
HER2 in Breast Cancer Stemness: A Negative Feedback Loop towards Trastuzumab Resistance


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. CD44, CD24 and ALDH Are the BC Stemness Markers
3. HER2 Is Eminent in BC Stemness
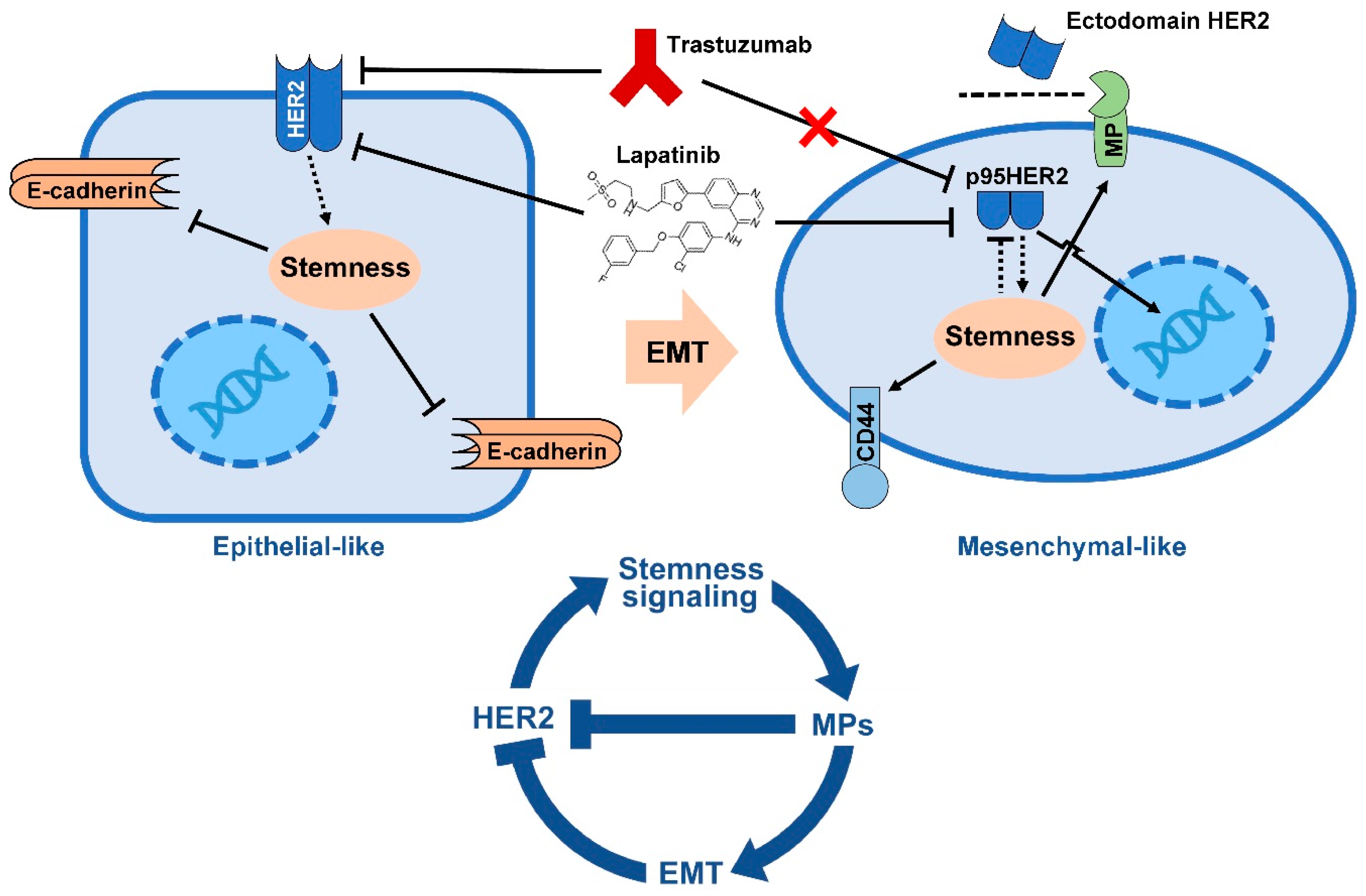
4. HER2 Blockade May Target BCSCs
5. EMT Drives Resistance to Trastuzumab
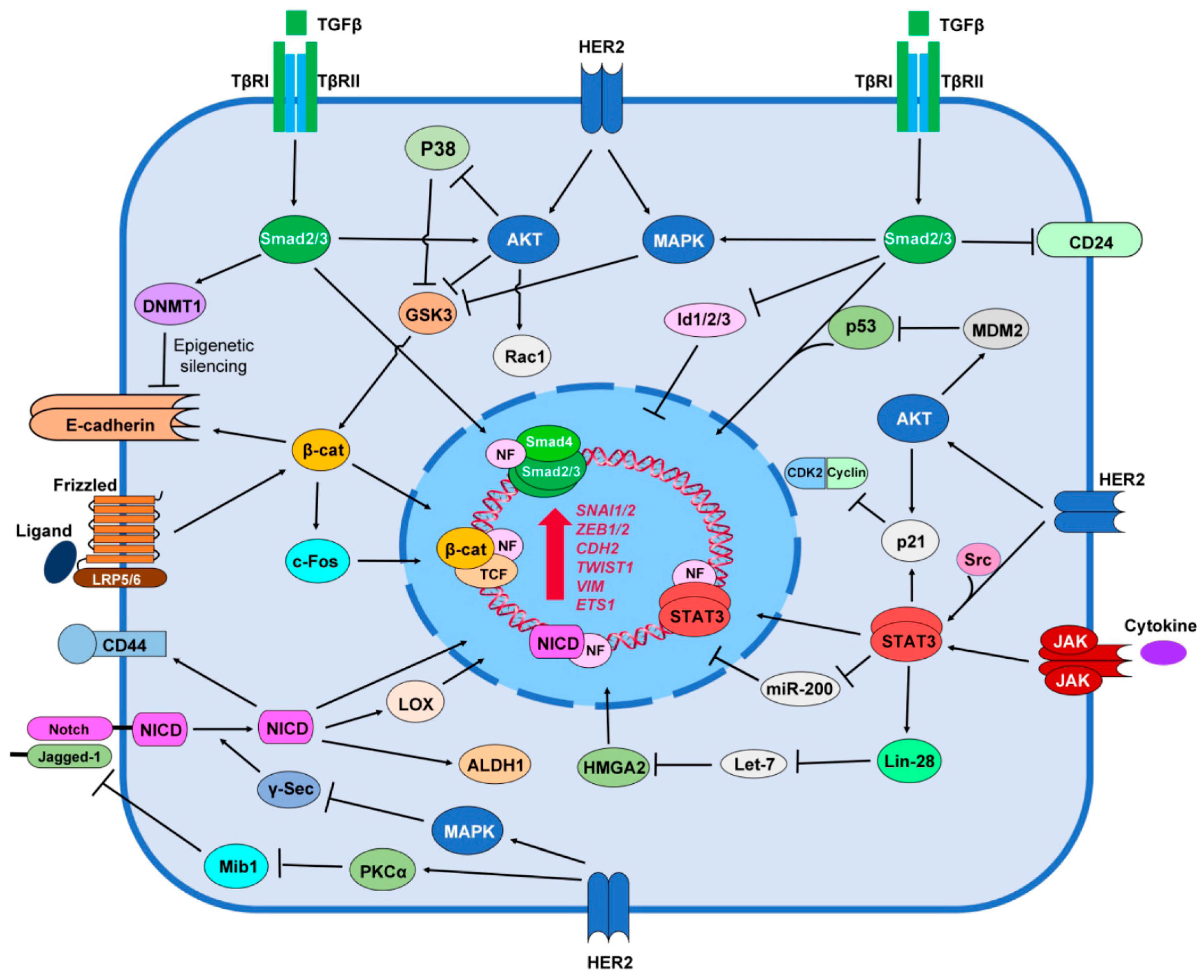
6. HER2 Promotes Stemness Signaling Pathways
6.1. TGF-β/Smad Signaling
6.2. Notch Signaling
6.3. Wnt/β-catenin Signaling
6.4. JAK/STAT Signaling
6.5. Hedgehog Signaling
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Weigelt, B.; Horlings, H.M.; Kreike, B.; Hayes, M.M.; Hauptmann, M.; Wessels, L.F.A.; de Jong, D.; Van de Vijver, M.J.; Van’t Veer, L.J.; Peterse, J.L. Refinement of breast cancer classification by molecular characterization of histological special types. J. Pathol. 2008, 216, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Tandon, A.K.; Clark, G.M.; Chamness, G.C.; Ullrich, A.; McGuire, W.L. HER-2/neu oncogene protein and prognosis in breast cancer. J. Clin. Oncol. 1989, 7, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Pegram, M.D.; Konecny, G.; Slamon, D.J. The molecular and cellular biology of HER2/neu gene amplification/overexpression and the clinical development of herceptin (trastuzumab) therapy for breast cancer. Cancer Treat. Res. 2000, 103, 57–75. [Google Scholar] [PubMed]
- Soady, K.J.; Kendrick, H.; Gao, Q.; Tutt, A.; Zvelebil, M.; Ordonez, L.D.; Quist, J.; Tan, D.W.-M.; Isacke, C.M.; Grigoriadis, A.; et al. Mouse mammary stem cells express prognostic markers for triple-negative breast cancer. Breast Cancer Res. 2015, 17. [Google Scholar] [CrossRef] [PubMed]
- Tiede, B.; Kang, Y. From milk to malignancy: The role of mammary stem cells in development, pregnancy and breast cancer. Cell Res. 2011, 21, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Nami, B.; Ghasemi-Dizgah, A.; Vaseghi, A. Overexpression of molecular chaperons GRP78 and GRP94 in CD44hi/CD24lo breast cancer stem cells. Bioimpacts 2016, 6, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.R.; Smigiel, J.M.; Junk, D.J.; Jackson, M.W. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Draffin, J.E.; McFarlane, S.; Hill, A.; Johnston, P.G.; Waugh, D.J.J. CD44 potentiates the adherence of metastatic prostate and breast cancer cells to bone marrow endothelial cells. Cancer Res. 2004, 64, 5702–5711. [Google Scholar] [CrossRef] [PubMed]
- Pham, P.V.; Phan, N.L.C.; Nguyen, N.T.; Truong, N.H.; Duong, T.T.; Le, D.V.; Truong, K.D.; Phan, N.K. Differentiation of breast cancer stem cells by knockdown of CD44: Promising differentiation therapy. J. Transl. Med. 2011, 9, 209. [Google Scholar] [CrossRef] [PubMed]
- Naor, D.; Sionov, R.V.; Ish-Shalom, D. CD44: Structure, function, and association with the malignant process. Adv. Cancer Res. 1997, 71, 241–319. [Google Scholar] [PubMed]
- Gao, A.C.; Lou, W.; Dong, J.T.; Isaacs, J.T. CD44 is a metastasis suppressor gene for prostatic cancer located on human chromosome 11p13. Cancer Res. 1997, 57, 846–849. [Google Scholar] [PubMed]
- Ponta, H.; Sherman, L.; Herrlich, P.A. CD44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell Biol. 2003, 4, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.; Zhu, H.; Chu, A.; Iida, N.; Zhang, L.; Hung, M.C. Interaction between the adhesion receptor, CD44, and the oncogene product, p185HER2, promotes human ovarian tumor cell activation. J. Biol. Chem. 1997, 272, 27913–27918. [Google Scholar] [CrossRef] [PubMed]
- Meran, S.; Luo, D.D.; Simpson, R.; Martin, J.; Wells, A.; Steadman, R.; Phillips, A.O. Hyaluronan facilitates transforming growth factor-beta1-dependent proliferation via CD44 and epidermal growth factor receptor interaction. J. Biol. Chem. 2011, 286, 17618–17630. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Park, Y.S.; Kim, H.J.; Kim, C.H.; Lim, S.W.; Huh, J.W.; Lee, J.H.; Kim, H.R. CD44 enhances the epithelial-mesenchymal transition in association with colon cancer invasion. Int. J. Oncol. 2012, 41, 211–218. [Google Scholar] [PubMed]
- Marotta, L.L.C.; Almendro, V.; Marusyk, A.; Shipitsin, M.; Schemme, J.; Walker, S.R.; Bloushtain-Qimron, N.; Kim, J.J.; Choudhury, S.A.; Maruyama, R.; et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(−) stem cell-like breast cancer cells in human tumors. J. Clin. Investig. 2011, 121, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Cremers, N.; Kroese, F.; Orend, G.; Chiquet-Ehrismann, R.; Uede, T.; Yagita, H.; Sleeman, J.P. CD24 expression causes the acquisition of multiple cellular properties associated with tumor growth and metastasis. Cancer Res. 2005, 65, 10783–10793. [Google Scholar] [CrossRef] [PubMed]
- Aigner, S.; Sthoeger, Z.M.; Fogel, M.; Weber, E.; Zarn, J.; Ruppert, M.; Zeller, Y.; Vestweber, D.; Stahel, R.; Sammar, M.; et al. CD24, a mucin-type glycoprotein, is a ligand for P-selectin on human tumor cells. Blood 1997, 89, 3385–3395. [Google Scholar] [PubMed]
- Friederichs, J.; Zeller, Y.; Hafezi-Moghadam, A.; Grone, H.J.; Ley, K.; Altevogt, P. The CD24/P-selectin binding pathway initiates lung arrest of human A125 adenocarcinoma cells. Cancer Res. 2000, 60, 6714–6722. [Google Scholar] [PubMed]
- Schabath, H.; Runz, S.; Joumaa, S.; Altevogt, P. CD24 affects CXCR4 function in pre-B lymphocytes and breast carcinoma cells. J. Cell Sci. 2006, 119, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.; Jang, K.; Lee, K.-M.; Kim, M.; Kim, J.; Yi, J.Y.; Noh, D.-Y.; Shin, I. CD24 enhances DNA damage-induced apoptosis by modulating NF-kappaB signaling in CD44-expressing breast cancer cells. Carcinogenesis 2011, 32, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Bretz, N.; Noske, A.; Keller, S.; Erbe-Hofmann, N.; Schlange, T.; Salnikov, A.V.; Moldenhauer, G.; Kristiansen, G.; Altevogt, P. CD24 promotes tumor cell invasion by suppressing tissue factor pathway inhibitor-2 (TFPI-2) in a c-Src-dependent fashion. Clin. Exp. Metastasis 2012, 29, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.; Liu, S.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.; Brocker, C.; Thompson, D.C.; Black, W.; Vasiliou, K.; Nebert, D.W.; Vasiliou, V. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum. Genom. 2011, 5, 283–303. [Google Scholar] [CrossRef]
- Hsu, L.C.; Chang, W.C.; Yoshida, A. Genomic structure of the human cytosolic aldehyde dehydrogenase gene. Genomics 1989, 5, 857–865. [Google Scholar] [CrossRef]
- Thomas, M.; Halsall, S.; Peters, T.J. Role of hepatic acetaldehyde dehydrogenase in alcoholism: Demonstration of persistent reduction of cytosolic activity in abstaining patients. Lancet 1982, 2, 1057–1058. [Google Scholar] [CrossRef]
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032. [Google Scholar] [PubMed]
- Ma, I.; Allan, A.L. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev. 2011, 7, 292–306. [Google Scholar] [CrossRef] [PubMed]
- Honeth, G.; Bendahl, P.-O.; Ringner, M.; Saal, L.H.; Gruvberger-Saal, S.K.; Lovgren, K.; Grabau, D.; Ferno, M.; Borg, A.; Hegardt, C. The CD44+/CD24− phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008, 10, R53. [Google Scholar] [CrossRef] [PubMed]
- Abraham, B.K.; Fritz, P.; McClellan, M.; Hauptvogel, P.; Athelogou, M.; Brauch, H. Prevalence of CD44+/CD24−/low cells in breast cancer may not be associated with clinical outcome but may favor distant metastasis. Clin. Cancer Res. 2005, 11, 1154–1159. [Google Scholar] [PubMed]
- Liu, H.; Patel, M.R.; Prescher, J.A.; Patsialou, A.; Qian, D.; Lin, J.; Wen, S.; Chang, Y.-F.; Bachmann, M.H.; Shimono, Y.; et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl. Acad. Sci. USA 2010, 107, 18115–18120. [Google Scholar] [CrossRef] [PubMed]
- Aktas, B.; Tewes, M.; Fehm, T.; Hauch, S.; Kimmig, R.; Kasimir-Bauer, S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009, 11, R46. [Google Scholar] [CrossRef] [PubMed]
- Balic, M.; Lin, H.; Young, L.; Hawes, D.; Giuliano, A.; McNamara, G.; Datar, R.H.; Cote, R.J. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin. Cancer Res. 2006, 12, 5615–5621. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulos, P.A.; Polioudaki, H.; Agelaki, S.; Kallergi, G.; Saridaki, Z.; Mavroudis, D.; Georgoulias, V. Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett. 2010, 288, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Nami, B.; Donmez, H.; Kocak, N. Tunicamycin-induced endoplasmic reticulum stress reduces in vitro subpopulation and invasion of CD44+/CD24− phenotype breast cancer stem cells. Exp. Toxicol. Pathol. 2016, 68, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Opyrchal, M.; Salisbury, J.L.; Iankov, I.; Goetz, M.P.; McCubrey, J.; Gambino, M.W.; Malatino, L.; Puccia, G.; Ingle, J.N.; Galanis, E.; et al. Inhibition of Cdk2 kinase activity selectively targets the CD44(+)/CD24(−)/Low stem-like subpopulation and restores chemosensitivity of SUM149PT triple-negative breast cancer cells. Int. J. Oncol. 2014, 45, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.-F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed]
