Ubiquitin Specific Peptidase 22 Regulates Histone H2B Mono-Ubiquitination and Exhibits Both Oncogenic and Tumor Suppressor Roles in Cancer


Abstract
1. Introduction
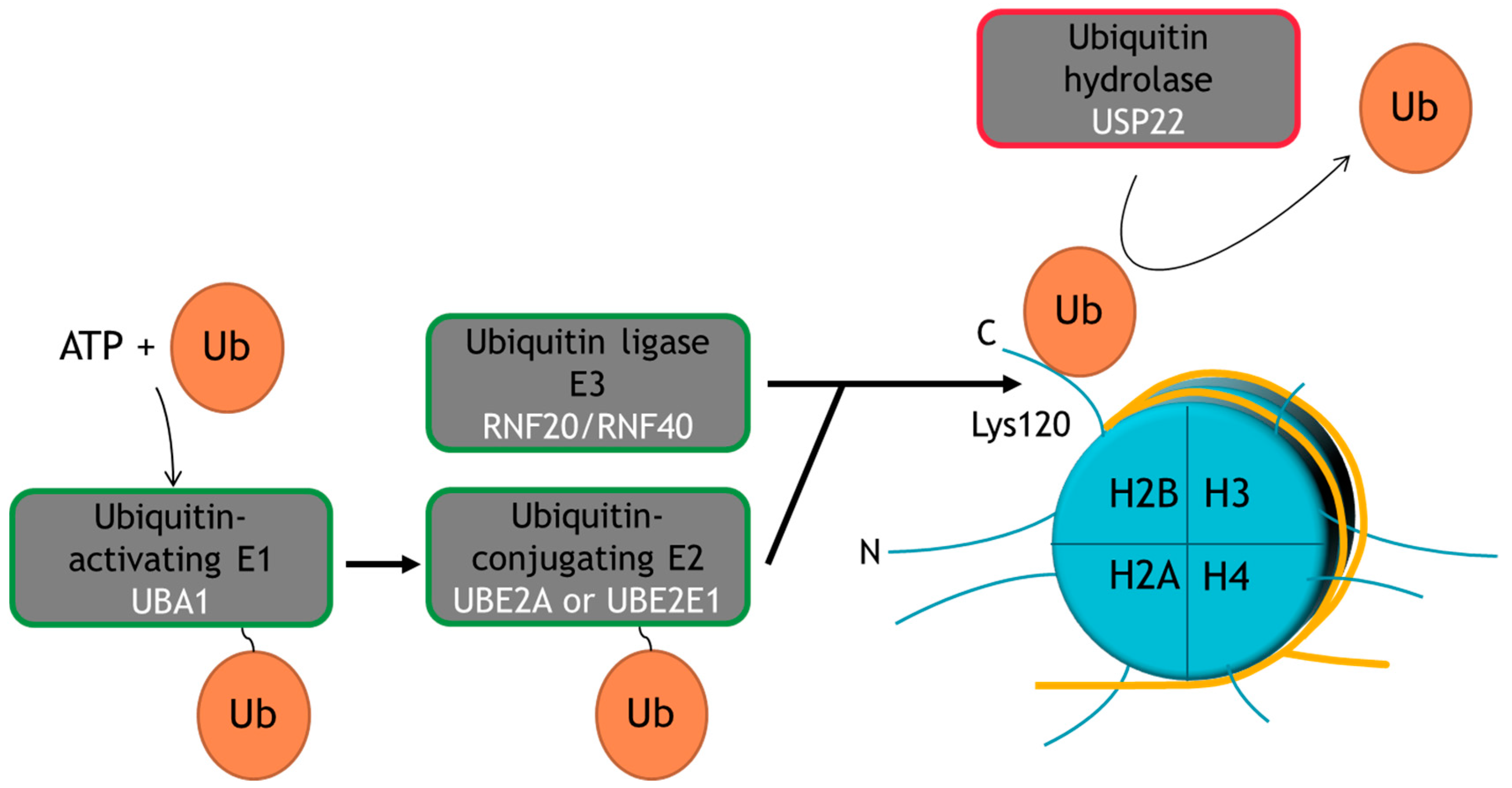
2. Ubiquitination Is a Multi-Functional PTM
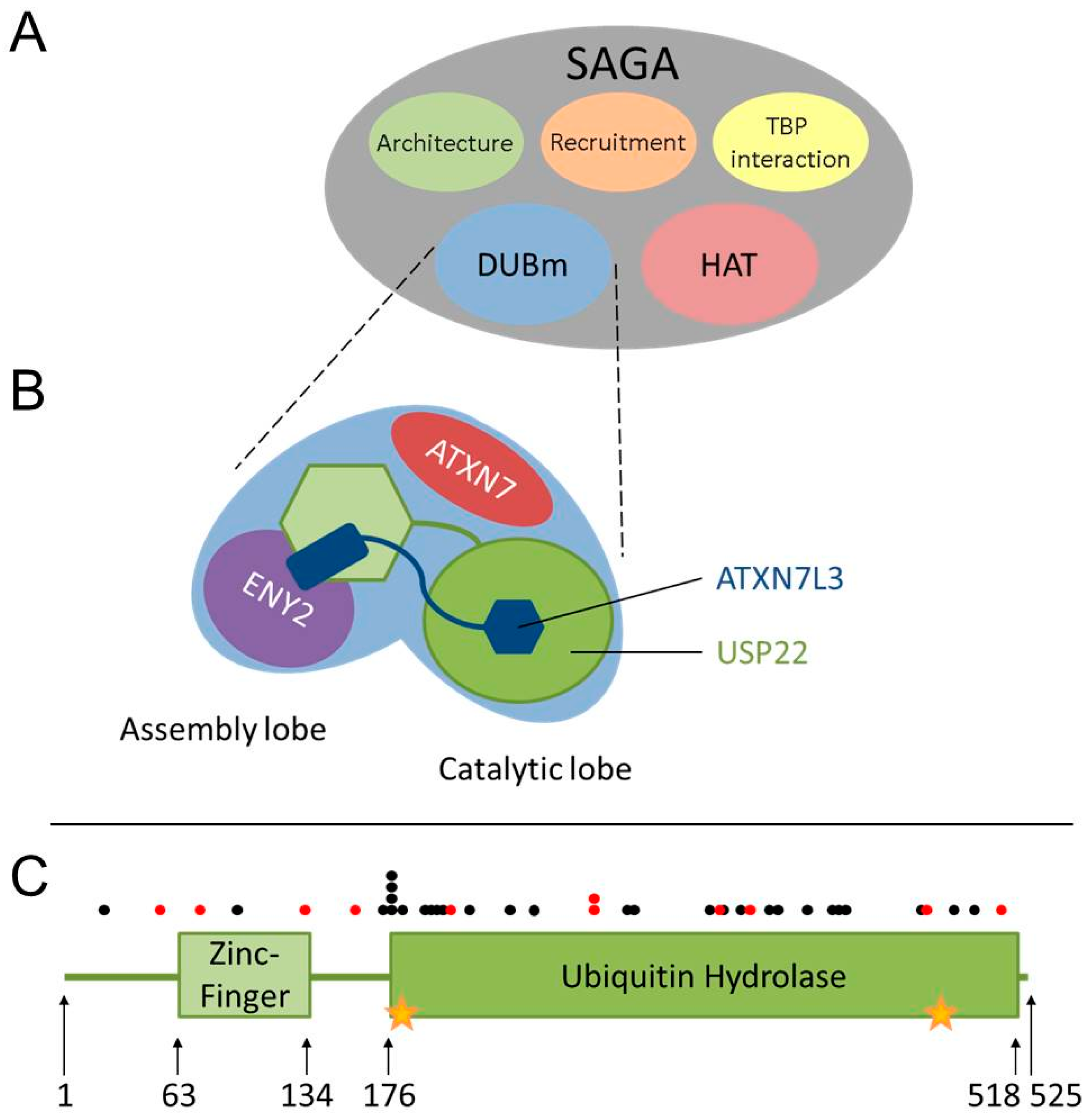
3. USP22 Is an Evolutionarily-Conserved Ubiquitin Hydrolase within the SAGA (Spt-Ada-Gcn5 acetyltransferase) Complex
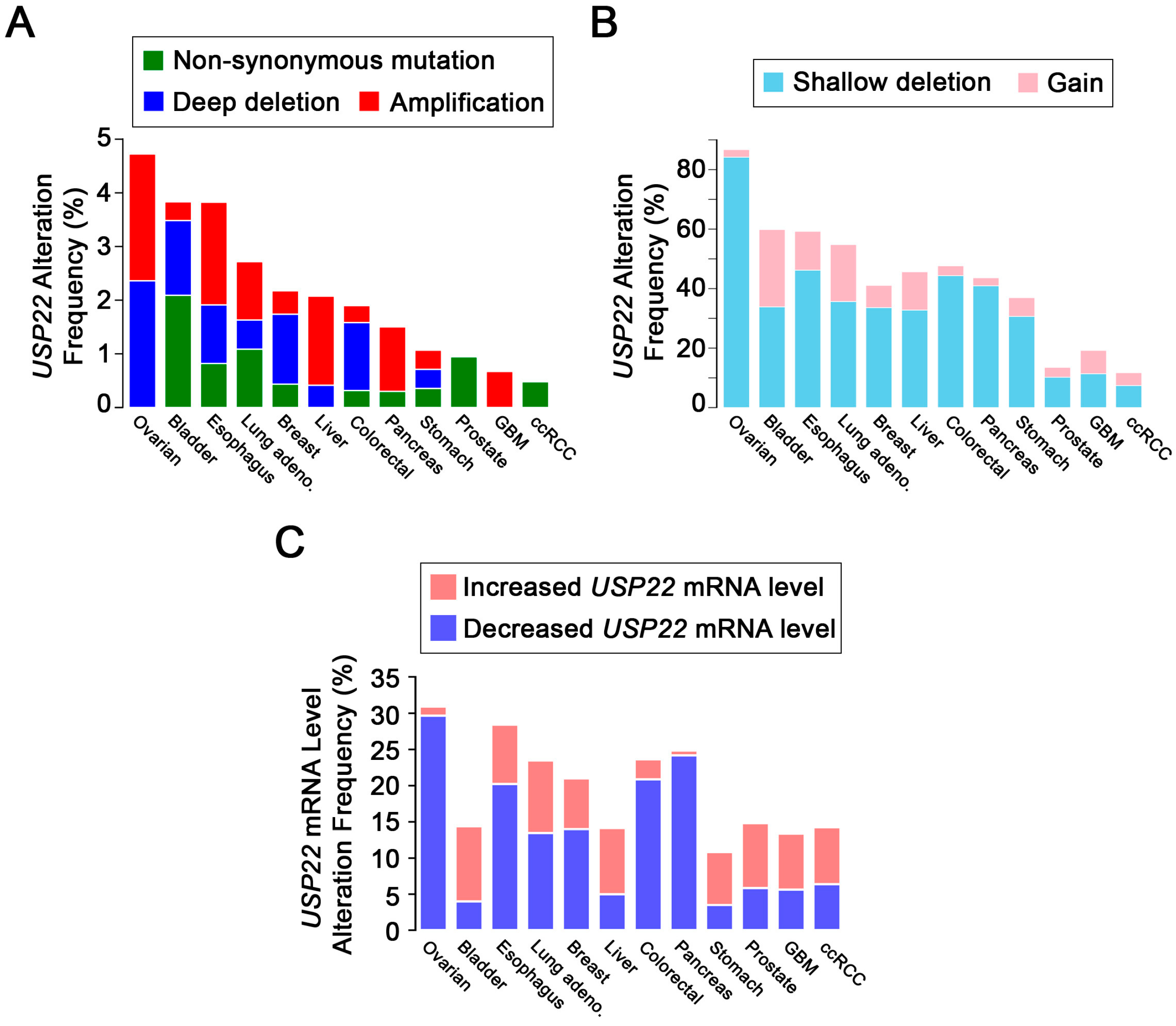
4. USP22 Expression Is Frequently Altered in Cancer
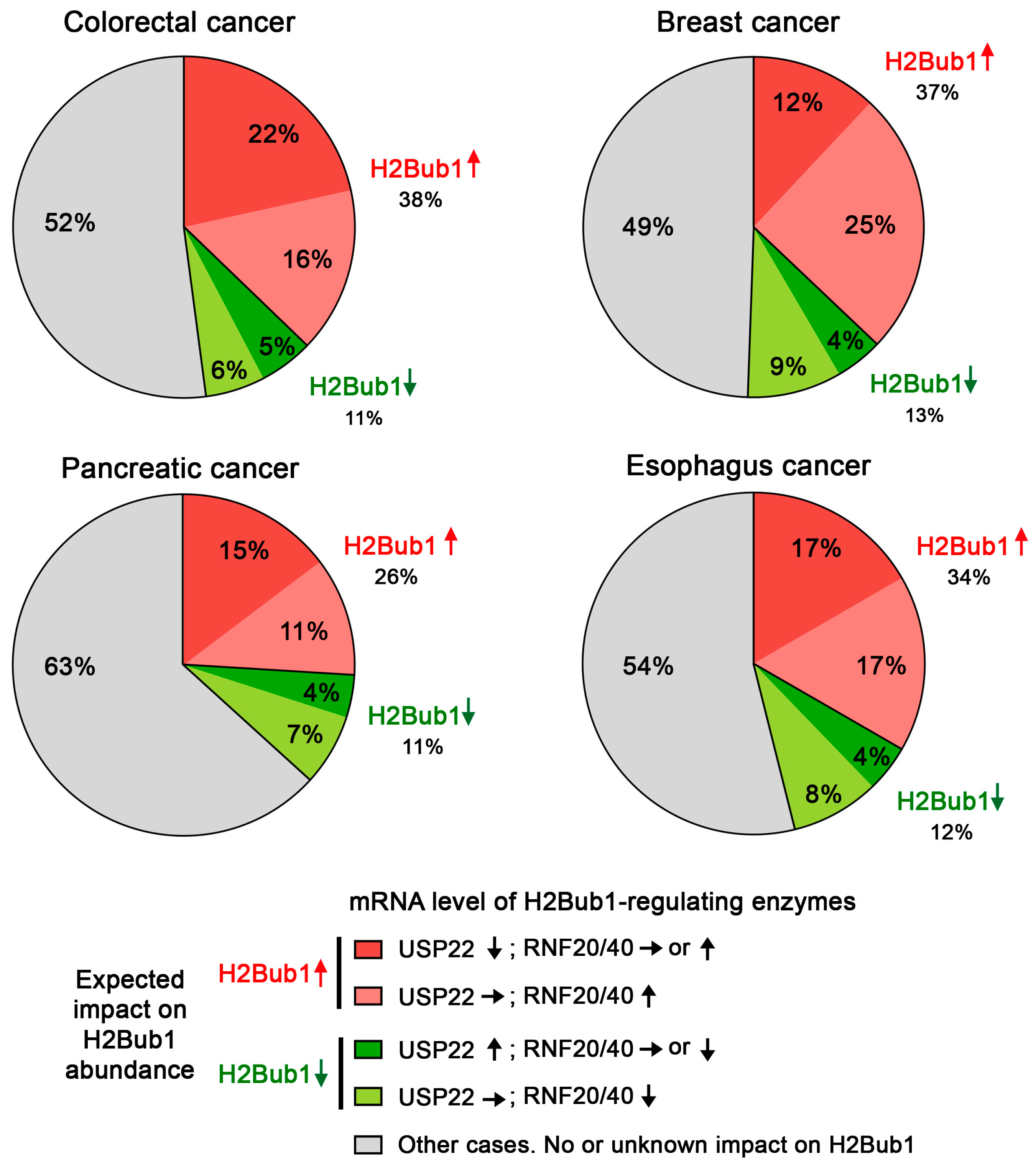
5. The Challenges of Evaluating USP22 Activity in Cancer
6. USP22 Is a Transcriptional Activator
7. USP22 Regulates Cell Death to Enhance Resistance to Chemotherapy
8. USP22 Modulates Cell Cycle Progression
9. Diminished USP22 Expression Compromises Genomic Stability
10. Summary and Conclusions
Acknowledgments
Conflicts of Interest
References
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Füllgrabe, J.; Kavanagh, E.; Joseph, B. Histone onco-modifications. Oncogene 2011, 30, 3391–3403. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications—miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.L.; Guppy, B.J.; Sawchuk, L.; Davie, J.R.; Mcmanus, K.J. Regulation of chromatin structure via histone post-translational modification and the link to carcinogenesis. Cancer Metastasis Rev. 2013, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Varthi, M.; Sykes, S.M.; Phillips, C.; Warzecha, C.; Zhu, W.; Wyce, A.; Thorne, A.W.; Berger, S.L.; McMahon, S.B. The putative cancer stem cell marker USP22 is a subunit of the human SAGA complex required for activated transcription and cell-cycle progression. Mol. Cell 2008, 29, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Pfeiffer, H.; Thorne, A.; McMahon, S.B. USP22, an hSAGA subunit and potential cancer stem cell marker, reverses the polycomb-catalyzed ubiquitylation of histone H2A. Cell Cycle 2008, 7, 1522–1524. