
Dysregulation of miRNA Expression in Cancer Associated Fibroblasts (CAFs) and Its Consequences on the Tumor Microenvironment


Abstract
:1. Introduction
2. Methods
- ([mir] OR [miR] OR [microRNA]) AND [cancer] AND ([caf] OR [cancer associated fibroblasts]) = 184;
- ([mir] OR [miR] OR [microRNA]) AND [cancer] AND ([hsc] OR [hepatic stellate cells]) = 64;
- ([mir] OR [miR] OR [microRNA]) AND [cancer] AND ([psc] OR [pancreatic stellate cells]) = 15.
3. Clinical Relevance of miR in CAF
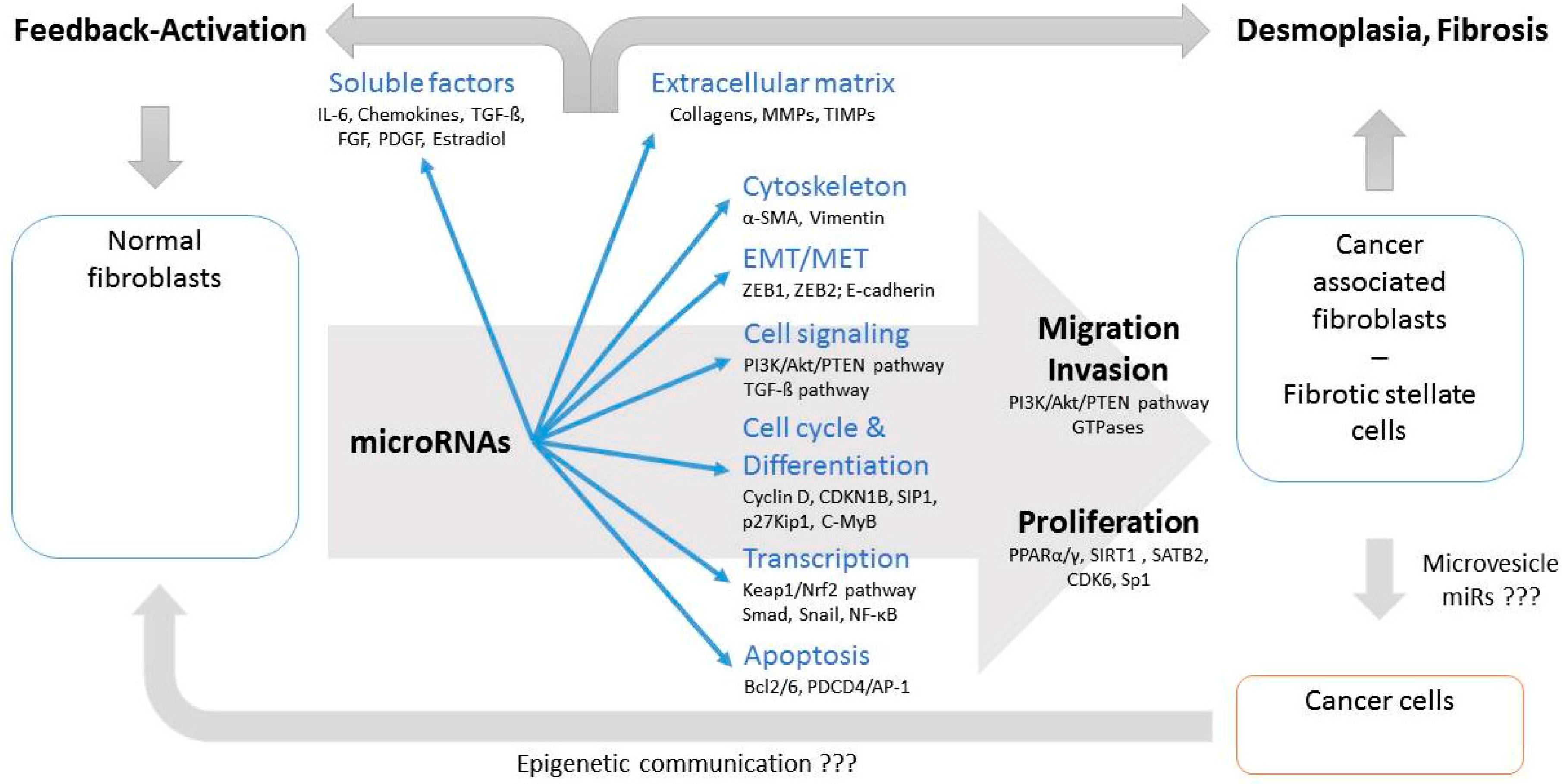
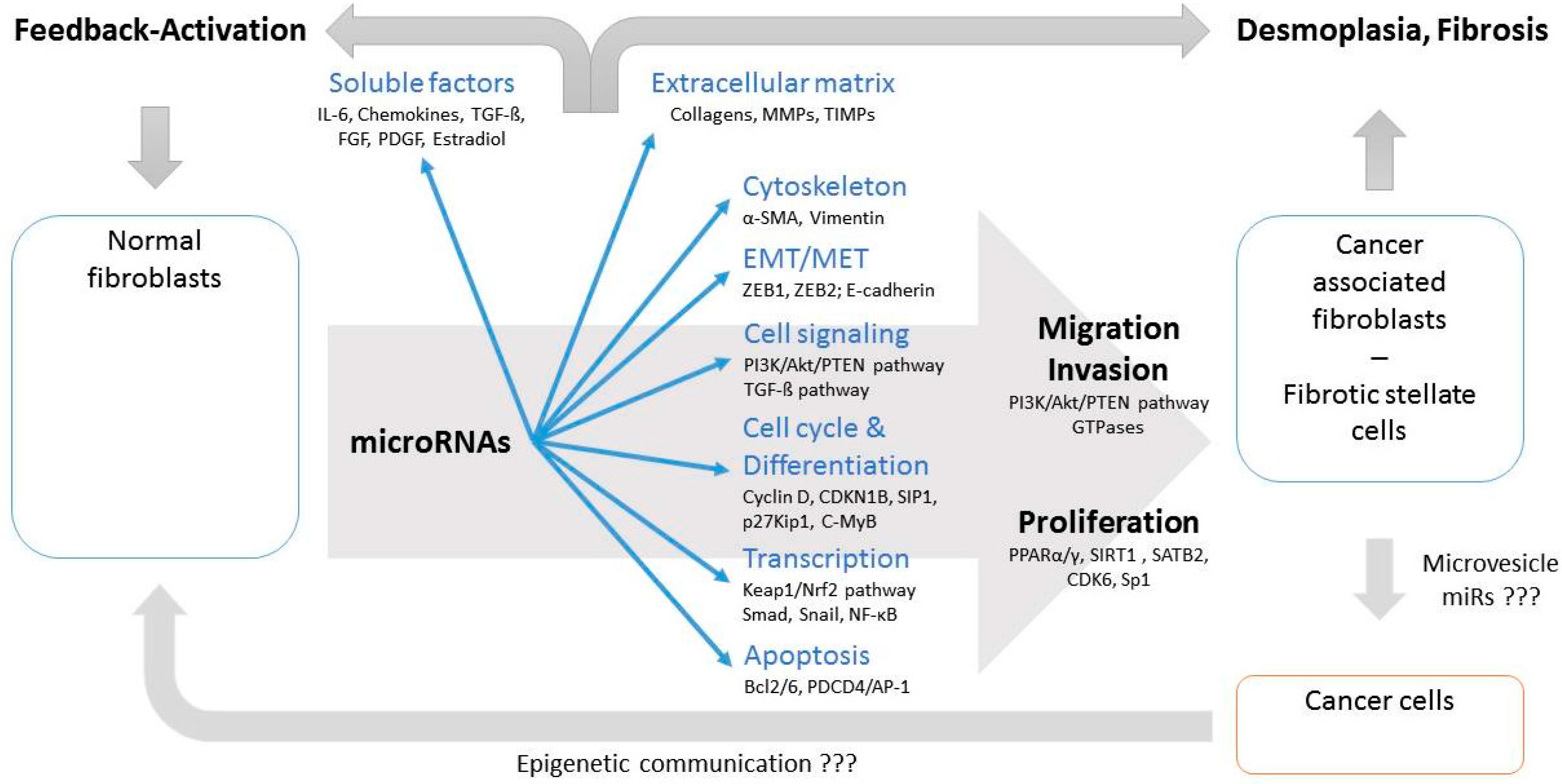
4. Functional Consequences of miRNA Dysregulation in CAFs
4.1. EMT/MET Switch
4.2. TGF-β Signaling
4.3. Extracellular Matrix, Migration and Invasion
4.4. Soluble Factors
4.5. Cell Cycle and Proliferation
5. MiR-Targeted Readouts for Cellular Functions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- De Vlieghere, E.; Verset, L.; Demetter, P.; Bracke, M.; De Wever, O. Cancer-associated fibroblasts as target and tool in cancer therapeutics and diagnostics. Virchows Archiv 2015, 467, 367–382. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Zhang, Y.; Jia, T.; Sun, Y. Molecular mechanism underlying the tumor-promoting functions of carcinoma-associated fibroblasts. Tumor Biol. 2015, 36, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Hou, Y.; Yang, G.; Wang, X.; Tang, S.; Du, Y.E.; Yang, L.; Yu, T.; Zhang, H.; Zhou, M.; et al. Stromal miR-200s contribute to breast cancer cell invasion through CAF activation and ECM remodeling. Cell Death Differ. 2016, 23, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Suresh, R.; Banerjee, S.; Bao, B.; Xu, Z.; Wilson, J.; Philip, P.A.; Apte, M.; Sarkar, F.H. Contribution of microRNAs in understanding the pancreatic tumor microenvironment involving cancer associated stellate and fibroblast cells. Am. J. Cancer Res. 2015, 5, 1251–1264. [Google Scholar] [PubMed]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. MiRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef] [PubMed]
- Altuvia, Y.; Landgraf, P.; Lithwick, G.; Elefant, N.; Pfeffer, S.; Aravin, A.; Brownstein, M.J.; Tuschl, T.; Margalit, H. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res. 2005, 33, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Haier, J.; Ströse, A.; Matuszcak, C.; Hummel, R. MiR clusters target cellular functional complexes by defining their degree of regulatory freedom. Cancer Metastasis Rev. 2016, 35, 289–322. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xu, X.; Zhu, J.; Zhang, S.; Wu, Y.; Wu, Y.; Zhao, K.; Xing, C.; Cao, J.; Zhu, H.; et al. MiR-31 affects colorectal cancer cells by inhibiting autophagy in cancer-associated fibroblasts. Oncotarget 2016, 7, 79617–79628. [Google Scholar] [CrossRef] [PubMed]
- Aprelikova, O.; Palla, J.; Hibler, B.; Yu, X.; Greer, Y.E.; Yi, M.; Stephens, R.; Maxwell, G.L.; Jazaeri, A.; Risinger, J.I.; et al. Silencing of miR-148a in cancer-associated fibroblasts results in WNT10B-mediated stimulation of tumor cell motility. Oncogene 2013, 32, 3246–3253. [Google Scholar] [CrossRef] [PubMed]
- Aprelikova, O.; Yu, X.; Palla, J.; Wei, B.R.; John, S.; Yi, M.; Stephens, R.; Simpson, R.M.; Risinger, J.I.; Jazaeri, A.; et al. The role of miR-31 and its target gene SATB2 in cancer-associated fibroblasts. Cell Cycle 2010, 9, 4387–4398. [Google Scholar] [CrossRef] [PubMed]