- Croker, A.K.; Goodale, D.; Chu, J.; Postenka, C.; Hedley, B.D.; Hess, D.A.; Allan, A.L. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J. Cell. Mol. Med. 2009, 13, 2236–2252. [Google Scholar] [CrossRef] [PubMed]
- Muleris, M.; Almeida, A.; Malfoy, B.; Dutrillaux, B. Assignment of v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2 (ERBB2) to human chromosome band 17q21.1 by in situ hybridization. Cytogenet. Cell Genet. 1997, 76, 34–35. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.; Yang-Feng, T.L.; Liao, Y.C.; Chen, E.; Gray, A.; McGrath, J.; Seeburg, P.H.; Libermann, T.A.; Schlessinger, J.; Francke, U. Tyrosine kinase receptor with extensive homology to EGF receptor shares chromosomal location with neu oncogene. Science 1985, 230, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, T.; Sudo, C.; Ogawara, H.; Toyoshima, K.; Yamamoto, T. The product of the human c-erbB-2 gene: A 185-kilodalton glycoprotein with tyrosine kinase activity. Science 1986, 232, 1644–1646. [Google Scholar] [CrossRef] [PubMed]
- Graus-Porta, D.; Beerli, R.R.; Daly, J.M.; Hynes, N.E. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997, 16, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- King, C.R.; Borrello, I.; Porter, L.; Comoglio, P.; Schlessinger, J. Ligand-independent tyrosine phosphorylation of EGF receptor and the erbB-2/neu proto-oncogene product is induced by hyperosmotic shock. Oncogene 1989, 4, 13–18. [Google Scholar] [PubMed]
- Guo, W.; Pylayeva, Y.; Pepe, A.; Yoshioka, T.; Muller, W.J.; Inghirami, G.; Giancotti, F.G. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 2006, 126, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Lenferink, A.E.; Busse, D.; Flanagan, W.M.; Yakes, F.M.; Arteaga, C.L. ErbB2/neu kinase modulates cellular p27(Kip1) and cyclin D1 through multiple signaling pathways. Cancer Res. 2001, 61, 6583–6591. [Google Scholar] [PubMed]
- Nelson, J.M.; Fry, D.W. Akt, MAPK (Erk1/2), and p38 act in concert to promote apoptosis in response to ErbB receptor family inhibition. J. Biol. Chem. 2001, 276, 14842–14847. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-Y.; Chen, H.; Hsieh, Y.-H.; Wang, Y.-N.; Chu, H.-J.; Chen, Y.-H.; Chen, H.-Y.; Chien, P.-J.; Ma, H.-T.; Tsai, H.-C.; et al. Nuclear ErbB2 enhances translation and cell growth by activating transcription of ribosomal RNA genes. Cancer Res. 2011, 71, 4269–4279. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Lien, H.-C.; Xia, W.; Chen, I.-F.; Lo, H.-W.; Wang, Z.; Ali-Seyed, M.; Lee, D.-F.; Bartholomeusz, G.; Ou-Yang, F.; et al. Binding at and transactivation of the COX-2 promoter by nuclear tyrosine kinase receptor ErbB-2. Cancer Cell 2004, 6, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Q.; Chen, X.Y.; Jiang, Y.Y.; Liu, J. Identification of novel nuclear localization signal within the ErbB-2 protein. Cell Res. 2005, 15, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Maher, M. Current and Emerging Treatment Regimens for HER2-Positive Breast Cancer. Pharm. Ther. 2014, 39, 206–212. [Google Scholar]
- Korkaya, H.; Paulson, A.; Iovino, F.; Wicha, M.S. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008, 27, 6120–6130. [Google Scholar] [CrossRef] [PubMed]
- Ithimakin, S.; Day, K.C.; Malik, F.; Zen, Q.; Dawsey, S.J.; Bersano-Begey, T.F.; Quraishi, A.A.; Ignatoski, K.W.; Daignault, S.; Davis, A.; et al. HER2 drives luminal breast cancer stem cells in the absence of HER2 amplification: Implications for efficacy of adjuvant trastuzumab. Cancer Res. 2013, 73, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Yao, H.; Liu, Q.; Chen, J.; Shi, J.; Su, F.; Song, E. Markers of tumor-initiating cells predict chemoresistance in breast cancer. PLoS ONE 2010, 5, e15630. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Voisin, V.; Bader, G.D.; Deng, T.; Pusztai, L.; Symmans, W.F.; Esteva, F.J.; Egan, S.E.; Zacksenhaus, E. Seventeen-gene signature from enriched Her2/Neu mammary tumor-initiating cells predicts clinical outcome for human HER2(+):ERα(−) breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 5832–5837. [Google Scholar] [CrossRef] [PubMed]
- Magnifico, A.; Albano, L.; Campaner, S.; Delia, D.; Castiglioni, F.; Gasparini, P.; Sozzi, G.; Fontanella, E.; Menard, S.; Tagliabue, E. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. Clin. Cancer Res. 2009, 15, 2010–2021. [Google Scholar] [CrossRef] [PubMed]
- Son, M.-Y.; Kim, J.; Han, H.-W.; Woo, S.-M.; Cho, Y.S.; Kang, Y.-K.; Han, Y.-M. Expression profiles of protein tyrosine kinase genes in human embryonic stem cells. Reproduction 2008, 136, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Schulz, T.C.; Sherrer, E.S.; Dauphin, D.S.; Shin, S.; Nelson, A.M.; Ware, C.B.; Zhan, M.; Song, C.-Z.; Chen, X.; et al. Self-renewal of human embryonic stem cells requires insulin-like growth factor-1 receptor and ERBB2 receptor signaling. Blood 2007, 110, 4111–4119. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, S.; Hu, Y.P.; Wang, J.; Hauser, J.; Conway, A.N.; Vinci, M.A.; Humphrey, L.; Zborowska, E.; Willson, J.K.V.; et al. Blockade of EGFR and ErbB2 by the novel dual EGFR and ErbB2 tyrosine kinase inhibitor GW572016 sensitizes human colon carcinoma GEO cells to apoptosis. Cancer Res. 2006, 66, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Pivot, X.; Manikhas, A.; Zurawski, B.; Chmielowska, E.; Karaszewska, B.; Allerton, R.; Chan, S.; Fabi, A.; Bidoli, P.; Gori, S.; et al. CEREBEL (EGF111438): A Phase III, Randomized, Open-label study of lapatinib plus capecitabine versus trastuzumab plus capecitabine in patients with human epidermal growth factor receptor 2-positive metastatic breast Cancer. J. Clin. Oncol. 2015, 33, 1564–1573. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-L.; Shen, Y.-W.; Li, S.-T.; Li, C.-L.; Zhang, L.-X.; Yang, J.; Lv, M.; Lin, Y.-Y.; Wang, X.; Yang, J. The efficiency and safety of trastuzumab and lapatinib added to neoadjuvant chemotherapy in Her2-positive breast cancer patients: A randomized meta-analysis. Onco Targets Ther. 2016, 9, 3233–3247. [Google Scholar] [CrossRef] [PubMed]
- Saleem, A.; Searle, G.E.; Kenny, L.M.; Huiban, M.; Kozlowski, K.; Waldman, A.D.; Woodley, L.; Palmieri, C.; Lowdell, C.; Kaneko, T.; et al. Lapatinib access into normal brain and brain metastases in patients with Her-2 overexpressing breast cancer. EJNMMI Res. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.-F.; Lin, Y.; Bratman, S.V.; Feng, W.; Kuo, A.H.; Scheeren, F.A.; Engreitz, J.M.; Varma, S.; West, R.B.; Diehn, M. Neuregulin autocrine signaling promotes self-renewal of breast tumor-initiating cells by triggering HER2/HER3 activation. Cancer Res. 2014, 74, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Farnie, G.; Johnson, R.L.; Williams, K.E.; Clarke, R.B.; Bundred, N.J. Lapatinib inhibits stem/progenitor proliferation in preclinical in vitro models of ductal carcinoma in situ (DCIS). Cell Cycle 2014, 13, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, Y.; Yasui, H.; Kakudo, K.; Nozaki, M. Lapatinib-resistant cancer cells possessing epithelial cancer stem cell properties develop sensitivity during sphere formation by activation of the ErbB/AKT/cyclin D2 pathway. Oncol. Rep. 2016, 36, 3058–3064. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, X.; Mao, Y.; Qu, Q.; Shen, K. Association of epithelial-mesenchymal transition with lapatinib resistance through multipe pathways activation in HER2-positive breast cancer. J. Clin. Oncol. 2014, 32, e11579. [Google Scholar]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.B.; Henner, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef] [PubMed]
- Fendly, B.M.; Winget, M.; Hudziak, R.M.; Lipari, M.T.; Napier, M.A.; Ullrich, A. Characterization of murine monoclonal antibodies reactive to either the human epidermal growth factor receptor or HER2/neu gene product. Cancer Res. 1990, 50, 1550–1558. [Google Scholar] [PubMed]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Lewis Phillips, G.D.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 2009, 15, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Lan, K.-H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Huang, W.-C.; Li, P.; Guo, H.; Poh, S.-B.; Brady, S.W.; Xiong, Y.; Tseng, L.-M.; Li, S.-H.; Ding, Z.; et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med. 2011, 17, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Nahta, R.; Takahashi, T.; Ueno, N.T.; Hung, M.-C.; Esteva, F.J. P27(kip1) down-regulation is associated with trastuzumab resistance in breast cancer cells. Cancer Res. 2004, 64, 3981–3986. [Google Scholar] [CrossRef] [PubMed]
- Le, X.-F.; Bedrosian, I.; Mao, W.; Murray, M.; Lu, Z.; Keyomarsi, K.; Lee, M.-H.; Zhao, J.; Bast, R.C.J. Anti-HER2 antibody trastuzumab inhibits CDK2-mediated NPAT and histone H4 expression via the PI3K pathway. Cell Cycle 2006, 5, 1654–1661. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Eichhorn, P.J.; Cortes, J.; Prudkin, L.; Aura, C.; Jimenez, J.; Chandarlapaty, S.; Serra, V.; Prat, A.; Ibrahim, Y.H.; et al. Cyclin E amplification/overexpression is a mechanism of trastuzumab resistance in HER2+ breast cancer patients. Proc. Natl. Acad. Sci. USA 2011, 108, 3761–3766. [Google Scholar] [CrossRef] [PubMed]