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Yang, Y.M.; Xu, H.; Dong, X.S. Increased expression of ubiquitin-specific protease 22 can promote cancer progression and predict therapy failure in human colorectal cancer. J. Gastroenterol. Hepatol. 2010, 25, 1800–1805. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, L.; Guo, T.; Wang, Y.; Yang, J. Decreased H2B monoubiquitination and overexpression of ubiquitin-specific protease enzyme 22 in malignant colon carcinoma. Hum. Pathol. 2015, 46, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.-D.; Cui, B.-B.; Sun, L.; Zheng, H.; Huang, Q.; Tong, J.-X.; Zhang, Q.-F. The Co-expression of USP22 and BMI-1 May Promote Cancer Progression and Predict Therapy Failure in Gastric Carcinoma. Cell Biochem. Biophys. 2011, 61, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Y.-P.; Chen, J.-H.; Yuan, S.-F.; Wang, L.; Zhang, J.-L.; Yao, Q.; Li, N.-L.; Bian, J.-F.; Fan, J.; et al. Prognostic significance of USP22 as an oncogene in papillary thyroid carcinoma. Tumor Biol. 2013, 34, 1635–1639. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Ning, Z.; Gao, W.; Ling, J.; Wang, A.; Luo, H.; Liang, Y.; Yan, Q.; Wang, Z. Ubiquitin-specific protease 22-induced autophagy is correlated with poor prognosis of pancreatic cancer. Oncol. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yang, Y. Aberrant Expression of USP22 is Associated With Liver Metastasis and Poor Prognosis of Colorectal Cancer. J. Surg. Oncol. 2011, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; Wang, A.; Liang, J.; Xie, Y. USP22 promotes the G1/S phase transition by upregulating FoxM1 expression via β-catenin nuclear localization and is associated with poor prognosis in stage II pancreatic ductal adenocarcinoma. Int. J. Oncol. 2014, 1594–1608. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Z.; Li, Y. USP22 nuclear expression is significantly associated with progression and unfavorable clinical outcome in human esophageal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2012, 138, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yao, L.; Zhang, X.; Ji, H.; Wang, L.; Sun, S.; Pang, D. Elevated expression of USP22 in correlation with poor prognosis in patients with invasive breast cancer. J. Cancer Res. Clin. Oncol. 2011, 137, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Shi, H.; Xie, Y.; Zhao, Z.; Li, S.; Chang, C.; Cheng, X.; Li, Y. Ubiquitin specific protease 22 promotes cell proliferation and tumor growth of epithelial ovarian cancer through synergy with transforming growth factor beta1. Oncol. Rep. 2015, 33, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Buetow, L.; Huang, D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016, 17, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Groen, E.J.N.; Gillingwater, T.H. UBA1: At the Crossroads of Ubiquitin Homeostasis and Neurodegeneration. Trends Mol. Med. 2015, 21, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Fierz, B.; Chatterjee, C.; McGinty, R.K.; Bar-Dagan, M.; Raleigh, D.P.; Muir, T.W. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat. Chem. Biol. 2011, 7, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.C.; Jason, L.; Ausió, J. The elusive structural role of ubiquitinated histones. Biochem. Cell Biol. 2002, 80, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.T.C.; Müller, C.W.; Vermeulen, M.; Müller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Kim, M.-S.; Shin, J.-M.; Park, T.-J.; Chung, H.-M.; Baek, K.-H. The expression patterns of deubiquitinating enzymes, USP22 and Usp22. Gene Expr. Patterns 2006, 6, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Harrow, J.; Harte, R.A.; Wallin, C.; Diekhans, M.; Maglott, D.R.; Searle, S.; Farrell, C.M.; Loveland, J.E.; Ruef, B.J.; et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009, 19, 1316–1323. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lang, G.; Ito, S.; Bonnet, J.; Metzger, E.; Sawatsubashi, S.; Suzuki, E.; Le Guezennec, X.; Stunnenberg, H.G.; Krasnov, A.; et al. A TFTC/STAGA Module Mediates Histone H2A and H2B Deubiquitination, Coactivates Nuclear Receptors, and Counteracts Heterochromatin Silencing. Mol. Cell 2008, 29, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Florens, L.; Swanson, S.K.; Washburn, M.P.; Workman, J.L. The deubiquitylation activity of Ubp8 is dependent upon Sgf11 and its association with the SAGA complex. Mol. Cell. Biol. 2005, 25, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Weake, V.M.; Lee, K.K.; Guelman, S.; Lin, C.-H.; Seidel, C.; Abmayr, S.M.; Workman, J.L. SAGA-mediated H2B deubiquitination controls the development of neuronal connectivity in the Drosophila visual system. EMBO J. 2008, 27, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Samara, N.L.; Datta, A.B.; Berndsen, C.E.; Zhang, X.; Yao, T.; Cohen, R.E.; Wolberger, C. Structural insights into the assembly and function of the SAGA deubiquitinating module. Science 2010, 328, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Köhler, A.; Zimmerman, E.; Schneider, M.; Hurt, E.; Zheng, N. Structural basis for assembly and activation of the heterotetrameric SAGA histone H2B deubiquitinase module. Cell 2010, 141, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Koutelou, E.; Hirsch, C.L.; Dent, S.Y.R. Multiple faces of the SAGA complex. Curr. Opin. Cell Biol. 2010, 22, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.T.; Haj-Yahya, M.; Ringel, A.E.; Bandi, P.; Brik, A.; Wolberger, C. Structural basis for histone H2B deubiquitination by the SAGA DUB module. Science 2016, 351, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Lang, G.; Bonnet, J.; Umlauf, D.; Karmodiya, K.; Koffler, J.; Stierle, M.; Devys, D.; Tora, L. The Tightly Controlled Deubiquitination Activity of the Human SAGA Complex Differentially Modifies Distinct Gene Regulatory Elements. Mol. Cell. Biol. 2011, 31, 3734–3744. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Kosinsky, R.L.; Wegwitz, F.; Hellbach, N.; Dobbelstein, M.; Mansouri, A.; Vogel, T.; Begus-Nahrmann, Y.; Johnsen, S.A. USP22 deficiency impairs intestinal epithelial lineage specification in vivo. Oncotarget 2015, 22, 37906–37918. [Google Scholar] [CrossRef] [PubMed]