- Verghese, E.T.; Drury, R.; Green, C.A.; Holliday, D.L.; Lu, X.; Nash, C.; Speirs, V.; Thorne, J.L.; Thygesen, H.H.; Zougman, A.; et al. MiR-26b is down-regulated in carcinoma associated fibroblasts from ER positive breast cancers leading to enhanced cell migration and invasion. J. Pathol. 2013, 231, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Baxter, E.W.; Chambers, P.A.; Green, C.A.; Hanby, A.M.; Hughes, T.A.; Nash, C.E.; Millican-Slater, R.A.; Stead, L.F.; Verghese, E.T.; et al. Down-Regulation of miR-92 in Breast Epithelial Cells and in Normal but Not Tumour Fibroblasts Contributes to Breast Carcinogenesis. PLoS ONE 2015, 10, e0139698. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Sun, Y.; Hou, Y.; Peng, Q.; Wang, L.; Luo, H.; Tang, X.; Zeng, Z.; Liu, M. MiRNA expression analysis of cancer-associated fibroblasts and normal fibroblasts in breast cancer. Int. J. Biochem. Cell Biol. 2012, 44, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Kurashige, J.; Mima, K.; Sawada, G.; Takahashi, Y.; Eguchi, H.; Sugimachi, K.; Mori, M.; Yanagihara, K.; Yashiro, M.; Hirakawa, K.; et al. Epigenetic modulation and repression of miR-200b by cancer-associated fibroblasts contribute to cancer invasion and peritoneal dissemination in gastric cancer. Carcinogenesis 2015, 36, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Omura, N.; Hong, S.M.; Vincent, A.; Walter, K.; Griffith, M.; Borges, M.; Goggins, M. Pancreatic cancers epigenetically silence SIP1 and hypomethylate and overexpress miR-200a/200b in association with elevated circulating miR-200a and miR-200b levels. Cancer Res. 2010, 70, 5226–5237. [Google Scholar] [CrossRef] [PubMed]
- Kadera, B.E.; Li, L.; Toste, P.A.; Wu, N.; Adams, C.; Dawson, D.W.; Donahue, T.R. MicroRNA-21 in pancreatic ductal adenocarcinoma tumor-associated fibroblasts promotes metastasis. PLoS ONE 2013, 8, e71978. [Google Scholar] [CrossRef] [PubMed]
- Donahue, T.R.; Nguyen, A.H.; Moughan, J.; Li, L.; Tatishchev, S.; Toste, P.; Farrell, J.J. Stromal microRNA-21 levels predict response to 5-fluorouracil in patients with pancreatic cancer. J. Surg. Oncol. 2014, 110, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Josson, S.; Gururajan, M.; Sung, S.Y.; Hu, P.; Shao, C.; Zhau, H.E.; Liu, C.; Lichterman, J.; Duan, P.; Li, Q.; et al. Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene 2015, 34, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, M.; Coppola, V.; Addario, A.; Patrizii, M.; Maugeri-Saccà, M.; Memeo, L.; Colarossi, C.; Francescangeli, F.; Biffoni, M.; Collura, D.; et al. Control of tumor and microenvironment cross-talk by miR-15a and miR-16 in prostate cancer. Oncogene 2011, 30, 4231–4242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, J.; Liu, Y.; Wu, W.; Li, X.; Wu, Y.; Chen, H.; Zhang, K.; Gu, L. MiR-101 represses lung cancer by inhibiting interaction of fibroblasts and cancer cells by down-regulating CXCL12. Biomed. Pharmacother. 2015, 74, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Doldi, V.; Callari, M.; Giannoni, E.; D'Aiuto, F.; Maffezzini, M.; Valdagni, R.; Chiarugi, P.; Gandellini, P.; Zaffaroni, N. Integrated gene and miRNA expression analysis of prostate cancer associated fibroblasts supports a prominent role for interleukin-6 in fibroblast activation. Oncotarget 2015, 6, 31441–31460. [Google Scholar] [PubMed]
