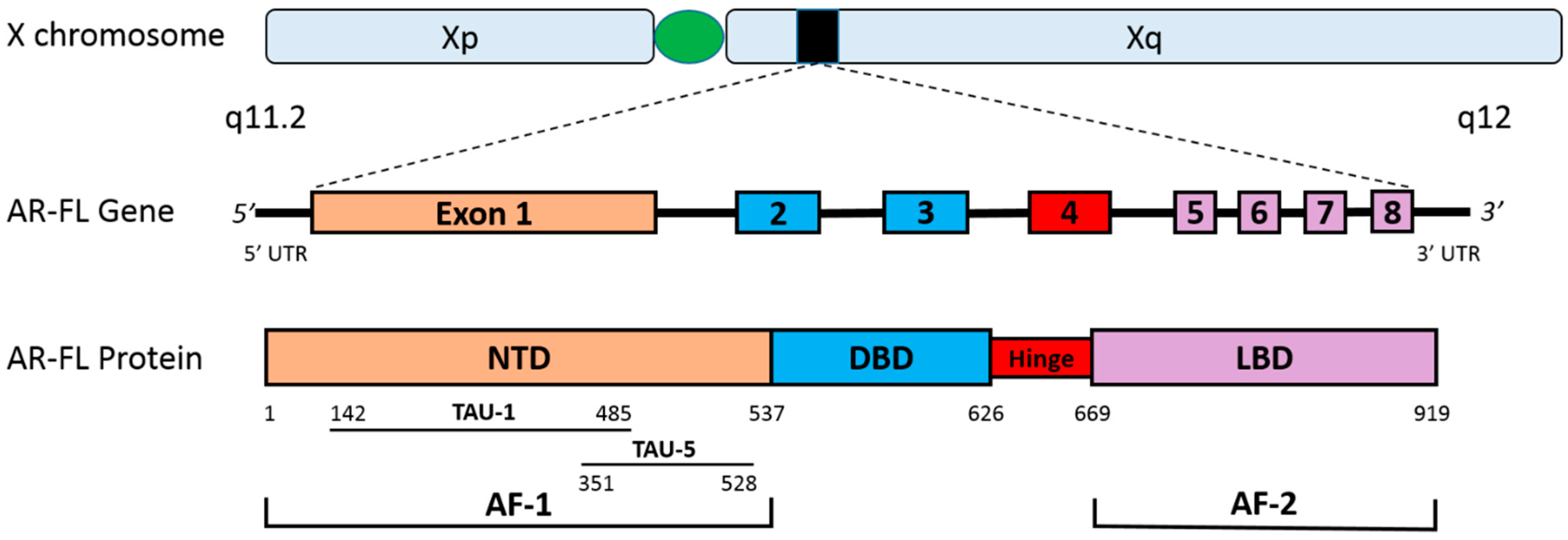
2. The Human Androgen Receptor
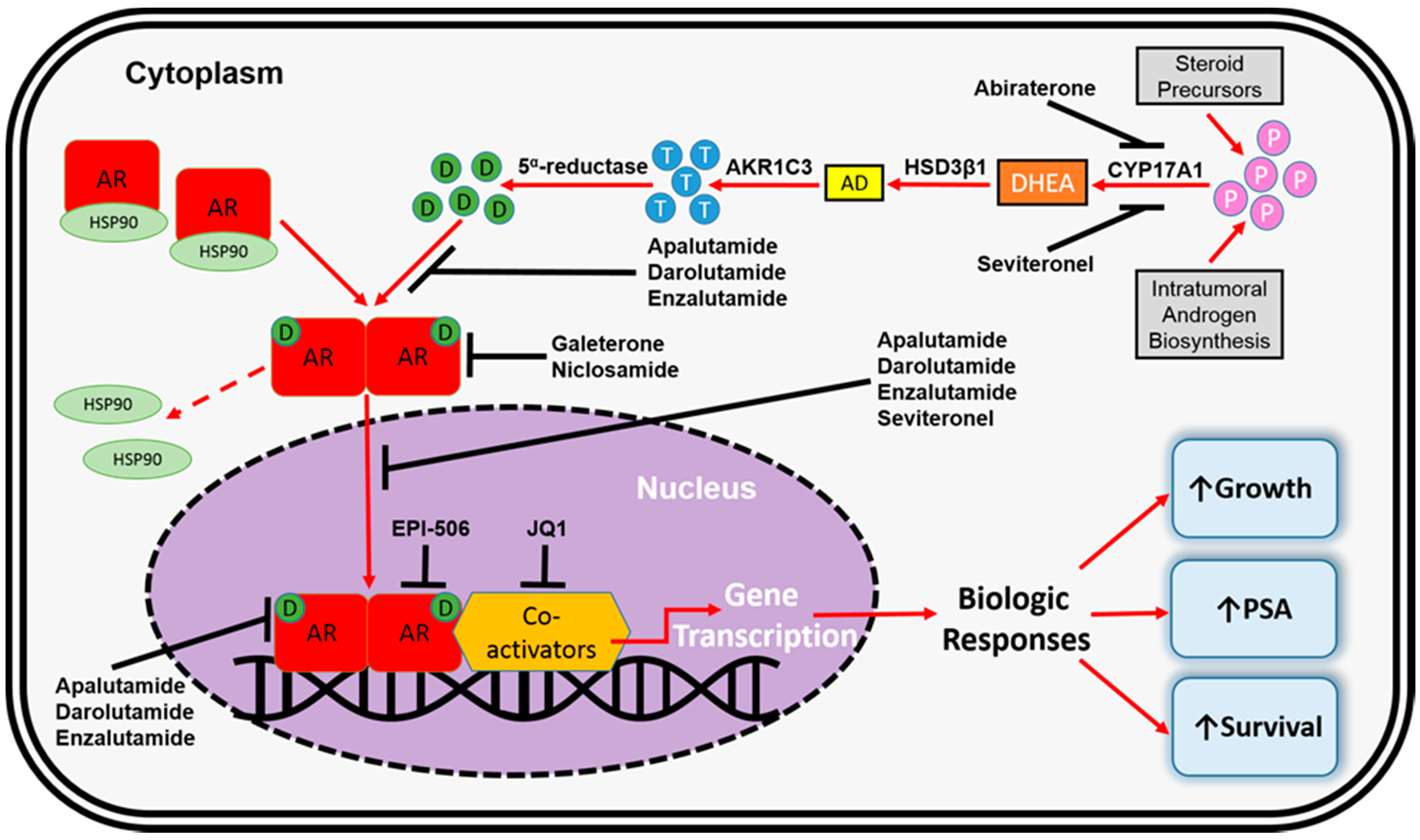
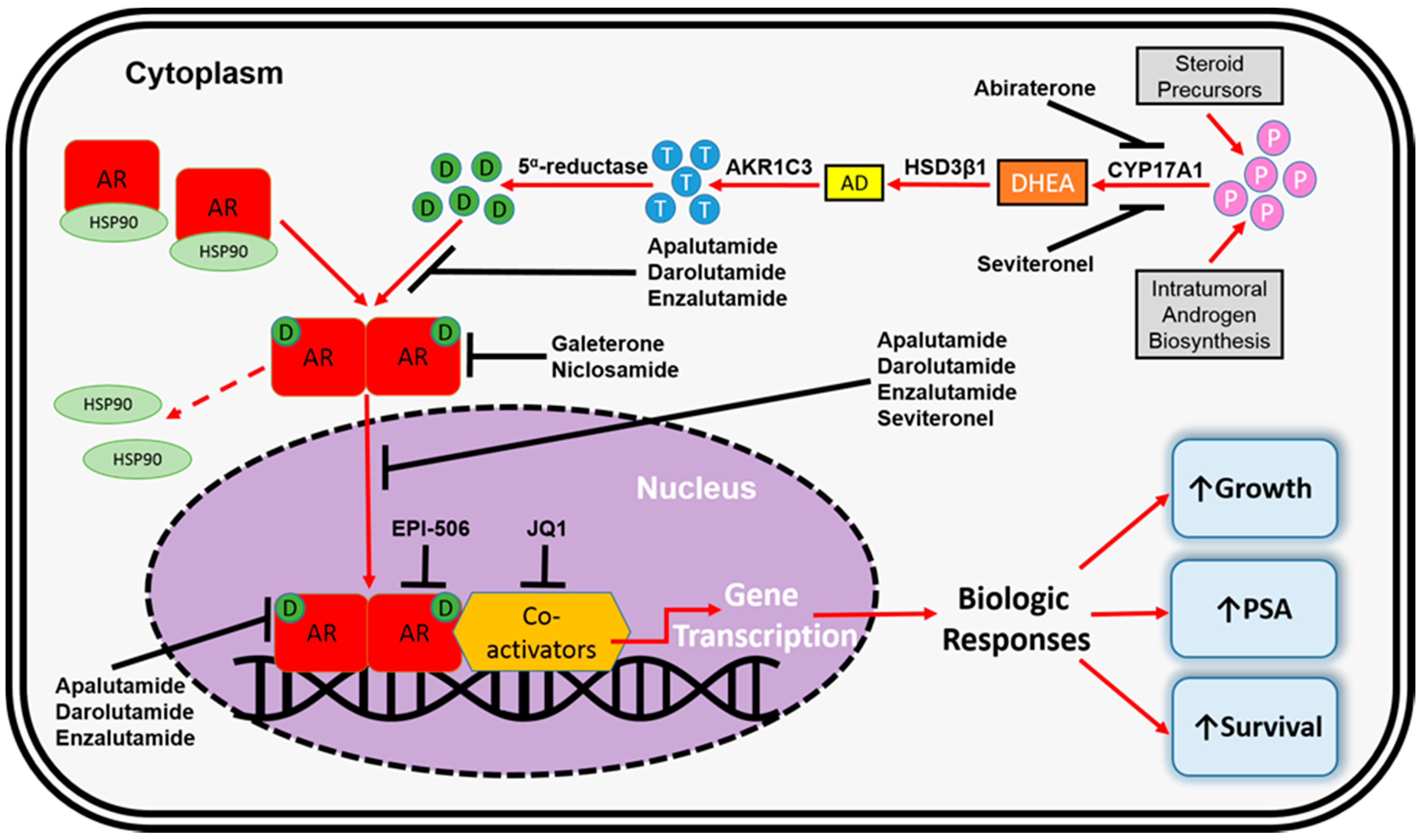
3. FDA-Approved Pharmacotherapeutics for mCRPC
4. AR-Dependent Resistance Mechanisms