- Klapper, L.N.; Waterman, H.; Sela, M.; Yarden, Y. Tumor-inhibitory antibodies to HER-2/ErbB-2 may act by recruiting c-Cbl and enhancing ubiquitination of HER-2. Cancer Res. 2000, 60, 3384–3388. [Google Scholar] [PubMed]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [PubMed]
- Raab, S.; Steinbacher, J.; Schmiedel, B.J.; Kousis, P.C.; Steinle, A.; Jung, G.; Grosse-Hovest, L.; Salih, H.R. Fc-optimized NKG2D-Fc constructs induce NK cell antibody-dependent cellular cytotoxicity against breast cancer cells independently of HER2/neu expression status. J. Immunol. 2014, 193, 4261–4272. [Google Scholar] [CrossRef] [PubMed]
- Esteva, F.J.; Valero, V.; Booser, D.; Guerra, L.T.; Murray, J.L.; Pusztai, L.; Cristofanilli, M.; Arun, B.; Esmaeli, B.; Fritsche, H.A.; et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Sato, N.; Sano, M.; Tabei, T.; Asaga, T.; Ando, J.; Fujii, H.; Yamamoto, N.; Kurosumi, M.; Inoue, K.; Kimura, M. Combination docetaxel and trastuzumab treatment for patients with HER-2-overexpressing metastatic breast cancer: A multicenter, phase-II study. Breast Cancer 2006, 13, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Marty, M.; Cognetti, F.; Maraninchi, D.; Snyder, R.; Mauriac, L.; Tubiana-Hulin, M.; Chan, S.; Grimes, D.; Anton, A.; Lluch, A.; et al. Randomized phase II trial of the efficacy and safety of trastuzumab combined with docetaxel in patients with human epidermal growth factor receptor 2-positive metastatic breast cancer administered as first-line treatment: The M77001 study group. J. Clin. Oncol. 2005, 23, 4265–4274. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Pienkowski, T.; Im, Y.-H.; Roman, L.; Tseng, L.-M.; Liu, M.-C.; Lluch, A.; Staroslawska, E.; de la Haba-Rodriguez, J.; Im, S.-A.; et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): A randomised multicentre, open-label, phase 2 trial. Lancet. Oncol. 2012, 13, 25–32. [Google Scholar] [CrossRef]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.L.; Cobleigh, M.A.; Tripathy, D.; Gutheil, J.C.; Harris, L.N.; Fehrenbacher, L.; Slamon, D.J.; Murphy, M.; Novotny, W.F.; Burchmore, M.; et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J. Clin. Oncol. 2002, 20, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Eiermann, W. Trastuzumab combined with chemotherapy for the treatment of HER2-positive metastatic breast cancer: Pivotal trial data. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2001, 12, S57–S62. [Google Scholar] [CrossRef]
- Dahabreh, I.J.; Linardou, H.; Siannis, F.; Fountzilas, G.; Murray, S. Trastuzumab in the adjuvant treatment of early-stage breast cancer: A systematic review and meta-analysis of randomized controlled trials. Oncologist 2008, 13, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Piccart-Gebhart, M.J.; Procter, M.; Leyland-Jones, B.; Goldhirsch, A.; Untch, M.; Smith, I.; Gianni, L.; Baselga, J.; Bell, R.; Jackisch, C.; et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N. Engl. J. Med. 2005, 353, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Mego, M.; Lee, B.; Anfossi, S.; Parker, C.A.; Alvarez, R.H.; Ueno, N.T.; Valero, V.; Cristofanilli, M.; Reuben, J.M. Epithelial-mesenchymal transition in patients with HER2+ metastatic breast cancer. J. Clin. Oncol. 2011, 29. Abstract 623. [Google Scholar] [CrossRef]
- Giordano, A.; Gao, H.; Anfossi, S.; Cohen, E.; Mego, M.; Lee, B.-N.; Tin, S.; De Laurentiis, M.; Parker, C.A.; Alvarez, R.H.; et al. Epithelial-mesenchymal transition and stem cell markers in patients with HER2-positive metastatic breast cancer. Mol. Cancer Ther. 2012, 11, 2526–2534. [Google Scholar] [CrossRef] [PubMed]
- Jenndahl, L.E.; Isakson, P.; Baeckstrom, D. c-erbB2-induced epithelial-mesenchymal transition in mammary epithelial cells is suppressed by cell-cell contact and initiated prior to E-cadherin downregulation. Int. J. Oncol. 2005, 27, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, G.M.A.; Akhtar, N.; Kannius-Janson, M.; Baeckstrom, D. Loss of E-cadherin expression is not a prerequisite for c-ERBB2-induced epithelial-mesenchymal transition. Int. J. Oncol. 2014, 45, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Ingthorsson, S.; Andersen, K.; Hilmarsdottir, B.; Maelandsmo, G.M.; Magnusson, M.K.; Gudjonsson, T. HER2 induced EMT and tumorigenicity in breast epithelial progenitor cells is inhibited by coexpression of EGFR. Oncogene 2016, 35, 4244–4255. [Google Scholar] [CrossRef] [PubMed]
- Rennstam, K.; Jonsson, G.; Tanner, M.; Bendahl, P.-O.; Staaf, J.; Kapanen, A.I.; Karhu, R.; Baldetorp, B.; Borg, A.; Isola, J. Cytogenetic characterization and gene expression profiling of the trastuzumab-resistant breast cancer cell line JIMT-1. Cancer Genet. Cytogenet. 2007, 172, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Barok, M.; Isola, J.; Palyi-Krekk, Z.; Nagy, P.; Juhasz, I.; Vereb, G.; Kauraniemi, P.; Kapanen, A.; Tanner, M.; Vereb, G.; et al. Trastuzumab causes antibody-dependent cellular cytotoxicity-mediated growth inhibition of submacroscopic JIMT-1 breast cancer xenografts despite intrinsic drug resistance. Mol. Cancer Ther. 2007, 6, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Koninki, K.; Barok, M.; Tanner, M.; Staff, S.; Pitkanen, J.; Hemmila, P.; Ilvesaro, J.; Isola, J. Multiple molecular mechanisms underlying trastuzumab and lapatinib resistance in JIMT-1 breast cancer cells. Cancer Lett. 2010, 294, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, C.; Corvigno, S.; Galgani, M.; Limite, G.; Nardone, A.; Veneziani, B.M. Combined inhibitory effect of formestane and herceptin on a subpopulation of CD44+/CD24low breast cancer cells. Cancer Sci. 2010, 101, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Oliveras-Ferraros, C.; Corominas-Faja, B.; Cufi, S.; Vazquez-Martin, A.; Martin-Castillo, B.; Iglesias, J.M.; Lopez-Bonet, E.; Martin, A.G.; Menendez, J.A. Epithelial-to-mesenchymal transition (EMT) confers primary resistance to trastuzumab (Herceptin). Cell Cycle 2012, 11, 4020–4032. [Google Scholar] [CrossRef] [PubMed]
- Cufí, S.; Corominas-Faja, B.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Dorca, J.; Bosch-Barrera, J.; Martin-Castillo, B.; Menendez, J.A. Metformin-induced preferential killing of breast cancer initiating CD44(+)CD24(−/low) cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget 2012, 3, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Oliveras-Ferraros, C.; Vazquez-Martin, A.; Martin-Castillo, B.; Cufi, S.; Del Barco, S.; Lopez-Bonet, E.; Brunet, J.; Menendez, J.A. Dynamic emergence of the mesenchymal CD44(pos)CD24(neg/low) phenotype in HER2-gene amplified breast cancer cells with de novo resistance to trastuzumab (Herceptin). Biochem. Biophys. Res. Commun. 2010, 397, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Bauerschmitz, G.J.; Ranki, T.; Kangasniemi, L.; Ribacka, C.; Eriksson, M.; Porten, M.; Herrmann, I.; Ristimaki, A.; Virkkunen, P.; Tarkkanen, M.; et al. Tissue-specific promoters active in CD44+CD24-/low breast cancer cells. Cancer Res. 2008, 68, 5533–5539. [Google Scholar] [CrossRef] [PubMed]
- Rennstam, K.; McMichael, N.; Berglund, P.; Honeth, G.; Hegardt, C.; Ryden, L.; Luts, L.; Bendahl, P.-O.; Hedenfalk, I. Numb protein expression correlates with a basal-like phenotype and cancer stem cell markers in primary breast cancer. Breast Cancer Res. Treat. 2010, 122, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, D.; Sabri, S.; Xu, Y.; Graham, K.; Bhatnagar, P.; Suresh, M.; Abdulkarim, B. Spontaneous Epithelial-Mesenchymal Transition and Resistance to HER-2-Targeted Therapies in HER-2-Positive Luminal Breast Cancer. PLoS ONE 2013, 8, e71987. [Google Scholar] [CrossRef] [PubMed]
- Reim, F.; Dombrowski, Y.; Ritter, C.; Buttmann, M.; Hausler, S.; Ossadnik, M.; Krockenberger, M.; Beier, D.; Beier, C.P.; Dietl, J.; et al. Immunoselection of breast and ovarian cancer cells with trastuzumab and natural killer cells: Selective escape of CD44high/CD24low/HER2low breast cancer stem cells. Cancer Res. 2009, 69, 8058–8066. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Rojo, F.; Ocana, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matias-Guiu, X.; Ramon y Cajal, S.; Arribas, J.; et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef] [PubMed]
- Sperinde, J.; Jin, X.; Banerjee, J.; Penuel, E.; Saha, A.; Diedrich, G.; Huang, W.; Leitzel, K.; Weidler, J.; Ali, S.M.; et al. Quantitation of p95HER2 in paraffin sections by using a p95-specific antibody and correlation with outcome in a cohort of trastuzumab-treated breast cancer patients. Clin. Cancer Res. 2010, 16, 4226–4235. [Google Scholar] [CrossRef] [PubMed]
- Arribas, J.; Baselga, J.; Pedersen, K.; Parra-Palau, J.L. p95HER2 and breast cancer. Cancer Res. 2011, 71, 1515–1519. [Google Scholar] [CrossRef] [PubMed]