- Yang, T.S.; Yang, X.H.; Chen, X.; Wang, X.D.; Hua, J.; Zhou, D.L.; Zhou, B.; Song, Z.S. MicroRNA-106b in cancer-associated fibroblasts from gastric cancer promotes cell migration and invasion by targeting PTEN. FEBS Lett. 2014, 588, 2162–2169. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Sakamoto, N.; Oue, N.; Yashiro, M.; Sentani, K.; Yanagihara, K.; Hirakawa, K.; Yasui, W. MicroRNA-143 regulates collagen type III expression in stromal fibroblasts of scirrhous type gastric cancer. Cancer Sci. 2014, 105, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Naito, Y.; Yasuno, K.; Tagawa, H.; Sakamoto, N.; Oue, N.; Yashiro, M.; Sentani, K.; Goto, K.; Shinmei, S.; Oo, H.Z.; et al. MicroRNA-145 is a potential prognostic factor of scirrhous type gastric cancer. Oncol. Rep. 2014, 32, 1720–1726. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Miyata, H.; Sugimura, K.; Fukuda, S.; Kanemuram, T.; Yamashita, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. miR-27 is associated with chemoresistance in esophageal cancer through transformation of normal fibroblasts to cancer-associated fibroblasts. Carcinogenesis 2015, 36, 894–903. [Google Scholar] [CrossRef] [PubMed]
- Yamamichi, N.; Shimomura, R.; Inada, K.; Sakurai, K.; Haraguchi, T.; Ozaki, Y.; Fujita, S.; Mizutani, T.; Furukawa, C.; Fujishiro, M.; et al. Locked nucleic acid in situ hybridization analysis of miR-21 expression during colorectal cancer development. Clin. Cancer Res. 2009, 15, 4009–4016. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Tang, N.; Wu, K.; Dai, W.; Ye, C.; Shi, J.; Zhang, J.; Ning, B.; Zeng, X.; Lin, Y. MiR-21 simultaneously regulates ERK1 signaling in HSC activation and hepatocyte EMT in hepatic fibrosis. PLoS ONE 2014, 9, e108005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zha, Y.; Hu, W.; Huang, Z.; Gao, Z.; Zang, Y.; Chen, J.; Dong, L.; Zhang, J. The autoregulatory feedback loop of microRNA-21/programmed cell death protein 4/activation protein-1 (MiR-21/PDCD4/AP-1) as a driving force for hepatic fibrosis development. J. Biol. Chem. 2013, 288, 37082–37093. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.J.; Ou-Yang, P.H.; Han, X.P. Profibrotic effect of miR-33a with Akt activation in hepatic stellate cells. Cell. Signal. 2014, 26, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wang, J.; Chen, Y.; Zhou, K.; Wen, J.; Wang, Y.; Zhou, Y.; Pan, W.; Cai, W. Up-regulation of miR-200b in biliary atresia patients accelerates proliferation and migration of hepatic stallate cells by activating PI3K/Akt signaling. Cell. Signal. 2014, 26, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Enomoto, M.; Fujii, H.; Sekiya, Y.; Yoshizato, K.; Ikeda, K.; et al. MicroRNA-221/222 upregulation indicates the activation of stellate cells and the progression of liver fibrosis. Gut 2012, 61, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Lakner, A.M.; Steuerwald, N.M.; Walling, T.L.; Ghosh, S.; Li, T.; McKillop, I.H.; Russo, M.W.; Bonkovsky, H.L.; Schrum, L.W. Inhibitory effects of microRNA 19b in hepatic stellate cell-mediated fibrogenesis. Hepatology 2012, 56, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Benz, F.; Vargas Cardenas, D.; Vucur, M.; Gautheron, J.; Schneider, A.; Hellerbrand, C.; Pottier, N.; Alder, J.; Tacke, F.; et al. MiR-30c and miR-193 are a part of the TGF-β-dependent regulatory network controlling extracellular matrix genes in liver fibrosis. J. Dig. Dis. 2015, 16, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Roderburg, C.; Luedde, M.; Vargas Cardenas, D.; Vucur, M.; Mollnow, T.; Zimmermann, H.W.; Koch, A.; Hellerbrand, C.; Weiskirchen, R.; Frey, N.; et al. MiR-133a mediates TGF-β-dependent derepression of collagen synthesis in hepatic stellate cells during liverfibrosis. J. Hepatol. 