4.1. AR Amplification and Overexpression
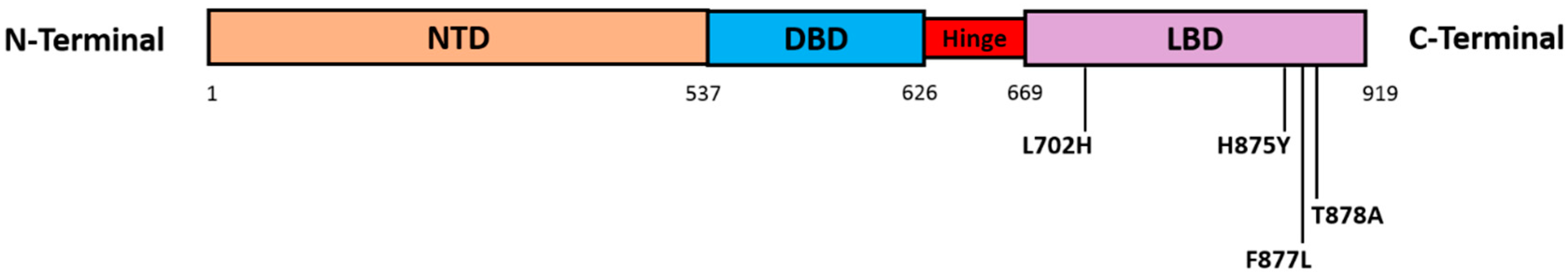
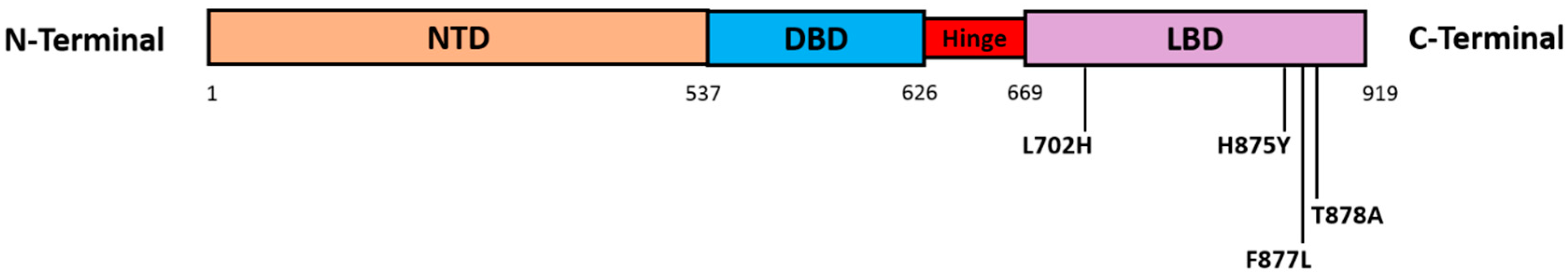
4.2. AR Point Mutations
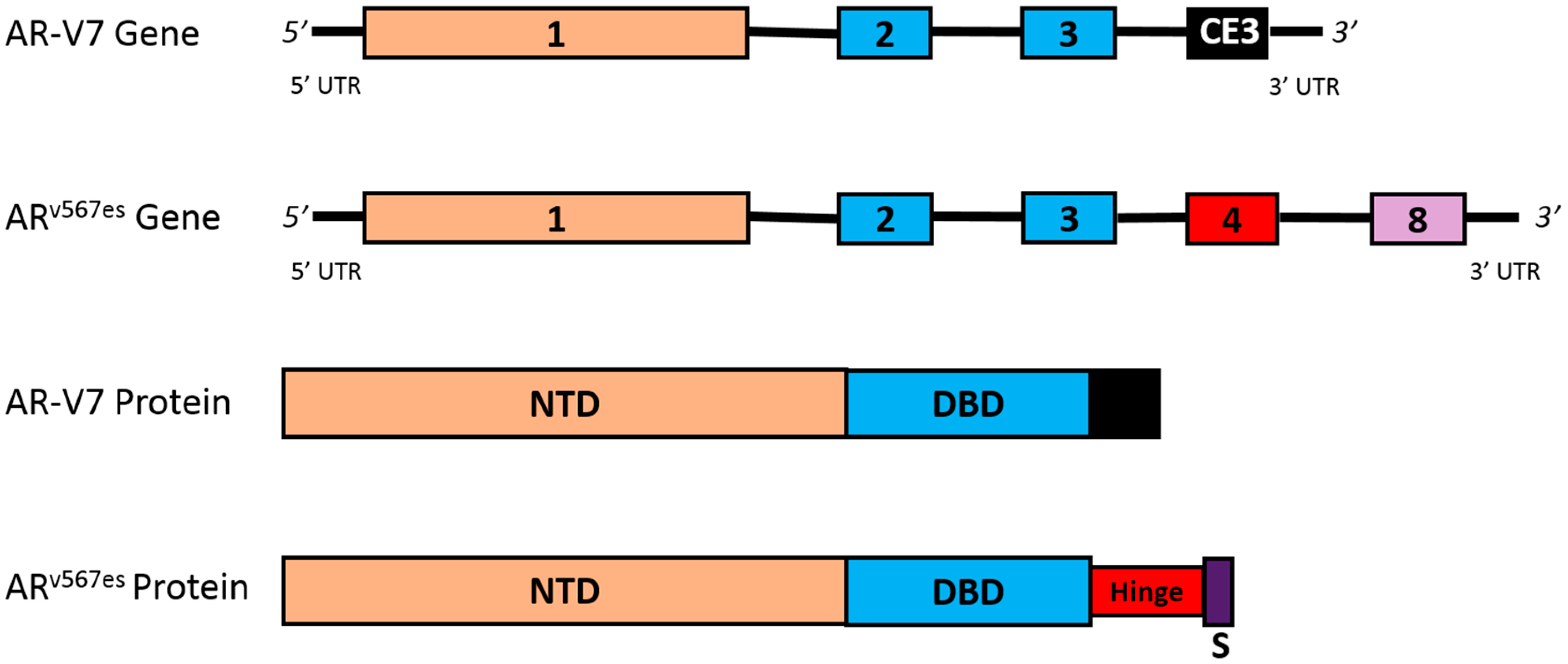
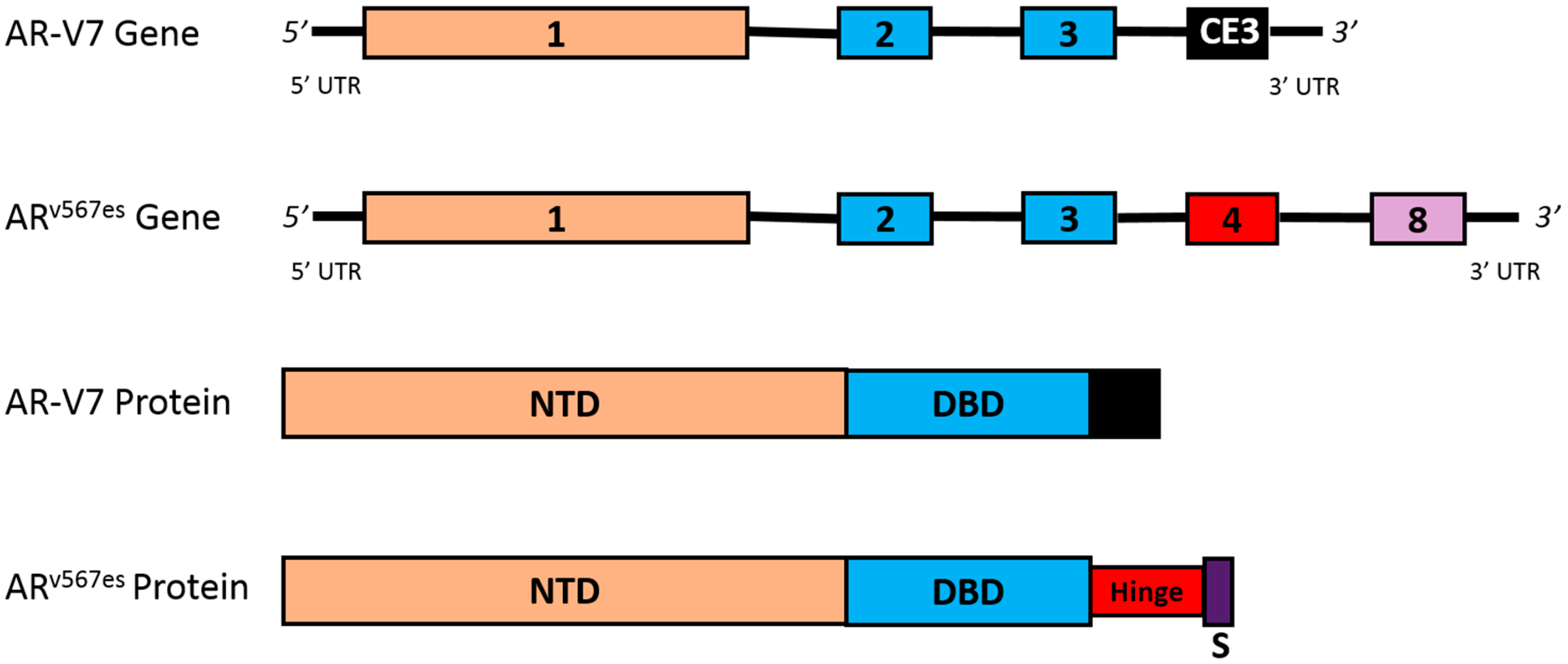
4.3. AR Splice Variants
4.4. Altered Steroidogenesis
5. AR-Independent Resistance Mechanisms
5.1. Glucocorticoid Receptor Activation
5.2. Immune-Mediated Resistance Mechanisms
5.3. Neuroendocrine Differentiation
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures 2017; American Cancer Society: Atlanta, GA, USA, 2017. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Bishop, K.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; et al. Seercancer Statistics Review, 1975–2012; National Cancer Institute: Bethesda, MD, USA, 2015.