- Codony-Servat, J.; Albanell, J.; Lopez-Talavera, J.C.; Arribas, J.; Baselga, J. Cleavage of the HER2 ectodomain is a pervanadate-activable process that is inhibited by the tissue inhibitor of metalloproteases-1 in breast cancer cells. Cancer Res. 1999, 59, 1196–1201. [Google Scholar] [PubMed]
- Yuan, C.-X.; Lasut, A.L.; Wynn, R.; Neff, N.T.; Hollis, G.F.; Ramaker, M.L.; Rupar, M.J.; Liu, P.; Meade, R. Purification of Her-2 extracellular domain and identification of its cleavage site. Protein Expr. Purif. 2003, 29, 217–222. [Google Scholar] [CrossRef]
- Sanderson, M.P.; Dempsey, P.J.; Dunbar, A.J. Control of ErbB signaling through metalloprotease mediated ectodomain shedding of EGF-like factors. Growth Factors 2006, 24, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.; Angelini, P.-D.; Laos, S.; Bach-Faig, A.; Cunningham, M.P.; Ferrer-Ramon, C.; Luque-Garcia, A.; Garcia-Castillo, J.; Parra-Palau, J.L.; Scaltriti, M.; et al. A naturally occurring HER2 carboxy-terminal fragment promotes mammary tumor growth and metastasis. Mol. Cell. Biol. 2009, 29, 3319–3331. [Google Scholar] [CrossRef] [PubMed]
- Ruff, M.; Leyme, A.; Le Cann, F.; Bonnier, D.; Le Seyec, J.; Chesnel, F.; Fattet, L.; Rimokh, R.; Baffet, G.; Theret, N. The disintegrin and metalloprotease ADAM12 is associated with TGF-beta-induced epithelial to mesenchymal transition. PLoS ONE 2015, 10, e0139179. [Google Scholar] [CrossRef] [PubMed]
- Duhachek-Muggy, S.; Qi, Y.; Wise, R.; Alyahya, L.; Li, H.; Hodge, J.; Zolkiewska, A. Metalloprotease-disintegrin ADAM12 actively promotes the stem cell-like phenotype in claudin-low breast cancer. Mol. Cancer 2017, 16, 32. [Google Scholar] [CrossRef] [PubMed]
- Pulyaeva, H.; Bueno, J.; Polette, M.; Birembaut, P.; Sato, H.; Seiki, M.; Thompson, E.W. MT1-MMP correlates with MMP-2 activation potential seen after epithelial to mesenchymal transition in human breast carcinoma cells. Clin. Exp. Metastasis 1997, 15, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Tester, A.M.; Ruangpanit, N.; Anderson, R.L.; Thompson, E.W. MMP-9 secretion and MMP-2 activation distinguish invasive and metastatic sublines of a mouse mammary carcinoma system showing epithelial-mesenchymal transition traits. Clin. Exp. Metastasis 2000, 18, 553–560. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, C.; McKie, N.; Buggy, Y.; Duggan, C.; Hill, A.D.K.; McDermott, E.; O’Higgins, N.; Duffy, M.J. Expression of ADAM-9 mRNA and protein in human breast cancer. Int. J. Cancer 2003, 105, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Wewer, U.M.; Zurakowski, D.; Pories, S.E.; Moses, M.A. ADAM 12 cleaves extracellular matrix proteins and correlates with cancer status and stage. J. Biol. Chem. 2004, 279, 51323–51330. [Google Scholar] [CrossRef] [PubMed]
- Lendeckel, U.; Kohl, J.; Arndt, M.; Carl-McGrath, S.; Donat, H.; Rocken, C. Increased expression of ADAM family members in human breast cancer and breast cancer cell lines. J. Cancer Res. Clin. Oncol. 2005, 131, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, Y.; Mochizuki, S.; Kodama, T.; Shimoda, M.; Ohtsuka, T.; Shiomi, T.; Chijiiwa, M.; Ikeda, T.; Kitajima, M.; Okada, Y. ADAM28 is overexpressed in human breast carcinomas: Implications for carcinoma cell proliferation through cleavage of insulin-like growth factor binding protein-3. Cancer Res. 2006, 66, 9913–9920. [Google Scholar] [CrossRef] [PubMed]
- McGowan, P.M.; Ryan, B.M.; Hill, A.D.K.; McDermott, E.; O’Higgins, N.; Duffy, M.J. ADAM-17 expression in breast cancer correlates with variables of tumor progression. Clin. Cancer Res. 2007, 13, 2335–2343. [Google Scholar] [CrossRef] [PubMed]
- Mullooly, M.; McGowan, P.M.; Kennedy, S.A.; Madden, S.F.; Crown, J.; O’ Donovan, N.; Duffy, M.J. ADAM10: A new player in breast cancer progression? Br. J. Cancer 2015, 113, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Köhrmann, A.; Kammerer, U.; Kapp, M.; Dietl, J.; Anacker, J. Expression of matrix metalloproteinases (MMPs) in primary human breast cancer and breast cancer cell lines: New findings and review of the literature. BMC Cancer 2009, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Benson, C.S.; Babu, S.D.; Radhakrishna, S.; Selvamurugan, N.; Ravi Sankar, B. Expression of matrix metalloproteinases in human breast cancer tissues. Dis. Markers 2013, 34, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, J.E.; Staren, E.D.; Appert, H.E. Matrix metalloproteinase expression in breast cancer. J. Surg. Res. 2003, 110, 383–392. [Google Scholar] [CrossRef]
- Merdad, A.; Karim, S.; Schulten, H.-J.; Dallol, A.; Buhmeida, A.; Al-Thubaity, F.; Gari, M.A.; Chaudhary, A.G.; Abuzenadah, A.M.; Al-Qahtani, M.H. Expression of matrix metalloproteinases (MMPs) in primary human breast cancer: MMP-9 as a potential biomarker for cancer invasion and metastasis. Anticancer Res. 2014, 34, 1355–1366. [Google Scholar] [PubMed]
- Liu, P.C.C.; Liu, X.; Li, Y.; Covington, M.; Wynn, R.; Huber, R.; Hillman, M.; Yang, G.; Ellis, D.; Marando, C.; et al. Identification of ADAM10 as a major source of HER2 ectodomain sheddase activity in HER2 overexpressing breast cancer cells. Cancer Biol. Ther. 2006, 5, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Feldinger, K.; Generali, D.; Kramer-Marek, G.; Gijsen, M.; Ng, T.B.; Wong, J.H.; Strina, C.; Cappelletti, M.; Andreis, D.; Li, J.-L.; et al. ADAM10 mediates trastuzumab resistance and is correlated with survival in HER2 positive breast cancer. Oncotarget 2014, 5, 6633–6646. [Google Scholar] [CrossRef] [PubMed]
- Gijsen, M.; King, P.; Perera, T.; Parker, P.J.; Harris, A.L.; Larijani, B.; Kong, A. HER2 phosphorylation is maintained by a PKB negative feedback loop in response to anti-HER2 herceptin in breast cancer. PLoS Biol. 2010, 8, e1000563. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, F.; McGowan, P.M.; Mullooly, M.; Murray, A.; Synnott, N.; O’Donovan, N.; Flanagan, L.; Tape, C.J.; Murphy, G.; Crown, J.; et al. Targeting ADAM-17 with an inhibitory monoclonal antibody has antitumour effects in triple-negative breast cancer cells. Br. J. Cancer 2015, 112, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Mullooly, M.; O’Donovan, N.; Sukor, S.; Crown, J.; Pierce, A.; McGowan, P.M. The ADAMs family of proteases: New biomarkers and therapeutic targets for cancer? Clin. Proteomics 2011, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.M.; He, C.; Li, Y.; Scherle, P.; Liu, X.; Marando, C.A.; Covington, M.B.; Yang, G.; Pan, M.; Turner, S.; et al. Conversion of an MMP-potent scaffold to an MMP-selective HER-2 sheddase inhibitor via scaffold hybridization and subtle P1’ permutations. Bioorg. Med. Chem. Lett. 2008, 18, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Duchnowska, R.; Sperinde, J.; Chenna, A.; Haddad, M.; Paquet, A.; Lie, Y.; Weidler, J.M.; Huang, W.; Winslow, J.; Jankowski, T.; et al. Quantitative measurements of tumoral p95HER2 protein expression in metastatic breast cancer patients treated with trastuzumab: Independent validation of the p95HER2 clinical cutoff. Clin. Cancer Res. 2014, 20, 2805–2813. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-W.; Cha, Y.; Paquet, A.; Huang, W.; Weidler, J.; Lie, Y.; Sherwood, T.; Bates, M.; Haddad, M.; Park, I.H.; et al. Correlation of HER2, p95HER2 and HER3 expression and treatment outcome of lapatinib plus capecitabine in her2-positive metastatic breast cancer. PLoS ONE 2012, 7, e39943. [Google Scholar] [CrossRef] [PubMed]
- Lipton, A.; Goodman, L.; Leitzel, K.; Cook, J.; Sperinde, J.; Haddad, M.; Kostler, W.J.; Huang, W.; Weidler, J.M.; Ali, S.; et al. HER3, p95HER2, and HER2 protein expression levels define multiple subtypes of HER2-positive metastatic breast cancer. Breast Cancer Res. Treat. 2013, 141, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Saez, R.; Molina, M.A.; Ramsey, E.E.; Rojo, F.; Keenan, E.J.; Albanell, J.; Lluch, A.; Garcia-Conde, J.; Baselga, J.; Clinton, G.M. p95HER-2 predicts worse outcome in patients with HER-2-positive breast cancer. Clin. Cancer Res. 2006, 12, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Watabe, T.; Miyazono, K. Roles of TGF-beta family signaling in stem cell renewal and differentiation. Cell Res. 2009, 19, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Rhee, L.; Chen, E.Y.; Van Tilburg, A. Intron-exon structure of the human transforming growth factor-beta precursor gene. Nucleic Acids Res. 1987, 15, 3188–3189. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, M.E.; Kobrin, M.S.; Silan, C.M.; Kingsley, D.M.; Justice, M.J.; Miller, D.A.; Ceci, J.D.; Lock, L.F.; Lee, A.; Buchberg, A.M. Chromosomal localization of seven members of the murine TGF-beta superfamily suggests close linkage to several morphogenetic mutant loci. Genomics 1990, 6, 505–520. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L.; Carcamo, J.; Zentella, A.; Doody, J.; Laiho, M.; Wang, X.F.; Massague, J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell 1992, 71, 1003–1014. [Google Scholar] [CrossRef]
- Yamashita, H.; ten Dijke, P.; Franzen, P.; Miyazono, K.; Heldin, C.H. Formation of hetero-oligomeric complexes of type I and type II receptors for transforming growth factor-beta. J. Biol. Chem. 1994, 269, 20172–20178. [Google Scholar] [PubMed]