2013, 58, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yi, J.; Ye, R.; Liu, J.; Duan, Q.; Xiao, J.; Liu, F. MiR-144 regulates transforming growth factor-β1 induced hepatic stellate cell activation in human fibrotic liver. Int. J. Clin. Exp. Pathol. 2015, 8, 3994–4000. [Google Scholar] [PubMed]
- Murakami, Y.; Toyoda, H.; Tanaka, M.; Kuroda, M.; Harada, Y.; Matsuda, F.; Tajima, A.; Kosaka, N.; Ochiya, T.; Shimotohno, K. The progression of liver fibrosis is related with overexpression of the miR-199 and 200 families. PLoS ONE 2011, 6, e16081. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Guo, Y.; Chen, B.; Dong, P.; Zheng, J. MicroRNA-17–5p activates hepatic stellate cells through targeting of Smad7. Lab. Investig. 2015, 95, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, W.; Guo, K.; Xiao, Y.; Wang, Y.; Fan, J. MiR-181b promotes hepatic stellate cells proliferation by targeting p27 and is elevated in the serum of cirrhosis patients. Biochem. Biophys. Res. Commun. 2012, 421, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Nouraee, N.; Van Roosbroeck, K.; Vasei, M.; Semnani, S.; Samaei, N.M.; Naghshvar, F.; Omidi, A.A.; Calin, G.A.; Mowla, S.J. Expression, tissue distribution and function of miR-21 in esophageal squamous cell carcinoma. PLoS ONE 2013, 8, e73009. [Google Scholar] [CrossRef] [PubMed]
- Al-Ansari, M.M.; Aboussekhra, A. MiR-146b-5p mediates p16-dependent repression of IL-6 and suppresses paracrine procarcinogenic effects of breast stromal fibroblasts. Oncotarget 2015, 6, 30006–30016. [Google Scholar] [PubMed]
- Roybal, J.D.; Zang, Y.; Ahn, Y.H.; Yang, Y.; Gibbons, D.L.; Baird, B.N.; Alvarez, C.; Thilaganathan, N.; Liu, D.D.; Saintigny, P.; et al. miR-200 Inhibits lung adenocarcinoma cell invasion and metastasis by targeting Flt1/VEGFR1. Mol. Cancer Res. 2011, 9, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Donnarumma, E.; Fiore, D.; Nappa, M.; Roscigno, G.; Adamo, A.; Iaboni, M.; Russo, V.; Affinito, A.; Puoti, I.; Quintavalle, C.; et al. Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget 2017, 8, 19592–19608. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Tan, W.; Cheng, S.; Wang, H.; Ye, S.; Yu, C.; He, Y.; Zeng, J.; Cen, J.; Hu, J.; et al. Upregulation of microRNA-126 in hepatic stellate cells may affect pathogenesis of liver fibrosis through the NF-κB pathway. DNA Cell Biol. 2015, 34, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Gandellini, P.; Giannoni, E.; Casamichele, A.; Taddei, M.L.; Callari, M.; Piovan, C.; Valdagni, R.; Pierotti, M.A.; Zaffaroni, N.; Chiarugi, P. miR-205 hinders the malignant interplay between prostate cancer cells and associated fibroblasts. Antioxid. Redox Signal. 2014, 20, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, M.; Guo, M.; Xie, Y.; Cong, Y.S. MiR-127 regulates cell proliferation and senescence by targeting BCL6. PLoS ONE 2013, 8, e80266. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; He, Y.; Ma, T.T.; Huang, C.; Zhang, L.; Li, J. Participation of miR-200a in TGF-β1-mediated hepatic stellate cell activation. Mol. Cell. Biochem. 2014, 388, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lu, Y.; Lin, Y.Y.; Zheng, Z.Y.; Fang, J.H.; He, S.; Zhuang, S.M. Vascular mimicry formation is promoted by paracrine TGF-β and SDF1 of cancer-associated fibroblasts and inhibited by miR-101 in hepatocellular carcinoma. Cancer Lett. 2016, 383, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Zhang, H.; Zhang, J.; Zhao, S.; Zheng, X.; Zhang, Z.; Zhu, J.; Chen, J.; Dong, L.; Zang, Y.; et al. MicroRNA-101 suppresses liver fibrosis by targeting the TGFβ signalling pathway. J. Pathol. 2014, 234, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ghazwani, M.; Zhang, Y.; Lu, J.; Li, J.; Fan, J.; Gandhi, C.R.; Li, S. MiR-122 regulates collagen production via targeting hepatic stellate cells and suppressing P4HA1 expression. J. Hepatol. 2013, 58, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, C.Q.; Zhang, Z.Q.; Yao, D.K.; Zhu, L. Loss of expression of miR-335 is implicated in hepatic stellate cell migration and activation. Exp. Cell Res. 2011, 317, 1714–1725. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, C.; Liu, Q.; Liu, B.; Song, C.; Zhu, S.; Wu, C.; Liu, S.; Yu, H.; Yao, D.; et al. The role of the miR-31/FIH1 pathway in TGF-β-induced liver fibrosis. Clin. Sci. 2015, 129, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Kabir, T.D.; Leigh, R.J.; Tasena, H.; Mellone, M.; Coletta, R.D.; Parkinson, E.K.; Prime, S.S.; Thomas, G.J.; Paterson, I.C.; Zhou, D.; et al. A miR-335/COX-2/PTEN axis regulates the secretory phenotype of senescent cancer-associated fibroblasts. Aging 2016, 8, 1608–1635. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Feng, L.; Li, Z.; Xu, G.; Fan, X. MicroRNA-21 activates hepatic stellate cells via PTEN/Akt signaling. Biomed. Pharmacother. 2013, 67, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Qin, X.; Yan, M.; Li, R.; Chen, G.; Zhang, J.; Chen, W. Cancer-associated fibroblasts promote cancer cell growth through a miR-7-RASSF2-PAR-4 axis in the tumor microenvironment. Oncotarget 2017, 8, 1290–1303. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.K.; Jiang, J.; Kim, T.H.; Li, Y.; Wang, S.S.; Torok, N.J.; Wu, J.; Zern, M.A. Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells, and their overexpression causes decreased stellate cell activation. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G101–G106. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.J.; Nabinger, S.C.; Vega, Z.; Sahu, S.S.; Alluri, R.K.; Abdul-Sater, Z.; Yu, Z.; Gore, J.; Nalepa, G.; Saxena, R.; et al. Pathophysiological role of microRNA-29 in pancreatic cancer stroma. Sci. Rep. 2015, 5, 11450. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Tiao, M.M.; Huang, L.T.; Chuang, J.H.; Kuo, K.C.; Yang, Y.L.; Wang, F.S. Activation of Mir-29a in Activated Hepatic Stellate Cells Modulates Its Profibrogenic Phenotype through Inhibition of Histone Deacetylases 4. PLoS ONE 2015, 10, e0136453. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Niu, X.; Wang, Y.; Kong, L.; Wang, R.; Zhang, Y.; Zhao, S.; Nan, Y. MiR-146a-5p suppresses activation and proliferation of hepatic stellate cells in nonalcoholic fibrosing steatohepatitis through directly targeting Wnt1 and Wnt5a. Sci. Rep. 2015, 5, 16163. [Google Scholar] [CrossRef] [PubMed]