- Nelson, P.S. Molecular states underlying androgen receptor activation: A framework for therapeutics targeting androgen signaling in prostate cancer. J. Clin. Oncol. 2012, 30, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, K.E.; Penning, T.M. Partners in crime: Deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol. Metab. 2010, 21, 315–324. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [PubMed]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Smith, M.R.; Fizazi, K.; Saad, F.; Mulders, P.F.; Sternberg, C.N.; Miller, K.; Logothetis, C.J.; Shore, N.D.; Small, E.J.; et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): Final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015, 16, 152–160. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Gelmann, E.P. Molecular biology of the androgen receptor. J. Clin. Oncol. 2002, 20, 3001–3015. [Google Scholar] [CrossRef] [PubMed]
- Claessens, F.; Denayer, S.; Van Tilborgh, N.; Kerkhofs, S.; Helsen, C.; Haelens, A. Diverse roles of androgen receptor (AR) domains in AR-mediated signaling. Nucl. Recept. Signal. 2008, 6, e008. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen receptor in prostate cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef] [PubMed]
- Jenster, G.; van der Korput, H.A.; Trapman, J.; Brinkmann, A.O. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J. Biol. Chem. 1995, 270, 7341–7346. [Google Scholar] [PubMed]
- Lonergan, P.E.; Tindall, D.J. Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 2011, 10, 20. [Google Scholar] [PubMed]
- Dehm, S.M.; Regan, K.M.; Schmidt, L.J.; Tindall, D.J. Selective role of an NH2-terminal WxxLF motif for aberrant androgen receptor activation in androgen depletion independent prostate cancer cells. Cancer Res. 2007, 67, 10067–10077. [Google Scholar] [CrossRef] [PubMed]
- Jentzmik, F.; Azoitei, A.; Zengerling, F.; Damjanoski, I.; Cronauer, M.V. Androgen receptor aberrations in the era of abiraterone and enzalutamide. World J. Urol. 2016, 34, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Saporita, A.J.; Zhang, Q.; Navai, N.; Dincer, Z.; Hahn, J.; Cai, X.; Wang, Z. Identification and characterization of a ligand-regulated nuclear export signal in androgen receptor. J. Biol. Chem. 2003, 278, 41998–42005. [Google Scholar] [CrossRef] [PubMed]
- Van Royen, M.E.; van Cappellen, W.A.; de Vos, C.; Houtsmuller, A.B.; Trapman, J. Stepwise androgen receptor dimerization. J. Cell Sci. 2012, 125, 1970–1979. [Google Scholar] [CrossRef] [PubMed]
- Oudard, S. Progress in emerging therapies for advanced prostate cancer. Cancer Treat. Rev. 2013, 39, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Tangen, C.M.; Hussain, M.H.; Lara, P.N., Jr.; Jones, J.A.; Taplin, M.E.; Burch, P.A.; Berry, D.; Moinpour, C.; Kohli, M.; et al. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; de Wit, R.; Berry, W.R.; Horti, J.; Pluzanska, A.; Chi, K.N.; Oudard, S.; Theodore, C.; James, N.D.; Turesson, I.; et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N. Engl. J. Med. 2004, 351, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef]
- Sweeney, C.J.; Chen, Y.H.; Carducci, M.; Liu, G.; Jarrard, D.F.; Eisenberger, M.; Wong, Y.N.; Hahn, N.; Kohli, M.; Cooney, M.M.; et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N. Engl. J. Med. 2015, 373, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Potter, G.A.; Barrie, S.E.; Jarman, M.; Rowlands, M.G. Novel steroidal inhibitors of human cytochrome P45017 alpha (17 alpha-hydroxylase-c17,20-lyase): Potential agents for the treatment of prostatic cancer. J. Med. Chem. 1995, 38, 2463–2471. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Reid, A.H.; Olmos, D.; de Bono, J.S. Antitumor activity with CYP17 blockade indicates that castration-resistant prostate cancer frequently remains hormone driven. Cancer Res. 2009, 69, 4937–4940. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N. Engl. J. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; de Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.S.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N. Engl. J. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.; Ouk, S.; Clegg, N.J.; Chen, Y.; Watson, P.A.; Arora, V.; Wongvipat, J.; Smith-Jones, P.M.; Yoo, D.; Kwon, A.; et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009, 324, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Shen, L.; et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomised open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fossa, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M.; et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Toren, P.J.; Kim, S.; Pham, S.; Mangalji, A.; Adomat, H.; Guns, E.S.; Zoubeidi, A.; Moore, W.; Gleave, M.E. Anticancer activity of a novel selective CYP17A1 inhibitor in preclinical models of castrate-resistant prostate cancer. Mol. Cancer Ther. 2015, 14, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Moilanen, A.M.; Riikonen, R.; Oksala, R.; Ravanti, L.; Aho, E.; Wohlfahrt, G.; Nykanen, P.S.; Tormakangas, O.P.; Palvimo, J.J.; Kallio, P.J. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci. Rep. 2015, 5, 12007. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, W.; Zhu, Y.; Nadiminty, N.; Schwartz, C.T.; Evans, C.P.; Gao, A.C. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2014, 20, 3198–3210. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.J.; Mawji, N.R.; Wang, J.; Wang, G.; Haile, S.; Myung, J.K.; Watt, K.; Tam, T.; Yang, Y.C.; Banuelos, C.A.; et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell 2010, 17, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.K.; Banuelos, C.A.; Fernandez, J.G.; Mawji, N.R.; Wang, J.; Tien, A.H.; Yang, Y.C.; Tavakoli, I.; Haile, S.; Watt, K.; et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J. Clin. Investig. 2013, 123, 2948–2960. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Beer, T.M.; Higano, C.S.; Anand, A.; Taplin, M.E.; Efstathiou, E.; Rathkopf, D.; Shelkey, J.; Yu, E.Y.; Alumkal, J.; et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: A phase 1–2 study. Lancet 2010, 375, 1437–1446. [Google Scholar] [CrossRef]