- Massague, J. TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, J.-A.; Kim, J.-H. Ras promotes transforming growth factor-beta (TGF-beta)-induced epithelial-mesenchymal transition via a leukotriene B4 receptor-2-linked cascade in mammary epithelial cells. J. Biol. Chem. 2014, 289, 22151–22160. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Kodaira, M.; Ito, A.; Okazaki, M.; Kawaguchi, N.; Hamada, Y.; Takada, Y.; Matsuura, N. Enhanced expression of integrin alphavbeta3 induced by TGF-beta is required for the enhancing effect of fibroblast growth factor 1 (FGF1) in TGF-beta-induced epithelial-mesenchymal transition (EMT) in mammary epithelial cells. PLoS ONE 2015, 10, e0137486. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.-F.; Georgoudaki, A.-M.; Lambut, L.; Johansson, J.; Tabor, V.; Hagikura, K.; Jin, Y.; Jansson, M.; Alexander, J.S.; Nelson, C.M.; et al. TGF-beta1-induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21-mediated chemotaxis. Oncogene 2016, 35, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, M.K.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. TGFβ/TNFα-nediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011, 71, 4707–4719. [Google Scholar] [CrossRef] [PubMed]
- Stankic, M.; Pavlovic, S.; Chin, Y.; Brogi, E.; Padua, D.; Norton, L.; Massague, J.; Benezra, R. TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013, 5, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Shirakihara, T.; Saitoh, M.; Miyazono, K. Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-beta. Mol. Biol. Cell 2007, 18, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Savagner, P.; Yamada, K.M.; Thiery, J.P. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J. Cell Biol. 1997, 137, 1403–1419. [Google Scholar] [CrossRef] [PubMed]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef]
- Thuault, S.; Valcourt, U.; Petersen, M.; Manfioletti, G.; Heldin, C.-H.; Moustakas, A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J. Cell Biol. 2006, 174, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Koinuma, D.; Tsutsumi, S.; Kamimura, N.; Taniguchi, H.; Miyazawa, K.; Sunamura, M.; Imamura, T.; Miyazono, K.; Aburatani, H. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor beta signaling. Mol. Cell. Biol. 2009, 29, 172–186. [Google Scholar] [CrossRef] [PubMed]
- Kowanetz, M.; Valcourt, U.; Bergstrom, R.; Heldin, C.-H.; Moustakas, A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein. Mol. Cell. Biol. 2004, 24, 4241–4254. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Cubillo, E.; Tobiume, K.; Shirakihara, T.; Fukuda, N.; Suzuki, H.; Shimizu, K.; Takehara, K.; Cano, A.; Saitoh, M.; et al. A role for Id in the regulation of TGF-beta-induced epithelial-mesenchymal transdifferentiation. Cell Death Differ. 2004, 11, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Papageorgis, P.; Lambert, A.W.; Ozturk, S.; Gao, F.; Pan, H.; Manne, U.; Alekseyev, Y.O.; Thiagalingam, A.; Abdolmaleky, H.M.; Lenburg, M.; et al. Smad signaling is required to maintain epigenetic silencing during breast cancer progression. Cancer Res. 2010, 70, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [PubMed]
- Burk, U.; Schubert, J.; Wellner, U.; Schmalhofer, O.; Vincan, E.; Spaderna, S.; Brabletz, T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008, 9, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Mayanagi, T.; Sobue, K. Dual roles of myocardin-related transcription factors in epithelial mesenchymal transition via slug induction and actin remodeling. J. Cell Biol. 2007, 179, 1027–1042. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hebert, M.C.; Zhang, Y.E. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002, 21, 3749–3759. [Google Scholar] [CrossRef] [PubMed]
- Papageorgis, P. TGFβ signaling in tumor initiation, epithelial-to-mesenchymal transition, and metastasis. J. Oncol. 2015, 2015, 587193. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Derynck, R. Cell size and invasion in TGF-beta-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 2007, 178, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-beta-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.-C. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Bachelder, R.E.; Yoon, S.-O.; Franci, C.; de Herreros, A.G.; Mercurio, A.M. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: Implications for the epithelial-mesenchymal transition. J. Cell Biol. 2005, 168, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, A.; Hussey, G.S.; Ray, P.S.; Jin, G.; Fox, P.L.; Howe, P.H. TGF-beta-mediated phosphorylation of hnRNP E1 induces EMT via transcript-selective translational induction of Dab2 and ILEI. Nat. Cell Biol. 2010, 12, 286–293. [Google Scholar] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, A.; Colletti, M.; Cozzolino, A.M.; Steindler, C.; Lunadei, M.; Mancone, C.; Tripodi, M. ERK5/MAPK is activated by TGFbeta in hepatocytes and required for the GSK-3beta-mediated Snail protein stabilization. Cell. Signal. 2008, 20, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, K.; Shirakihara, T.; Nakano, A.; Imamura, T.; Miyazono, K.; Saitoh, M. Role of Ras signaling in the induction of snail by transforming growth factor-beta. J. Biol. Chem. 2009, 284, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Morel, A.-P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Eaton, E.N.; Li, S.H.-J.; Chaffer, C.L.; Reinhardt, F.; Kah, K.-J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Buijs, J.T.; van der Horst, G.; van den Hoogen, C.; Cheung, H.; de Rooij, B.; Kroon, J.; Petersen, M.; van Overveld, P.G.M.; Pelger, R.C.M.; van der Pluijm, G. The BMP2/7 heterodimer inhibits the human breast cancer stem cell subpopulation and bone metastases formation. Oncogene 2012, 31, 2164–2174. [Google Scholar] [CrossRef] [PubMed]
- Santibanez, J.F. JNK mediates TGF-beta1-induced epithelial mesenchymal transdifferentiation of mouse transformed keratinocytes. FEBS Lett. 2006, 580, 5385–5391. [Google Scholar] [CrossRef] [PubMed]
- Tavares, A.L.P.; Mercado-Pimentel, M.E.; Runyan, R.B.; Kitten, G.T. TGF beta-mediated RhoA expression is necessary for epithelial-mesenchymal transition in the embryonic chick heart. Dev. Dyn. 2006, 235, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.E. The functional crosstalk between HER2 tyrosine kinase and TGF-β signaling in breast cancer malignancy. J. Signal Transduct. 2011, 2011, 804236. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Wang, S.; Dumont, N.; Yi, J.Y.; Koh, Y.; Arteaga, C.L. Overexpression of HER2 (erbB2) in human breast epithelial cells unmasks transforming growth factor beta-induced cell motility. J. Biol. Chem. 2004, 279, 24505–24513. [Google Scholar] [CrossRef] [PubMed]
- Siegel, P.M.; Shu, W.; Cardiff, R.D.; Muller, W.J.; Massague, J. Transforming growth factor beta signaling impairs Neu-induced mammary tumorigenesis while promoting pulmonary metastasis. Proc. Natl. Acad. Sci. USA 2003, 100, 8430–8435. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, R.S.; Koh, Y.; Roebuck, L.R.; Sanders, M.E.; Brantley-Sieders, D.; Gorska, A.E.; Moses, H.L.; Arteaga, C.L. Increased malignancy of Neu-induced mammary tumors overexpressing active transforming growth factor beta1. Mol. Cell. Biol. 2003, 23, 8691–8703. [Google Scholar] [CrossRef] [PubMed]
- Seton-Rogers, S.E.; Lu, Y.; Hines, L.M.; Koundinya, M.; LaBaer, J.; Muthuswamy, S.K.; Brugge, J.S. Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Ren, J.Q.; Li, H.; Kong, Z.L.; Zhu, H.G. Downregulation of wild-type p53 protein by HER-2/neu mediated PI3K pathway activation in human breast cancer cells: Its effect on cell proliferation and implication for therapy. Cell Res. 2004, 14, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, Y.; Ren, X.; Tsuyada, A.; Li, A.; Liu, L.J.; Wang, S.E. Context-dependent bidirectional regulation of the MutS homolog 2 by transforming growth factor beta contributes to chemoresistance in breast cancer cells. Mol. Cancer Res. 2010, 8, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.; Arteaga, C.L.; Wang, S.E. When tumor suppressor TGFβ meets the HER2 (ERBB2) oncogene. J. Mammary Gland Biol. Neoplasia 2011, 16, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.T.; Zlobin, A.; Osipo, C. Notch-EGFR/HER2 bidirectional crosstalk in breast cancer. Front. Oncol. 2014, 4, 360. [Google Scholar] [CrossRef] [PubMed]
- Rebay, I.; Fehon, R.G.; Artavanis-Tsakonas, S. Specific truncations of Drosophila Notch define dominant activated and dominant negative forms of the receptor. Cell 1993, 74, 319–329. [Google Scholar] [CrossRef]
- Rebay, I.; Fleming, R.J.; Fehon, R.G.; Cherbas, L.; Cherbas, P.; Artavanis-Tsakonas, S. Specific EGF repeats of Notch mediate interactions with Delta and Serrate: Implications for Notch as a multifunctional receptor. Cell 1991, 67, 687–699. [Google Scholar] [CrossRef]