- Maubach, G.; Lim, M.C.; Chen, J.; Yang, H.; Zhuo, L. MiRNA studies in in vitro and in vivo activated hepatic stellate cells. World J. Gastroenterol. 2011, 17, 2748–2773. [Google Scholar] [CrossRef] [PubMed]
- Takikawa, T.; Masamune, A.; Hamada, S.; Nakano, E.; Yoshida, N.; Shimosegawa, T. MiR-210 regulates the interaction between pancreatic cancer cells and stellate cells. Biochem. Biophys. Res. Commun. 2013, 437, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ma, N.; Zhao, R.; Wu, G.; Zhang, Y.; Qiao, Y.; Han, D.; Xu, Y.; Xiang, Y.; Yan, B.; et al. Overexpression of miR-483–5p/3p cooperate to inhibit mouse liver fibrosis by suppressing the TGF-βstimulated HSCs in transgenic mice. J. Cell. Mol. Med. 2014, 18, 966–974. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Li, H.; Zhang, M.; Xu, J.; Lu, Y.; Zheng, Y.; Qian, J.; Chang, J.T.; Yang, J.; Yi, Q. p38 MAPK inhibits breast cancer metastasis through regulation of stromal expansion. Int. J. Cancer 2015, 136, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Xie, J.; Liu, F.; He, J.; He, J. MicroRNA-19b reduces hepatic stellate cell proliferation by targeting GRB2 in hepatic fibrosis models in vivo and in vitro as part of the inhibitory effect of estradiol. J. Cell. Biochem. 2015, 116, 2455–2464. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.; Su, J.; Wang, Y.; Feng, H.; Dai, X.; Yuan, Y.; Chen, X.; Yao, W. Pancreatic cancer-secreted miR-155 implicates in the conversion from normal fibroblasts to cancer-associated fibroblasts. Cancer Sci. 2015, 106, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wan, R.; Hu, G.; Yang, L.; Xiong, J.; Wang, F.; Shen, J.; He, S.; Guo, X.; Ni, J.; et al. MiR-15b and miR-16 induce the apoptosis of rat activated pancreatic stellate cells by targeting Bcl-2 in vitro. Pancreatology 2012, 12, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.J.; Pan, Q.; Jiang, B.; Chen, G.Y.; Li, D.G. Effects of upregulated expression of microRNA-16 on biological properties of culture-activated hepatic stellate cells. Apoptosis 2009, 14, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Huang, C.; Sun, X.; Long, X.R.; Lv, X.W.; Li, J. MicroRNA-146a modulates TGF-beta1-induced hepatic stellate cell proliferation by targeting SMAD4. Cell. Signal. 2012, 24, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chen, Y.; Wu, S.; He, J.; Lou, L.; Ye, W.; Wang, J. microRNA-34a and microRNA-34c promote the activation of human hepatic stellate cells by targeting peroxisome proliferator-activated receptor γ. Mol. Med. Rep. 2015, 11, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Wang, J.; Lu, H.; Zhang, G.; Liu, Y.; Wang, J.; Zhang, Y.; Shang, H.; Ji, H.; Chen, X.; et al. MicroRNA-130a and -130b enhance activation of hepatic stellate cells by suppressing PPARγ expression: A rat fibrosis model study. Biochem. Biophys. Res. Community 2015, 465, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Takeshita, F.; Hino, Y.; Fukunaga, S.; Kudo, Y.; Tamaki, A.; Matsunaga, J.; Takahashi, R.U.; Takata, T.; Shimamoto, A.; et al. MiR-22 represses cancer progression by inducing cellular senescence. J. Cell Biol. 2011, 193, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Hu, J.F.; Liu, B.H.; Wu, C.Q.; Yu, H.Y.; Yao, D.K.; Zhu, L. MiR-9a-5p regulates proliferation and migration of hepatic stellate cells under pressure through inhibition of SIRT1. World J. Gastroenerol. 