- Schrader, A.J.; Boegemann, M.; Ohlmann, C.H.; Schnoeller, T.J.; Krabbe, L.M.; Hajili, T.; Jentzmik, F.; Stoeckle, M.; Schrader, M.; Herrmann, E.; et al. Enzalutamide in castration-resistant prostate cancer patients progressing after docetaxel and abiraterone. Eur. Urol. 2014, 65, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Noonan, K.L.; North, S.; Bitting, R.L.; Armstrong, A.J.; Ellard, S.L.; Chi, K.N. Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Ann. Oncol. 2013, 24, 1802–1807. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Bishop, A.C.; Alyamani, M.; Garcia, J.A.; Dreicer, R.; Bunch, D.; Liu, J.; Upadhyay, S.K.; Auchus, R.J.; Sharifi, N. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature 2015, 523, 347–351. [Google Scholar] [CrossRef] [PubMed]
- Crona, D.J.; Milowsky, M.I.; Whang, Y.E. Androgen receptor targeting drugs in castration-resistant prostate cancer and mechanisms of resistance. Clin. Pharmacol. Ther. 2015, 98, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N. A road map to comprehensive androgen receptor axis targeting for castration-resistant prostate cancer. Cancer Res. 2013, 73, 4599–4605. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Sawyers, C.L. Biology of progressive, castration-resistant prostate cancer: Directed therapies targeting the androgen-receptor signaling axis. J. Clin. Oncol. 2005, 23, 8253–8261. [Google Scholar] [CrossRef] [PubMed]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.; Krishna, N.S.; Grigor, K.M.; Bartlett, J.M. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br. J. Cancer 2003, 89, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Taplin, M.E.; Bubley, G.J.; Shuster, T.D.; Frantz, M.E.; Spooner, A.E.; Ogata, G.K.; Keer, H.N.; Balk, S.P. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med. 1995, 332, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Welsbie, D.S.; Tran, C.; Baek, S.H.; Chen, R.; Vessella, R.; Rosenfeld, M.G.; Sawyers, C.L. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004, 10, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Loriot, Y.; Beraldi, E.; Zhang, F.; Wyatt, A.W.; Al Nakouzi, N.; Mo, F.; Zhou, T.; Kim, Y.; Monia, B.P.; et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cancer cell growth. Clin. Cancer Res. 2015, 21, 1675–1687. [Google Scholar] [CrossRef] [PubMed]
- Mostaghel, E.A.; Marck, B.T.; Plymate, S.R.; Vessella, R.L.; Balk, S.; Matsumoto, A.M.; Nelson, P.S.; Montgomery, R.B. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: Induction of steroidogenesis and androgen receptor splice variants. Clin. Cancer Res. 2011, 17, 5913–5925. [Google Scholar] [CrossRef] [PubMed]
- Romanel, A.; Gasi Tandefelt, D.; Conteduca, V.; Jayaram, A.; Casiraghi, N.; Wetterskog, D.; Salvi, S.; Amadori, D.; Zafeiriou, Z.; Rescigno, P.; et al. Plasma AR and abiraterone-resistant prostate cancer. Sci. Transl. Med. 2015, 7, 312re310. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Wetterskog, D.; Sharabiani, M.T.A.; Grande, E.; Fernandez-Perez, M.P.; Jayaram, A.; Salvi, S.; Castellano, D.; Romanel, A.; Lolli, C.; et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: A multi-institution correlative biomarker study. Ann. Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Azad, A.A.; Volik, S.V.; Wyatt, A.W.; Haegert, A.; Le Bihan, S.; Bell, R.H.; Anderson, S.A.; McConeghy, B.; Shukin, R.; Bazov, J.; et al. Androgen receptor gene aberrations in circulating cell-free DNA: Biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin. Cancer Res. 2015, 21, 2315–2324. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, A.W.; Azad, A.A.; Volik, S.V.; Annala, M.; Beja, K.; McConeghy, B.; Haegert, A.; Warner, E.W.; Mo, F.; Brahmbhatt, S.; et al. Genomic alterations in cell-free DNA and enzalutamide resistance in castration-resistant prostate cancer. JAMA Oncol. 2016, 2, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, I.; Day, T.K.; Tilley, W.D.; Selth, L.A. Androgen receptor signaling in castration-resistant prostate cancer: A lesson in persistence. Endocr. Relat. Cancer 2016, 23, T179–T197. [Google Scholar] [CrossRef] [PubMed]
- Boudadi, K.; Antonarakis, E.S. Resistance to novel antiandrogen therapies in metastatic castration-resistant prostate cancer. Clin. Med. Insights Oncol. 2016, 10, 1–9. [Google Scholar] [PubMed]
- Cai, C.; Chen, S.; Ng, P.; Bubley, G.J.; Nelson, P.S.; Mostaghel, E.A.; Marck, B.; Matsumoto, A.M.; Simon, N.I.; Wang, H.; et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011, 71, 6503–6513. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.J.; Sowalsky, A.G.; Gao, S.; Cai, C.; Voznesensky, O.; Schaefer, R.; Loda, M.; True, L.D.; Ye, H.; Troncoso, P.; et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin. Cancer Res. 2015, 21, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, G.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimont, B.; et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Korn, J.M.; Gao, X.; Rakiec, D.P.; Ruddy, D.A.; Doshi, S.; Yuan, J.; Kovats, S.G.; Kim, S.; Cooke, V.G.; et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discov. 2013, 3, 1030–1043. [Google Scholar] [CrossRef] [PubMed]