- Fitzgerald, K.; Greenwald, I. Interchangeability of Caenorhabditis elegans DSL proteins and intrinsic signalling activity of their extracellular domains in vivo. Development 1995, 121, 4275–4282. [Google Scholar] [PubMed]
- Henderson, S.T.; Gao, D.; Christensen, S.; Kimble, J. Functional domains of LAG-2, a putative signaling ligand for LIN-12 and GLP-1 receptors in Caenorhabditis elegans. Mol. Biol. Cell 1997, 8, 1751–1762. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998, 393, 382–386. [Google Scholar] [PubMed]
- Mumm, J.S.; Schroeter, E.H.; Saxena, M.T.; Griesemer, A.; Tian, X.; Pan, D.J.; Ray, W.J.; Kopan, R. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch. Mol. Cell 2000, 5, 197–206. [Google Scholar] [CrossRef]
- Brou, C.; Logeat, F.; Gupta, N.; Bessia, C.; LeBail, O.; Doedens, J.R.; Cumano, A.; Roux, P.; Black, R.A.; Israel, A. A novel proteolytic cleavage involved in Notch signaling: The role of the disintegrin-metalloprotease TACE. Mol. Cell 2000, 5, 207–216. [Google Scholar] [CrossRef]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, R605–R615. [Google Scholar] [CrossRef] [PubMed]
- Bouras, T.; Pal, B.; Vaillant, F.; Harburg, G.; Asselin-Labat, M.-L.; Oakes, S.R.; Lindeman, G.J.; Visvader, J.E. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell 2008, 3, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Grudzien, P.; Lo, S.; Albain, K.S.; Robinson, P.; Rajan, P.; Strack, P.R.; Golde, T.E.; Miele, L.; Foreman, K.E. Inhibition of Notch signaling reduces the stem-like population of breast cancer cells and prevents mammosphere formation. Anticancer Res. 2010, 30, 3853–3867. [Google Scholar] [CrossRef] [PubMed]
- Farnie, G.; Clarke, R.B.; Spence, K.; Pinnock, N.; Brennan, K.; Anderson, N.G.; Bundred, N.J. Novel cell culture technique for primary ductal carcinoma in situ: Role of Notch and epidermal growth factor receptor signaling pathways. J. Natl. Cancer Inst. 2007, 99, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Harrison, H.; Farnie, G.; Howell, S.J.; Rock, R.E.; Stylianou, S.; Brennan, K.R.; Bundred, N.J.; Clarke, R.B. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010, 70, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Investig. 2007, 117, 3988–4002. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Storci, G.; Giovannini, C.; Pandolfi, S.; Pianetti, S.; Taffurelli, M.; Santini, D.; Ceccarelli, C.; Chieco, P.; Bonafe, M. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells 2007, 25, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Mo, Y.; Li, M.-T.; Zou, S.-W.; Cheng, Z.-L.; Sun, Y.-P.; Xiong, Y.; Guan, K.-L.; Lei, Q.-Y. NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. J. Clin. Investig. 2014, 124, 5453–5465. [Google Scholar] [CrossRef] [PubMed]
- Mamaeva, V.; Niemi, R.; Beck, M.; Ozliseli, E.; Desai, D.; Landor, S.; Gronroos, T.; Kronqvist, P.; Pettersen, I.K.N.; McCormack, E.; et al. Inhibiting Notch activity in breast cancer stem cells by glucose functionalized nanoparticles carrying gamma-secretase inhibitors. Mol. Ther. 2016, 24, 926–936. [Google Scholar] [CrossRef] [PubMed]
- McGowan, P.M.; Simedrea, C.; Ribot, E.J.; Foster, P.J.; Palmieri, D.; Steeg, P.S.; Allan, A.L.; Chambers, A.F. Notch1 inhibition alters the CD44hi/CD24lo population and reduces the formation of brain metastases from breast cancer. Mol. Cancer Res. 2011, 9, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.-K.; Kaushik, N.; Suh, Y.; Yoo, K.-C.; Cui, Y.-H.; Kim, M.-J.; Lee, H.-J.; Kim, I.-G.; Lee, S.-J. Radiation driven epithelial-mesenchymal transition is mediated by Notch signaling in breast cancer. Oncotarget 2016, 7, 53430–53442. [Google Scholar] [CrossRef] [PubMed]
- Azzam, D.J.; Zhao, D.; Sun, J.; Minn, A.J.; Ranganathan, P.; Drews-Elger, K.; Han, X.; Picon-Ruiz, M.; Gilbert, C.A.; Wander, S.A.; et al. Triple negative breast cancer initiating cell subsets differ in functional and molecular characteristics and in γ-secretase inhibitor drug responses. EMBO Mol. Med. 2013, 5, 1502–1522. [Google Scholar] [CrossRef] [PubMed]
- Noseda, M.; McLean, G.; Niessen, K.; Chang, L.; Pollet, I.; Montpetit, R.; Shahidi, R.; Dorovini-Zis, K.; Li, L.; Beckstead, B.; et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ. Res. 2004, 94, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertran, E.; Perez-Pomares, J.M.; Diez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisua-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef] [PubMed]
- Niessen, K.; Fu, Y.; Chang, L.; Hoodless, P.A.; McFadden, D.; Karsan, A. Slug is a direct Notch target required for initiation of cardiac cushion cellularization. J. Cell Biol. 2008, 182, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.G.; Niessen, K.; Kulic, I.; Raouf, A.; Eaves, C.; Pollet, I.; Karsan, A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J. Exp. Med. 2007, 204, 2935–2948. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Zhao, X.; Zhang, X.; Luo, M.; Zuo, X.; Huang, S.; Wang, Y.; Gu, S.; Zhao, X. Notch1 signaling regulates the epithelial-mesenchymal transition and invasion of breast cancer in a Slug-dependent manner. Mol. Cancer 2015, 14, 28. [Google Scholar] [CrossRef] [PubMed]
- Sahlgren, C.; Gustafsson, M.V.; Jin, S.; Poellinger, L.; Lendahl, U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 6392–6397. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Subramanyam, D.; Dey, D.; Kumar, R.V.; Rangarajan, A. Cooperation of Notch and Ras/MAPK signaling pathways in human breast carcinogenesis. Mol. Cancer 2009, 8, 128. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Xin, X.; Liu, L.; Tutunea-Fatan, E.; Rodriguez-Torres, M.; Vincent, K.; Postovit, L.-M.; Hess, D.; Lala, P.K. COX-2 induces breast cancer stem cells via EP4/PI3K/AKT/NOTCH/WNT axis. Stem Cells 2016, 34, 2290–2305. [Google Scholar] [CrossRef] [PubMed]
- Osipo, C.; Patel, P.; Rizzo, P.; Clementz, A.G.; Hao, L.; Golde, T.E.; Miele, L. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a gamma-secretase inhibitor. Oncogene 2008, 27, 5019–5032. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.; Yang, W.; Oh, S.; Nam, K.; Lee, K.; Noh, D.; Shin, I. HER2 stabilizes survivin while concomitantly down-regulating survivin gene transcription by suppressing Notch cleavage. Biochem. J. 2013, 451, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Pandya, K.; Wyatt, D.; Gallagher, B.; Shah, D.; Baker, A.; Bloodworth, J.; Zlobin, A.; Pannuti, A.; Green, A.; Ellis, I.O.; et al. PKCα attenuates Jagged-1-mediated Notch signaling in ErbB-2 positive breast cancer to reverse trastuzumab resistance. Clin. Cancer Res. 2016, 22, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Pandya, K.; Meeke, K.; Clementz, A.G.; Rogowski, A.; Roberts, J.; Miele, L.; Albain, K.S.; Osipo, C. Targeting both Notch and ErbB-2 signalling pathways is required for prevention of ErbB-2-positive breast tumour recurrence. Br. J. Cancer 2011, 105, 796–806. [Google Scholar] [CrossRef] [PubMed]
- Rattner, A.; Hsieh, J.C.; Smallwood, P.M.; Gilbert, D.J.; Copeland, N.G.; Jenkins, N.A.; Nathans, J. A family of secreted proteins contains homology to the cysteine-rich ligand-binding domain of frizzled receptors. Proc. Natl. Acad. Sci. USA 1997, 94, 2859–2863. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.-C.; Lee, L.; Zhang, L.; Wefer, S.; Brown, K.; DeRossi, C.; Wines, M.E.; Rosenquist, T.; Holdener, B.C. Mesd encodes an LRP5/6 chaperone essential for specification of mouse embryonic polarity. Cell 2003, 112, 355–367. [Google Scholar] [CrossRef]
- Mikels, A.J.; Nusse, R. Wnts as ligands: Processing, secretion and reception. Oncogene 2006, 25, 7461–7468. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Zeng, X.; Liu, C.; Zhang, X.; Harada, Y.; Chang, Z.; He, X. A mechanism for Wnt coreceptor activation. Mol. Cell 2004, 13, 149–156. [Google Scholar] [CrossRef]
- Behrens, J.; Jerchow, B.A.; Wurtele, M.; Grimm, J.; Asbrand, C.; Wirtz, R.; Kuhl, M.; Wedlich, D.; Birchmeier, W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science 1998, 280, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Seeling, J.M.; Miller, J.R.; Gil, R.; Moon, R.T.; White, R.; Virshup, D.M. Regulation of beta-catenin signaling by the B56 subunit of protein phosphatase 2A. Science 1999, 283, 2089–2091. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.-H.; Seeling, J.M.; Hill, V.; Yochum, A.; Virshup, D.M. Casein kinase I phosphorylates and destabilizes the beta-catenin degradation complex. Proc. Natl. Acad. Sci. USA 2002, 99, 1182–1187. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Wang, J.; Liu, B.; Pan, W.; Farr, G.H., 3rd; Flynn, C.; Yuan, H.; Takada, S.; Kimelman, D.; Li, L.; et al. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol. Cell 2001, 7, 801–809. [Google Scholar] [CrossRef]
- Brannon, M.; Gomperts, M.; Sumoy, L.; Moon, R.T.; Kimelman, D. A beta-catenin/XTcf-3 complex binds to the siamois promoter to regulate dorsal axis specification in Xenopus. Genes Dev. 1997, 11, 2359–2370. [Google Scholar] [CrossRef] [PubMed]