2015, 21, 9900–9915. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Tao, H.; Hu, W.; Liu, L.P.; Shi, K.H.; Deng, Z.Y.; Li, J. MicroRNA-200a controls Nrf2 activation by target Keap1 in hepatic stellate cell proliferation and fibrosis. Cell. Signal. 2014, 26, 2381–2389. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Nakano, E.; Hamada, S.; Takikawa, T.; Yoshida, N.; Shimosegawa, T. Alteration of the microRNA expression profile during the activation of pancreatic stellate cells. Scand. J. Gastroenterol. 2014, 49, 323–331. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Alteration in Clinical Specimens | microRNA | Cancer Entity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Breast | Ovarian | Endometrial | Esophageal | Gastric | Colorectal | Pancreatic | Prostate | Mixed Entities | ||
| upregulated | miR-21 | ↑ | ↑ | ↑ | ||||||
| miR-31 | ↑ | ↑ | ||||||||
| miR-92 | ↑ | |||||||||
| miR-221 | ↑ | |||||||||
| miR-409 | ↑ | |||||||||
| miR-155 | ↑ | |||||||||
| downregulated | miR-15 | ↓ | ||||||||
| miR-16 | ↓ | |||||||||
| miR-26b | ↓ | |||||||||
| miR-31 | ↓ | ↓ | ||||||||
| miR-101 | ↓ | |||||||||
| miR-106b | ↓ | |||||||||
| miR-141 | ↓ | ↓ | ||||||||
| miR-148a | ↓ | |||||||||
| miR-200 | ↓abc* | ↓b* | ↓ab* | ↓bc* | ||||||
| miR-205 | ↓ | |||||||||
| miR-214 | ↓ | |||||||||
| miR-342 | ↓ | |||||||||
| miR-let7g | ↓ | |||||||||
| (A) | ||||||||||||||
| MiR with Cluster | Cancer Entities | Regulation of miR in CAFs | Interaction in CAFs | Cellular Consequences | Pathway | Targets | ||||||||
| Migration | Invasion | Adhesion | Growth | Proliferation, Progession | Differentiation | Chemoresistance | Apoptosis | Methylation | ||||||
| 15, 16 | PC | ↓ | α-SMA ↑ | ↑ | ↑ | ↑ | p-AKT, p-ERK ↑ | Fgf-2 and its receptor Fgfr1 ↑ | ||||||
| 27a/b | EOC | ↑ | α-SMA ↑ | ↑ | ↓ | TGF-β ↑ | ||||||||
| 92 | BC | ↓ | ↑ | |||||||||||
| 106b | GC | ↑ | α-SMA ↑ | ↑ | ↑ | ↑ | TGF-β ↑ | PTEN | ||||||
| 101 | BC, LC | ↓↑ | α-SMA ↑, IL-6 ↑ | ↓↑ | ↓↑ | ↓ | ↓↑ | ↓ | ↑ | ↓ | PI3K-AKT ↓, TGF-β ↑ | CXCL 12 ↓ | ||
| 143, 145 | GC | ↑ | α-SMA ↑, Collagen Typ III ↑ | ↑ | ↑ | ↑ | TGF-β /SMAD signaling ↑ | |||||||
| 200a, 200b | BC, LC, PaC, GC | ↓↑ | α-SMA ↓↑, IL-6 ↑ | ↓↑ | ↓↑ | ↓↑ | ↓↑ | ↓↑ | ↓ | TGF-β ↑↓ | ZEB1, ZEB2 ↑; Flt 1 ↓; SIP1 | |||
| 221 | BC, PaC | ↑ | α-SMA ↑, IL-6 ↑ | ↑ | ↑ | ↑ | TGF-β ↑ | NF-κB, K-Ras ↑ | ||||||
| 214 | OC | ↓ | cytokines ↑ | ↑ | ↑ | ↑ | CCL5 | |||||||
| 127 | BC | ↓ | ↑ | p53/p21 ↑ | BCL6 oncogene ↑ | |||||||||
| 133b | PC | ↑ | α-SMA, IL-6 ↑, Collagen 1A1 | ↑ | TGF-β ↑ | |||||||||
| 141, 200c | BC | ↓↑ | α-SMA ↑, IL-6 ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | TGF-β ↑ | Fli-1, TCF12 | |||
| 342 | BC | ↓ | α-SMA ↑, IL-6 ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | ↑ | TGF-β ↑ | ||||
| 365 | BC | ↓ | IL-6 ↑ | ↑ | ↑ | p38 MAPK ↓ | NF-κB p65 ↑ | |||||||
| 409 | PC | ↑ | α-SMA, EMT ↑, extracelluar vesikel (EV) release ↑ | ↑ | ↑ | Ras suppressor 1, stromal antigen 2 | ||||||||
| (B) | ||||||||||||||
| MiR with Cluster | PSC/HSC | Regulation of miR in PSC/HSC | Interaction in Cells | Cellular Consequences | Pathway | Targets | ||||||||