- Atanassov, B.S.; Mohan, R.D.; Lan, X.; Kuang, X.; Lu, Y.; Lin, K.; McIvor, E.; Li, W.; Zhang, Y.; Florens, L.; et al. ATXN7L3 and ENY2 Coordinate Activity of Multiple H2B Deubiquitinases Important for Cellular Proliferation and Tumor Growth. Mol. Cell 2016, 62, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Haddad, D.; Li, C.; Le, M.X.; Ling, A.K.; So, C.C.; Nepal, R.M.; Gommerman, J.L.; Yu, K.; Ketela, T.; et al. The SAGA Deubiquitination Module Promotes DNA Repair and Class Switch Recombination through ATM and DNAPK-Mediated γH2AX Formation. Cell Rep. 2016, 15, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Glinsky, G.V.; Berezovska, O.; Glinskii, A.B. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J. Clin. Investig. 2005, 115, 1503–1521. [Google Scholar] [CrossRef] [PubMed]
- Glinsky, G.V. Genomic Models of Metastatic Cancer: Functional Analysis of Death-from-Cancer Signature Genes Reveals Aneuploid, Anoikis-Resistant, Metastasis-Enabling Phenotype with Altered Cell Cycle Control and Activated PcG Protein Chromatin Silencing Pathway. Cell Cycle 2006, 5, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Urasaki, Y.; Heath, L.; Xu, C.W. Coupling of Glucose Deprivation with Impaired Histone H2B Monoubiquitination in Tumors. PLoS ONE 2012, 7, e36775. [Google Scholar] [CrossRef] [PubMed]
- Prenzel, T.; Begus-Nahrmann, Y.; Kramer, F.; Hennion, M.; Hsu, C.; Gorsler, T.; Hintermair, C.; Eick, D.; Kremmer, E.; Simons, M.; et al. Estrogen-dependent gene transcription in human breast cancer cells relies upon proteasome-dependent monoubiquitination of histone H2B. Cancer Res. 2011, 71, 5739–5753. [Google Scholar] [CrossRef] [PubMed]