- Brunner, E.; Peter, O.; Schweizer, L.; Basler, K. pangolin encodes a Lef-1 homologue that acts downstream of Armadillo to transduce the Wingless signal in Drosophila. Nature 1997, 385, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.Y.; Hens, J.; Andl, T.; Kairo, A.; Yamaguchi, T.P.; Brisken, C.; Glick, A.; Wysolmerski, J.J.; Millar, S.E. Canonical WNT signaling promotes mammary placode development and is essential for initiation of mammary gland morphogenesis. Development 2004, 131, 4819–4829. [Google Scholar] [CrossRef] [PubMed]
- Veltmaat, J.M.; Van Veelen, W.; Thiery, J.P.; Bellusci, S. Identification of the mammary line in mouse by Wnt10b expression. Dev. Dyn. 2004, 229, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Van Amerongen, R.; Bowman, A.N.; Nusse, R. Developmental stage and time dictate the fate of Wnt/beta-catenin-responsive stem cells in the mammary gland. Cell Stem Cell 2012, 11, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.; Ablett, M.P.; Spence, K.; Landberg, G.; Sims, A.H.; Clarke, R.B. Wnt pathway activity in breast cancer sub-types and stem-like cells. PLoS ONE 2013, 8, e67811. [Google Scholar] [CrossRef] [PubMed]
- Khalil, S.; Tan, G.A.; Giri, D.D.; Zhou, X.K.; Howe, L.R. Activation status of Wnt/ss-catenin signaling in normal and neoplastic breast tissues: Relationship to HER2/neu expression in human and mouse. PLoS ONE 2012, 7, e33421. [Google Scholar] [CrossRef] [PubMed]
- Woodward, W.A.; Chen, M.S.; Behbod, F.; Alfaro, M.P.; Buchholz, T.A.; Rosen, J.M. WNT/β-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc. Natl. Acad. Sci. USA 2007, 104, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ginther, C.; Kim, J.; Mosher, N.; Chung, S.; Slamon, D.; Vadgama, J.V. Expression of Wnt3 activates Wnt/beta-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol. Cancer Res. 2012, 10, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Eger, A.; Stockinger, A.; Schaffhauser, B.; Beug, H.; Foisner, R. Epithelial mesenchymal transition by c-Fos estrogen receptor activation involves nuclear translocation of beta-catenin and upregulation of beta-catenin/lymphoid enhancer binding factor-1 transcriptional activity. J. Cell Biol. 2000, 148, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lu, Z.; Hay, E.D. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol. Int. 2002, 26, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Brenot, A.; Lawson, D.A.; Linneman, J.; Van Kappel, E.C.; Wong, K.; de Sauvage, F.; Klein, O.D.; Werb, Z. Lgr5 expressing cells are sufficient and necessary for postnatal mammary gland organogenesis. Cell Rep. 2013, 3, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Oskarsson, T.; Acharyya, S.; Zhang, X.H.-F.; Vanharanta, S.; Tavazoie, S.F.; Morris, P.G.; Downey, R.J.; Manova-Todorova, K.; Brogi, E.; Massagué, J. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat. Med. 2011, 17, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Tang, H.; Kong, Y.; Xie, X.; Chen, J.; Song, C.; Liu, X.; Ye, F.; Li, N.; Wang, N.; et al. LGR5 promotes breast cancer progression and maintains stem-like cells through activation of Wnt/beta-catenin signaling. Stem Cells 2015, 33, 2913–2924. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-W.; Su, Y.-J.; Hsiao, M.; Wei, K.-C.; Lin, W.-H.; Liang, C.-L.; Chen, S.-C.; Lee, J.-L. Diverse targets of beta-catenin during the epithelial-mesenchymal transition define cancer stem cells and predict disease relapse. Cancer Res. 2015, 75, 3398–3410. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, J.A.; Adriance, M.C.; McConnell, E.J.; Thompson, M.C.; Pockaj, B.; Gendler, S.J. ErbB-beta-catenin complexes are associated with human infiltrating ductal breast and murine mammary tumor virus (MMTV)-Wnt-1 and MMTV-c-Neu transgenic carcinomas. J. Biol. Chem. 2002, 277, 22692–22698. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ma, Q.; Ren, Y.; He, J.; Zhang, Y.; Zhang, Y.; Chen, W. Geldanamycin destabilizes HER2 tyrosine kinase and suppresses Wnt/beta-catenin signaling in HER2 overexpressing human breast cancer cells. Oncol. Rep. 2007, 17, 89–96. [Google Scholar] [PubMed]
- Yamaguchi, H.; Chang, S.-S.; Hsu, J.L.; Hung, M.-C. Signaling cross-talk in the resistance to HER family receptor targeted therapy. Oncogene 2014, 33, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Khramtsov, A.I.; Khramtsova, G.F.; Tretiakova, M.; Huo, D.; Olopade, O.I.; Goss, K.H. Wnt/β-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am. J. Pathol. 2010, 176, 2911–2920. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; Young, B.; Abramovitz, M.; Bouzyk, M.; Barwick, B.; De, P.; Leyland-Jones, B. Differential activation of Wnt-β-catenin pathway in triple negative breast cancer increases MMP7 in a PTEN dependent manner. PLoS ONE 2013, 8, e77425. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Boulton, T.G.; Farruggella, T.; Ip, N.Y.; Davis, S.; Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Barbieri, G.; Pellegrini, S. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 1994, 263, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Argetsinger, L.S.; Campbell, G.S.; Yang, X.; Witthuhn, B.A.; Silvennoinen, O.; Ihle, J.N.; Carter-Su, C. Identification of JAK2 as a growth hormone receptor-associated tyrosine kinase. Cell 1993, 74, 237–244. [Google Scholar] [CrossRef]
- Heim, M.H.; Kerr, I.M.; Stark, G.R.; Darnell, J.E.J. Contribution of STAT SH2 groups to specific interferon signaling by the Jak-STAT pathway. Science 1995, 267, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Ziemiecki, A.; Wilks, A.F.; Harpur, A.G.; Sadowski, H.B.; Gilman, M.Z.; Darnell, J.E. Polypeptide signalling to the nucleus through tyrosine phosphorylation of Jak and Stat proteins. Nature 1993, 366, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Vinkemeier, U.; Cohen, S.L.; Moarefi, I.; Chait, B.T.; Kuriyan, J.; Darnell, J.E.J. DNA binding of in vitro activated Stat1 alpha, Stat1 beta and truncated Stat1: Interaction between NH2-terminal domains stabilizes binding of two dimers to tandem DNA sites. EMBO J. 1996, 15, 5616–5626. [Google Scholar] [PubMed]
- Wang, X.; Wang, G.; Zhao, Y.; Liu, X.; Ding, Q.; Shi, J.; Ding, Y.; Wang, S. STAT3 mediates resistance of CD44(+)CD24(-/low) breast cancer stem cells to tamoxifen in vitro. J. Biomed. Res. 2012, 26, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Tweardy, D.J.; Zhang, M.; Zhang, X.; Landua, J.; Petrovic, I.; Bu, W.; Roarty, K.; Hilsenbeck, S.G.; Rosen, J.M.; et al. STAT3 signaling is activated preferentially in tumor-initiating cells in claudin-low models of human breast cancer. Stem Cells 2014, 32, 2571–2582. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Hutzen, B.; Lee, H.-F.; Peng, Z.; Wang, W.; Zhao, C.; Lin, H.-J.; Sun, D.; Li, P.-K.; Li, C.; et al. Evaluation of STAT3 signaling in ALDH+ and ALDH+/CD44+/CD24− subpopulations of breast cancer cells. PLoS ONE 2013, 8, e82821. [Google Scholar] [CrossRef] [PubMed]
- Dave, B.; Landis, M.D.; Tweardy, D.J.; Chang, J.C.; Dobrolecki, L.E.; Wu, M.-F.; Zhang, X.; Westbrook, T.F.; Hilsenbeck, S.G.; Liu, D.; et al. Selective small molecule Stat3 inhibitor reduces breast cancer tumor-initiating cells and improves recurrence free survival in a human-xenograft model. PLoS ONE 2012, 7, e30207. [Google Scholar] [CrossRef]
- So, J.Y.; Smolarek, A.K.; Salerno, D.M.; Maehr, H.; Uskokovic, M.; Liu, F.; Suh, N. Targeting CD44-STAT3 signaling by Gemini vitamin D analog leads to inhibition of invasion in basal-like breast cancer. PLoS ONE 2013, 8, e54020. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Vargas, H.; Ouzounova, M.; Le Calvez-Kelm, F.; Lambert, M.-P.; McKay-Chopin, S.; Tavtigian, S.V.; Puisieux, A.; Matar, C.; Herceg, Z. Methylome analysis reveals Jak-STAT pathway deregulation in putative breast cancer stem cells. Epigenetics 2011, 6, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Yao, Q.; Liu, Y.; Du, S.; Liu, A.; Guo, Z.; Sun, A.; Ruan, J.; Chen, L.; Ye, C.; Yuan, Y. IL-6-induced epithelial-mesenchymal transition promotes the generation of breast cancer stem-like cells analogous to mammosphere cultures. Int. J. Oncol. 2012, 40, 1171–1179. [Google Scholar] [PubMed]
- Zhang, F.; Li, C.; Halfter, H.; Liu, J. Delineating an oncostatin M-activated STAT3 signaling pathway that coordinates the expression of genes involved in cell cycle regulation and extracellular matrix deposition of MCF-7 cells. Oncogene 2003, 22, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Schwarzbauer, J.E. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene 2014, 33, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Azimi, I.; Faville, R.A.; Peters, A.A.; Jalink, K.; Putney, J.W.J.; Goodhill, G.J.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Induction of epithelial-mesenchymal transition (EMT) in breast cancer cells is calcium signal dependent. Oncogene 2014, 33, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Olayioye, M.A.; Beuvink, I.; Horsch, K.; Daly, J.M.; Hynes, N.E. ErbB receptor-induced activation of stat transcription factors is mediated by Src tyrosine kinases. J. Biol. Chem. 1999, 274, 17209–17218. [Google Scholar] [CrossRef] [PubMed]