| Migration | Growth | Development, Movement | Proliferation, Progession, Activation | Fibrosis, Apoptosis | ||||||||||
| 15, 16 | PSC | ↓ | ↑ | BCL-2 ↑ | ||||||||||
| 27a/b | HSC | ↑ | ↑ | retinoid X receptor alpha | ||||||||||
| 17, 19b | HSC | ↑↓ | Collagen Type I and α-SMA ↑↓, expression of α1(I) and α2(I) procollagen in mRNAs ↑ | ↓ | ↓ | ↑ | TGF-β 1 ↑↓ | SMAD7 ↓, TGF-β 2 rezeptor and SMAD 3, GRB2 | ||||||
| 101 | HSC | ↑ | ↓ | ↓ | TGF-β ↓ | TβRI/KLF6 ↓ | ||||||||
| 143 | PSC | ↑ | ↑ | ↑ | Smad 2/4 | p39 MAP kinase & extracell.-signal–regulated kinase | ||||||||
| 200a, 200b | HSC | ↓↑ | α-SMA , EMT process ↓ | ↑ | ↓ | ↓↑ | ↑ | Wnt/β-catenin, TGF-β , PI3K/Akt ↑, Hh pathway | FOG2 ↓-regulation, Keap1/Nrf2 ↑, Gli2 ↓ | |||||
| 221, 222 | HSC, PSC | ↑ | α1Collagen and α-SMA ↑ | ↑ | ↑ | ↑ | Smad 2/5, NF-κB | p40 MAP kinase & extracell.-signal–regulated kinase | ||||||
| 214 | HSC | ↓ | Collagen Type I ↓ | ↑ | Cox-2 protein expression, NF-κB ↑ | |||||||||
| 29a | HSC, PSC | ↓↑ | Collagen Type I | ↑ | ↑ | ↑ | ↑ | TGF-β1 (SMAD3 dependant) ↑ | HDAC4 ↑, Cox-2 protein expression, NF-κB ↑ | |||||
| 34c | HSC | ↑ | α-SMA ↑ | PPARγ ↓ | ||||||||||
| 122 | HSC | ↓ | collagen maturation and ECM production ↑ | ↑ | P4HA1 ↓ | |||||||||
| 130b | HSC | ECM ↑ | ↑ | PPARy ↓ | ||||||||||
| 144 | HSC | ↓ | α-SMA ↑ | ↑ | TGF- β1 ↑ | |||||||||
| miR family | 8 | 15 | 17 | 27 | 29 | 33 | 34 | 101 | |||||||||
| chromosomes | 12 | 1 | 13 | 3 | 17 | 13 | 7 | x | 9 | 19 | 7 | 1 | 17 | 1 | 11 | 1 | 9 |
| members of miR-clusters | 141 200c | 200a 200b 429 | 15a, 16-1 | 15b, 16-2 | 195 497 | 17 18a 19a 19b-1 20a 92a-1 | 25 93 106b | 18b 19b-2 20b 92a-2 106a 363 | 23b 24-1 27b 3074 | 23a 27a 24-2 | 29a 29b-1 | 29b-2 29c | 33a 33b 6777 | 34a | 34b 34c | 101-1 3671 | mir-101-precursor-9 |
| miR family | 122 | 127 | 130 | 133 | 143/ 145 | 144 | 154 | 181 | 214 | 221 | 342 | 365 | |||||
| chromosomes | 18 | 14 | 11 | 22 | 18 | 20 | 6 | 5 | 17 | 14 | 1 | 9 | 1 | x | 14 | 16 | 17 |
| members of miR-clusters | 122 3591 | 127 136 337 431 433 432 665 | 130a | 130b 301b | 1-2 133a-1 | 133a-2 | 133b 206 | 143 145 | 144 451a 451b 4732 | 134 154 323b 365 377 409 410 412 485 496 541 656 | 181a-1 181b-1 | 181a-2 181b-2 | 199a-2 214 3120 | 221 222 | 151b 342 | 193b 365a | 365b 4725 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schoepp, M.; Ströse, A.J.; Haier, J. Dysregulation of miRNA Expression in Cancer Associated Fibroblasts (CAFs) and Its Consequences on the Tumor Microenvironment. Cancers 2017, 9, 54. https://doi.org/10.3390/cancers9060054
Schoepp M, Ströse AJ, Haier J. Dysregulation of miRNA Expression in Cancer Associated Fibroblasts (CAFs) and Its Consequences on the Tumor Microenvironment. Cancers. 2017; 9(6):54. https://doi.org/10.3390/cancers9060054
Chicago/Turabian StyleSchoepp, Maren, Anda Jana Ströse, and Jörg Haier. 2017. "Dysregulation of miRNA Expression in Cancer Associated Fibroblasts (CAFs) and Its Consequences on the Tumor Microenvironment" Cancers 9, no. 6: 54. https://doi.org/10.3390/cancers9060054
APA StyleSchoepp, M., Ströse, A. J., & Haier, J. (2017). Dysregulation of miRNA Expression in Cancer Associated Fibroblasts (CAFs) and Its Consequences on the Tumor Microenvironment. Cancers, 9(6), 54. https://doi.org/10.3390/cancers9060054