- Melling, N.; Grimm, N.; Simon, R.; Stahl, P.; Bokemeyer, C.; Terracciano, L.; Sauter, G.; Izbicki, J.R.; Marx, A.H. Loss of H2Bub1 Expression is Linked to Poor Prognosis in Nodal Negative Colorectal Cancers. Pathol. Oncol. Res. 2016, 22, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Dickson, K.-A.; Cole, A.J.; Gill, A.J.; Clarkson, A.; Gard, G.B.; Chou, A.; Kennedy, C.J.; Henderson, B.R.; Fereday, S.; Traficante, N.; et al. The RING finger domain E3 ubiquitin ligases BRCA1 and the RNF20/RNF40 complex in global loss of the chromatin mark histone H2B monoubiquitination (H2Bub1) in cell line models and primary high-grade serous ovarian cancer. Hum. Mol. Genet. 2016, 25, ddw362. [Google Scholar] [CrossRef] [PubMed]
- Di Croce, L.; Helin, K. Transcriptional regulation by Polycomb group proteins. Nat. Struct. Mol. Biol. 2013, 20, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jones, A.E.; Wu, W.; Kim, J.; Kang, Y.; Bi, X.; Gu, Y.; Popov, I.K.; Renfrow, M.B.; Vassylyeva, M.N.; et al. Role of remodeling and spacing factor 1 in histone H2A ubiquitination-mediated gene silencing. Proc. Natl. Acad. Sci. USA 2017, 114, E7949–E7958. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, J.; Wang, C.-Y.; Baptista, T.; Vincent, S.D.; Hsiao, W.-C.; Stierle, M.; Kao, C.-F.; Tora, L.; Devys, D. The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes Dev. 2014, 28, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y.; Shema, E.; Moyal, L.; Oren, M. RNF20-RNF40: A ubiquitin-driven link between gene expression and the DNA damage response. FEBS Lett. 2011, 585, 2795–2802. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Li, L.; Zhou, B.; Qin, Z.; Dou, Y. H2B Ubiquitylation Promotes RNA Pol II Processivity via PAF1 and pTEFb. Mol. Cell 2014, 54, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Pavri, R.; Zhu, B.; Li, G.; Trojer, P.; Mandal, S.; Shilatifard, A.; Reinberg, D. Histone H2B Monoubiquitination Functions Cooperatively with FACT to Regulate Elongation by RNA Polymerase II. Cell 2006, 125, 703–717. [Google Scholar] [CrossRef] [PubMed]