- DeArmond, D.; Brattain, M.G.; Jessup, J.M.; Kreisberg, J.; Malik, S.; Zhao, S.; Freeman, J.W. Autocrine-mediated ErbB-2 kinase activation of STAT3 is required for growth factor independence of pancreatic cancer cell lines. Oncogene 2003, 22, 7781–7795. [Google Scholar] [CrossRef] [PubMed]
- Hawthorne, V.S.; Huang, W.-C.; Neal, C.L.; Tseng, L.-M.; Hung, M.-C.; Yu, D. ErbB2-mediated Src and STAT3 activation leads to transcriptional ppregulation of p21(Cip1) and chemoresistance in breast cancer cells. Mol. Cancer Res. 2009, 7, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Quaglino, E.; Maritano, D.; Pannellini, T.; Riera, L.; Cavallo, F.; Forni, G.; Musiani, P.; Chiarle, R.; Poli, V. Stat3 is required for anchorage-independent growth and metastasis but not for mammary tumor development downstream of the ErbB-2 oncogene. Mol. Carcinog. 2010, 49, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Yang, X.-Y.; Glass, O.; Lei, G.; Osada, T.; Dave, S.S.; Morse, M.A.; Clay, T.M.; Lyerly, H.K. HER2 overexpression elicits a pro-inflammatory IL-6 autocrine signaling loop that is critical for tumorigenesis. Cancer Res. 2011, 71, 4380–4391. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Giehl, N.; Wu, Y.; Vadgama, J.V. STAT3 activation in HER2-overexpressing breast cancer promotes epithelial-mesenchymal transition and cancer stem cell traits. Int. J. Oncol. 2014, 44, 403–411. [Google Scholar] [PubMed]
- Espinoza, I.; Pochampally, R.; Xing, F.; Watabe, K.; Miele, L. Notch signaling: Targeting cancer stem cells and epithelial-to-mesenchymal transition. Onco. Targets. Ther. 2013, 6, 1249–1259. [Google Scholar] [PubMed]
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Lanske, B.; Karaplis, A.C.; Lee, K.; Luz, A.; Vortkamp, A.; Pirro, A.; Karperien, M.; Defize, L.H.; Ho, C.; Mulligan, R.C.; et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 1996, 273, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Parmantier, E.; Lynn, B.; Lawson, D.; Turmaine, M.; Namini, S.S.; Chakrabarti, L.; McMahon, A.P.; Jessen, K.R.; Mirsky, R. Schwann cell-derived Desert hedgehog controls the development of peripheral nerve sheaths. Neuron 1999, 23, 713–724. [Google Scholar] [CrossRef]
- Alcedo, J.; Ayzenzon, M.; Von Ohlen, T.; Noll, M.; Hooper, J.E. The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell 1996, 86, 221–232. [Google Scholar] [CrossRef]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.R.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol. Cell. Biol. 1990, 10, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Ruppert, J.M.; Bigner, S.H.; Vogelstein, B. The GLI gene is a member of the Kruppel family of zinc finger proteins. Nature 1988, 332, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-H.; Wilson, C.W.; Li, Y.-J.; Law, K.K.L.; Lu, C.-S.; Gacayan, R.; Zhang, X.; Hui, C.; Chuang, P.-T. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.O.-L.; Zhang, X.; Ribeiro, A.; Mo, R.; Makino, S.; Puviindran, V.; Law, K.K.L.; Briscoe, J.; Hui, C.-C. The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci. Signal. 2009, 2, ra29. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog signaling in the maintenance of cancer stem cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Tang, H.; Xiao, Q.; He, M.; Zhao, L.; Fu, Y.; Wu, H.; Yu, Z.; Jiang, Q.; Yan, Y.; et al. The Hedgehog signaling pathway is associated with poor prognosis in breast cancer patients with the CD44+/CD24 phenotype. Mol. Med. Rep. 2016, 14, 5261–5270. [Google Scholar] [CrossRef] [PubMed]
- Shipitsin, M.; Campbell, L.L.; Argani, P.; Weremowicz, S.; Bloushtain-Qimron, N.; Yao, J.; Nikolskaya, T.; Serebryiskaya, T.; Beroukhim, R.; Hu, M.; et al. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007, 11, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Nakamura, M.; Kameda, C.; Kubo, M.; Sato, N.; Kuroki, S.; Tanaka, M.; Katano, M. The Hedgehog signaling pathway plays an essential role in maintaining the CD44+CD24-/low subpopulation and the side population of breast cancer cells. Anticancer Res. 2009, 29, 2147–2157. [Google Scholar] [PubMed]
- Yang, N.; Zhou, T.-C.; Lei, X.; Wang, C.; Yan, M.; Wang, Z.-F.; Liu, W.; Wang, J.; Ming, K.-H.; Wang, B.-C.; et al. Inhibition of Sonic Hedgehog signaling pathway by thiazole antibiotic thiostrepton attenuates the CD44+/CD24− stem-like population and sphere-forming capacity in triple-negative breast cancer. Cell. Physiol. Biochem. 2016, 38, 1157–1170. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Fu, Y.; Yan, Y.; Xiao, Q.; Wu, H.; Yao, W.; Zhao, H.; Zhao, L.; Jiang, Q.; Yu, Z.; et al. The Hedgehog signalling pathway mediates drug response of MCF-7 mammosphere cells in breast cancer patients. Clin. Sci. 2015, 129, 809–822. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhang, N.; Wang, X.; Li, Y.; Qi, W.; Zhang, H.; Li, Z.; Yang, Q. Hedgehog pathway is involved in nitidine chloride induced inhibition of epithelial-mesenchymal transition and cancer stem cells-like properties in breast cancer cells. Cell Biosci. 2016, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Ma, W.; Mao, J.; Yu, X.; Hou, Z.; Fan, S.; Song, B.; Wang, H.; Li, J.; Kang, L.; et al. Salinomycin exerts anticancer effects on human breast carcinoma MCF-7 cancer stem cells via modulation of Hedgehog signaling. Chem. Biol. Interact. 2015, 228, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-Z.; Yan, Y.-Y.; He, M.; Xiao, Q.-H.; Yao, W.-F.; Zhao, L.; Wu, H.-Z.; Yu, Z.-J.; Zhou, M.-Y.; Lv, M.-T.; et al. Salinomycin induces selective cytotoxicity to MCF-7 mammosphere cells through targeting the Hedgehog signaling pathway. Oncol. Rep. 2016, 35, 912–922. [Google Scholar] [CrossRef] [PubMed]
- Memmi, E.M.; Sanarico, A.G.; Giacobbe, A.; Peschiaroli, A.; Frezza, V.; Cicalese, A.; Pisati, F.; Tosoni, D.; Zhou, H.; Tonon, G.; et al. p63 Sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 3499–3504. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Qu, Y.; Yu-Rice, Y.; Johnson, J.; Cui, X. FOXC1-induced Gli2 activation: A non-canonical pathway contributing to stemness and anti-Hedgehog resistance in basal-like breast cancer. Mol. Cell. Oncol. 2016, 3, e1131668. [Google Scholar] [CrossRef] [PubMed]
- Benvenuto, M.; Masuelli, L.; De Smaele, E.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Di Stefano, E.; Frajese, G.V.; Orlandi, A.; et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget 2016, 7, 9250–9270. [Google Scholar] [PubMed]
- Matevossian, A.; Resh, M.D. Hedgehog Acyltransferase as a target in estrogen receptor positive, HER2 amplified, and tamoxifen resistant breast cancer cells. Mol. Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.; Lu, Y.; Teng, K.; Nuova, G.; Li, X.; Shapiro, C.L.; Majumder, S. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012, 72, 5048–5059. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.; Regl, G.; Hofbauer, S.W.; Altenhofer, P.; Achatz, G.; Dlugosz, A.; Schnidar, H.; Greil, R.; Hartmann, T.N.; Aberger, F. Hedgehog/GLI and PI3K signaling in the initiation and maintenance of chronic lymphocytic leukemia. Oncogene 2015, 34, 5341–5351. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Caiping, S.; Qing, Z.; Xiaojing, W. Sonic hedgehog-Gli1 signals promote epithelial-mesenchymal transition in ovarian cancer by mediating PI3K/AKT pathway. Med. Oncol. 2015, 32, 368. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Nanta, R.; Sharma, J.; Gunewardena, S.; Singh, K.P.; Shankar, S.; Srivastava, R.K. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget 2015, 6, 32039–32060. [Google Scholar] [PubMed]
- Kebenko, M.; Drenckhan, A.; Gros, S.J.; Jucker, M.; Grabinski, N.; Ewald, F.; Grottke, A.; Schultze, A.; Izbicki, J.R.; Bokemeyer, C.; et al. ErbB2 signaling activates the Hedgehog pathway via PI3K-Akt in human esophageal adenocarcinoma: Identification of novel targets for concerted therapy concepts. Cell. Signal. 2015, 27, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinases as breast cancer drivers and therapeutic targets. Front. Biosci. 2015, 20, 1144–1163. [Google Scholar]
- Cathcarta, J.; Pulkoski-Grossa, A.; Cao, J. Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [PubMed]
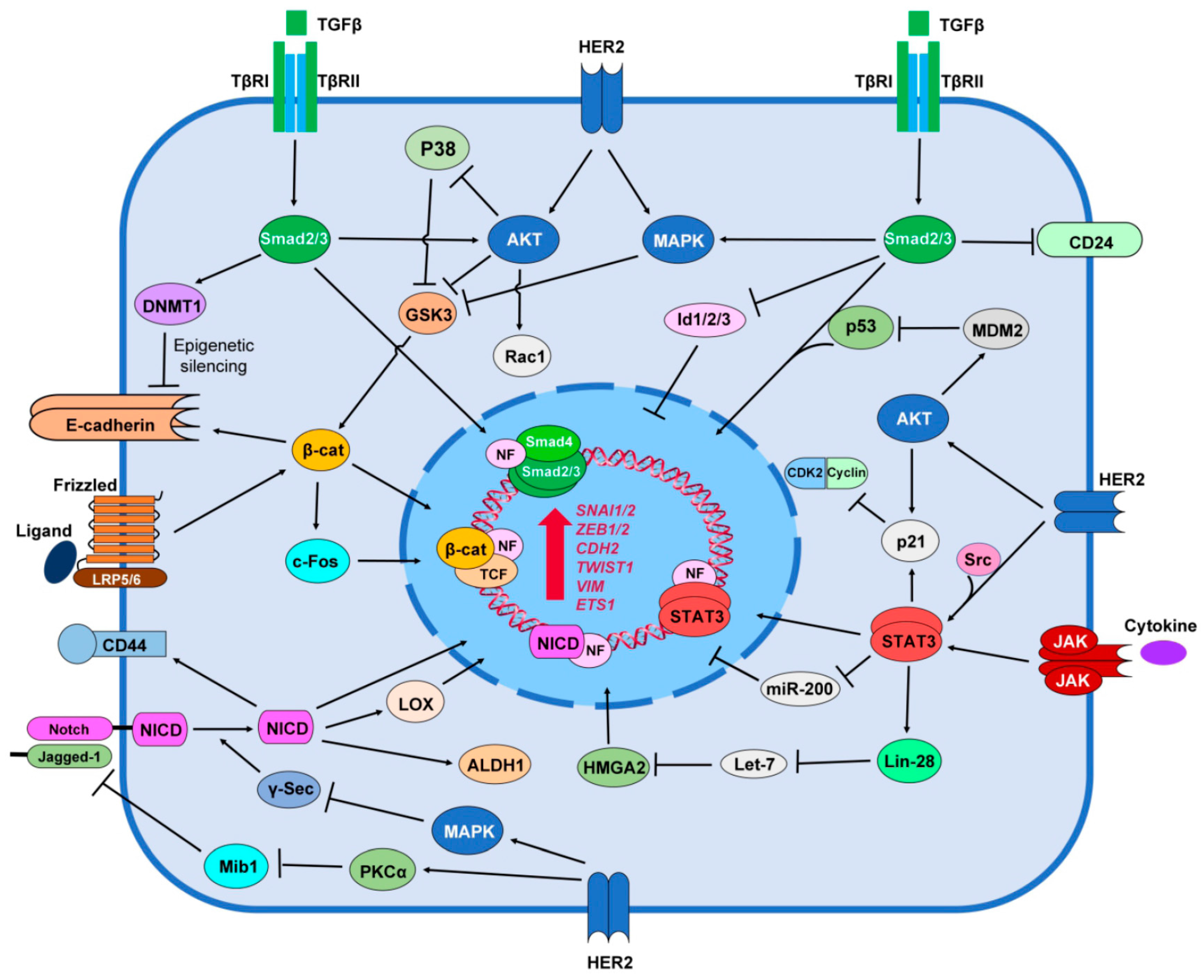
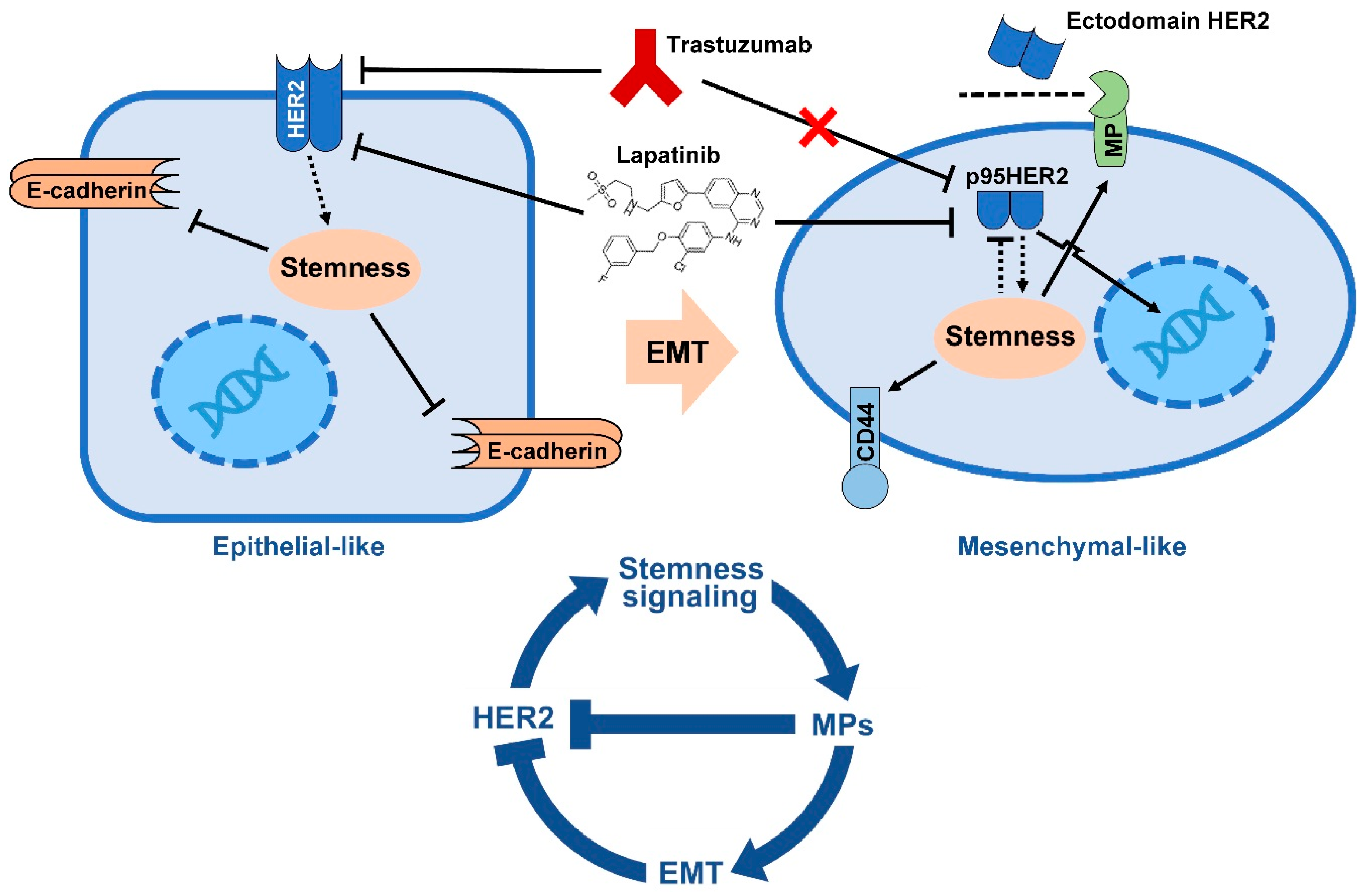
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nami, B.; Wang, Z. HER2 in Breast Cancer Stemness: A Negative Feedback Loop towards Trastuzumab Resistance. Cancers 2017, 9, 40. https://doi.org/10.3390/cancers9050040
Nami B, Wang Z. HER2 in Breast Cancer Stemness: A Negative Feedback Loop towards Trastuzumab Resistance. Cancers. 2017; 9(5):40. https://doi.org/10.3390/cancers9050040
Chicago/Turabian StyleNami, Babak, and Zhixiang Wang. 2017. "HER2 in Breast Cancer Stemness: A Negative Feedback Loop towards Trastuzumab Resistance" Cancers 9, no. 5: 40. https://doi.org/10.3390/cancers9050040
APA StyleNami, B., & Wang, Z. (2017). HER2 in Breast Cancer Stemness: A Negative Feedback Loop towards Trastuzumab Resistance. Cancers, 9(5), 40. https://doi.org/10.3390/cancers9050040