- Balbas, M.D.; Evans, M.J.; Hosfield, D.J.; Wongvipat, J.; Arora, V.K.; Watson, P.A.; Chen, Y.; Greene, G.L.; Shen, Y.; Sawyers, C.L.; et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife 2013, 2, e00499. [Google Scholar] [CrossRef] [PubMed]
- Moore, W.R.; Norris, N.J.; Wardell, S.; Eisner, J.R.; Hoekstra, W.J.; Schotzinger, R.J.; McDonnell, D.P. Direct effects of the selective CYP17 lyase (L) inhibitor, VT-464, on the androgen receptor (AR) and its oral activity in an F876L tumor mouse xenograft model. J. Clin. Oncol. 2015, 33, 263. [Google Scholar] [CrossRef]
- Norris, J.D.; Ellison, S.J.; Baker, J.G.; Stagg, D.B.; Wardell, S.E.; Park, S.; Alley, H.M.; Baldi, R.M.; Yllanes, A.; Andreano, K.J.; et al. Androgen receptor antagonism drives cytochrome P450 17A1 inhibitor efficacy in prostate cancer. J. Clin. Investig. 2017, 127, 2326–2338. [Google Scholar] [CrossRef] [PubMed]
- Carreira, S.; Romanel, A.; Goodall, J.; Grist, E.; Ferraldeschi, R.; Miranda, S.; Prandi, D.; Lorente, D.; Frenel, J.S.; Pezaro, C.; et al. Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med. 2014, 6, 254ra125. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Luo, J. Decoding the androgen receptor splice variants. Transl. Androl. Urol. 2013, 2, 178–186. [Google Scholar] [PubMed]
- Chan, S.C.; Dehm, S.M. Constitutive activity of the androgen receptor. Adv. Pharmacol. 2014, 70, 327–366. [Google Scholar] [PubMed]
- Hu, R.; Dunn, T.A.; Wei, S.; Isharwal, S.; Veltri, R.W.; Humphreys, E.; Han, M.; Partin, A.W.; Vessella, R.L.; Isaacs, W.B. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009, 69, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Ware, K.E.; Garcia-Blanco, M.A.; Armstrong, A.J.; Dehm, S.M. Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocr. Relat. Cancer 2014, 21, T87–T103. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Investig. 2010, 120, 2715–2730. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Lu, C.; Mostaghel, E.A.; Yegnasubramanian, S.; Gurel, M.; Tannahill, C.; Edwards, J.; Isaacs, W.B.; Nelson, P.S.; Bluemn, E.; et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012, 72, 3457–3462. [Google Scholar] [CrossRef] [PubMed]
- Hornberg, E.; Ylitalo, E.B.; Crnalic, S.; Antti, H.; Stattin, P.; Widmark, A.; Bergh, A.; Wikstrom, P. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PLoS ONE 2011, 6, e19059. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, M.; Lu, C.; Chen, Y.; Paller, C.J.; Carducci, M.A.; Eisenberger, M.A.; Luo, J.; Antonarakis, E.S. Serial blood-based analysis of AR-V7 in men with advanced prostate cancer. Ann. Oncol. 2015, 26, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Nakazawa, M.; Nadal, R.; Paller, C.J.; Denmeade, S.R.; Carducci, M.A.; et al. Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration-resistant prostate cancer. JAMA Oncol. 2015, 1, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Armstrong, C.; Zhu, Y.; Lou, W.; Gao, A.C. Niclosamide enhances abiraterone treatment via inhibition of androgen receptor variants in castration resistant prostate cancer. Oncotarget 2016, 7, 32210–32220. [Google Scholar] [CrossRef] [PubMed]
- Kwegyir-Afful, A.K.; Ramalingam, S.; Purushottamachar, P.; Ramamurthy, V.P.; Njar, V.C. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget 2015, 6, 27440–27460. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Banuelos, C.A.; Tavakoli, I.; Tien, A.H.; Caley, D.P.; Mawji, N.R.; Li, Z.; Wang, J.; Yang, Y.C.; Imamura, Y.; Yan, L.; et al. Sintokamide a is a novel antagonist of androgen receptor that uniquely binds activation function-1 in its amino-terminal domain. J. Biol. Chem. 2016, 291, 22231–22243. [Google Scholar] [CrossRef] [PubMed]
- Heemers, H.V.; Tindall, D.J. Androgen receptor (AR) coregulators: A diversity of functions converging on and regulating the ar transcriptional complex. Endocr. Rev. 2007, 28, 778–808. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Wilder-Romans, K.; Dommeti, V.L.; Krishnamurthy, P.M.; Apel, I.J.; Escara-Wilke, J.; Plymate, S.R.; Navone, N.M.; Wang, S.; Feng, F.Y.; et al. BET bromodomain inhibitors enhance efficacy and disrupt resistance to ar antagonists in the treatment of prostate cancer. Mol. Cancer Res. 2016, 14, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.C.; Selth, L.A.; Li, Y.; Nyquist, M.D.; Miao, L.; Bradner, J.E.; Raj, G.V.; Tilley, W.D.; Dehm, S.M. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res. 2015, 43, 5880–5897. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lou, W.; Zhu, Y.; Yang, J.C.; Nadiminty, N.; Gaikwad, N.W.; Evans, C.P.; Gao, A.C. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. 2015, 75, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Sung, C.C.; Schultz, N.; Danila, D.C.; He, B.; Eedunuri, V.K.; Fleisher, M.; Sander, C.; Sawyers, C.L.; Scher, H.I. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012, 72, 6142–6152. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.B.; Mostaghel, E.A.; Vessella, R.; Hess, D.L.; Kalhorn, T.F.; Higano, C.S.; True, L.D.; Nelson, P.S. Maintenance of intratumoral androgens in metastatic prostate cancer: A mechanism for castration-resistant tumor growth. Cancer Res. 2008, 68, 4447–4454. [Google Scholar] [CrossRef] [PubMed]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006, 66, 2815–2825. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Li, R.; Kuri, B.; Lotan, Y.; Roehrborn, C.G.; Liu, J.; Vessella, R.; Nelson, P.S.; Kapur, P.; Guo, X.; et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell 2013, 154, 1074–1084. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Casadio, V.; Conteduca, V.; Burgio, S.L.; Menna, C.; Bianchi, E.; Rossi, L.; Carretta, E.; Masini, C.; Amadori, D.; et al. Circulating cell-free AR and CYP17A1 copy number variations may associate with outcome of metastatic castration-resistant prostate cancer patients treated with abiraterone. Br. J. Cancer 2015, 112, 1717–1724. [Google Scholar] [CrossRef] [PubMed]
- Buttigliero, C.; Tucci, M.; Bertaglia, V.; Vignani, F.; Bironzo, P.; Di Maio, M.; Scagliotti, G.V. Understanding and overcoming the mechanisms of primary and acquired resistance to abiraterone and enzalutamide in castration resistant prostate cancer. Cancer Treat. Rev. 2015, 41, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Chism, D.D.; De Silva, D.; Whang, Y.E. Mechanisms of acquired resistance to androgen receptor targeting drugs in castration-resistant prostate cancer. Expert Rev. Anticancer Ther. 2014, 14, 1369–1378. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Cheng, H.; Lin, D.; Liu, L.; Yang, O.; Jia, L.; Fazli, L.; Gleave, M.E.; Wang, Y.; Rennie, P.; et al. The expression of glucocorticoid receptor is negatively regulated by active androgen receptor signaling in prostate tumors. Int. J. Cancer 2015, 136, E27–E38. [Google Scholar] [CrossRef] [PubMed]
- Arora, V.K.; Schenkein, E.; Murali, R.; Subudhi, S.K.; Wongvipat, J.; Balbas, M.D.; Shah, N.; Cai, L.; Efstathiou, E.; Logothetis, C.; et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell 2013, 155, 1309–1322. [Google Scholar] [CrossRef] [PubMed]
- Isikbay, M.; Otto, K.; Kregel, S.; Kach, J.; Cai, Y.; Vander Griend, D.J.; Conzen, S.D.; Szmulewitz, R.Z. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm. Cancer 2014, 5, 72–89. [Google Scholar] [CrossRef] [PubMed]
- Kroon, J.; Puhr, M.; Buijs, J.T.; van der Horst, G.; Hemmer, D.M.; Marijt, K.A.; Hwang, M.S.; Masood, M.; Grimm, S.; Storm, G.; et al. Glucocorticoid receptor antagonism reverts docetaxel resistance in human prostate cancer. Endocr. Relat. Cancer 2016, 23, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; van den Eertwegh, A.J.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184–043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef]
- Gevensleben, H.; Dietrich, D.; Golletz, C.; Steiner, S.; Jung, M.; Thiesler, T.; Majores, M.; Stein, J.; Uhl, B.; Muller, S.; et al. The immune checkpoint regulator PD-L1 is highly expressed in aggressive primary prostate cancer. Clin. Cancer Res. 2016, 22, 1969–1977. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.L.; Sio, A.; Angeles, A.; Roberts, M.E.; Azad, A.A.; Chi, K.N.; Zoubeidi, A. PD-L11 is highly expressed in enzalutamide resistant prostate cancer. Oncotarget 2015, 6, 234–242. [Google Scholar] [PubMed]
- Graff, J.N.; Alumkal, J.J.; Drake, C.G.; Thomas, G.V.; Redmond, W.L.; Farhad, M.; Cetnar, J.P.; Ey, F.S.; Bergan, R.C.; Slottke, R.; et al. Early evidence of anti-PD-1 activity in enzalutamide-resistant prostate cancer. Oncotarget 2016, 7, 52810–52817. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S. Defining and targeting the oncogenic drivers of neuroendocrine prostate cancer. Cancer Cell 2016, 29, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.; Varambally, S.; et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, A.M.; Shen, L.; Tapia, E.L.; Lu, J.F.; Chen, H.C.; Zhang, J.; Wu, G.; Wang, X.; Troncoso, P.; Corn, P.; et al. Combined tumor suppressor defects characterize clinically defined aggressive variant prostate cancers. Clin. Cancer Res. 2016, 22, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Sood, A.; Rahimi, H.A.; Wang, W.; Gupta, N.; Hicks, J.; Mosier, S.; Gocke, C.D.; Epstein, J.I.; Netto, G.J.; et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin. Cancer Res. 2014, 20, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbe, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.C.; Wongvipat, J.; Ku, S.Y.; Gao, D.; Cao, Z.; et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Rickman, D.S.; Park, K.; Chae, S.S.; Sboner, A.; MacDonald, T.Y.; Wang, Y.; Sheikh, K.L.; Terry, S.; Tagawa, S.T.; et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, J.M.; Beltran, H.; Park, K.; MacDonald, T.Y.; Robinson, B.D.; Tagawa, S.T.; Perner, S.; Bismar, T.A.; Erbersdobler, A.; Dhir, R.; et al. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia 2013, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-Myc drives neuroendocrine prostate cancer initiated from human prostate epithelial cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crona, D.J.; Whang, Y.E. Androgen Receptor-Dependent and -Independent Mechanisms Involved in Prostate Cancer Therapy Resistance. Cancers 2017, 9, 67. https://doi.org/10.3390/cancers9060067
Crona DJ, Whang YE. Androgen Receptor-Dependent and -Independent Mechanisms Involved in Prostate Cancer Therapy Resistance. Cancers. 2017; 9(6):67. https://doi.org/10.3390/cancers9060067
Chicago/Turabian StyleCrona, Daniel J., and Young E. Whang. 2017. "Androgen Receptor-Dependent and -Independent Mechanisms Involved in Prostate Cancer Therapy Resistance" Cancers 9, no. 6: 67. https://doi.org/10.3390/cancers9060067
APA StyleCrona, D. J., & Whang, Y. E. (2017). Androgen Receptor-Dependent and -Independent Mechanisms Involved in Prostate Cancer Therapy Resistance. Cancers, 9(6), 67. https://doi.org/10.3390/cancers9060067