- Henry, K.W.; Wyce, A.; Lo, W.-S.; Duggan, L.J.; Emre, N.C.T.; Kao, C.-F.; Pillus, L.; Shilatifard, A.; Osley, M.A.; Berger, S.L. Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev. 2003, 17, 2648–2663. [Google Scholar] [CrossRef] [PubMed]
- Wyce, A.; Xiao, T.; Whelan, K.A.; Kosman, C.; Walter, W.; Eick, D.; Hughes, T.R.; Krogan, N.J.; Strahl, B.D.; Berger, S.L. H2B Ubiquitylation Acts as a Barrier to Ctk1 Nucleosomal Recruitment Prior to Removal by Ubp8 within a SAGA-Related Complex. Mol. Cell 2007, 27, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Schrecengost, R.S.; Dean, J.L.; Goodwin, J.F.; Schiewer, M.J.; Urban, M.W.; Stanek, T.J.; Sussman, R.T.; Hicks, J.L.; Birbe, R.C.; Draganova-Tacheva, R.A.; et al. USP22 regulates oncogenic signaling pathways to drive lethal cancer progression. Cancer Res. 2014, 74, 272–286. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-L.; Jiang, S.-H.; Yang, Y.-M.; Xu, H.; Liu, J.-L.; Wang, X.S. USP22 Acts as an Oncogene by the Activation of BMI-1-Mediated INK4a / ARF Pathway and Akt Pathway. Cell Biochem. Biophys. 2012, 229–235. [Google Scholar] [CrossRef]
- Li, L.; Osdal, T.; Ho, Y.; Chun, S.; McDonald, T.; Agarwal, P.; Lin, A.; Chu, S.; Qi, J.; Li, L.; et al. SIRT1 Activation by a c-MYC Oncogenic Network Promotes the Maintenance and Drug Resistance of Human FLT3-ITD Acute Myeloid Leukemia Stem Cells. Cell Stem Cell 2014, 15, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Atanassov, B.S.; Dent, S.Y.R. USP22 regulates cell proliferation by deubiquitinating the transcriptional regulator FBP1. EMBO Rep. 2011, 12, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.H.; Yu, Y.; Du, C.; Fu, H.; Wang, J.; Tian, Y. RNA interference-mediated USP22 gene silencing promotes human brain glioma apoptosis and induces cell cycle arrest. Oncol. Lett. 2013, 5, 1290–1294. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Liu, Y.L.; Yang, Y.M.; Dong, X.S. Knock-down of ubiquitin-specific protease 22 by micro-RNA interference inhibits colorectal cancer growth. Int. J. Colorectal Dis. 2012, 27, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Yang, H.; Kong, Q.; Li, J.; Lee, S.-M.; Gao, B.; Dong, H.; Wei, J.; Song, J.; Zhang, D.D.; et al. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol. Cell 2012, 46, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Xu, X.; Zhou, X.; Liu, J.; Gong, Z.; Wu, P.; Li, W. USP22 transcriptional activity is negatively regulated by the histone deacetylase inhibitor trichostatin A. Mol. Med. Rep. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Armour, S.M.; Bennett, E.J.; Braun, C.R.; Zhang, X.-Y.; McMahon, S.B.; Gygi, S.P.; Harper, J.W.; Sinclair, D.A. A high-confidence interaction map identifies SIRT1 as a mediator of acetylation of USP22 and the SAGA coactivator complex. Mol. Cell. Biol. 2013, 33, 1487–1502. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The Role of Autophagy in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Gómez, V.E.; Giovannetti, E.; Peters, G.J. Unraveling the complexity of autophagy: Potential therapeutic applications in Pancreatic Ductal Adenocarcinoma. Semin. Cancer Biol. 2015, 35, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Jang, B. The Role of Autophagy in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2015, 16, 26629–26643. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Ni, Z.; He, J.; Jiang, S.; Li, X.; He, J.; Gong, W.; Zheng, L.; Chen, S.; Li, B.; et al. LncRNA HULC triggers autophagy via stabilizing Sirt1 and attenuates the chemosensitivity of HCC cells. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Tan, C.; Qiu, Q.; Kong, S.; Yang, H.; Zhao, F.; Liu, Z.; Li, J.; Kong, Q.; Gao, B.; et al. Ubiquitin-specific protease 22 is a deubiquitinase of CCNB1. Cell Discov. 2015, 1, 15028. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.H.; Stoeber, K. The cell cycle and cancer. J. Pathol. 2012, 226, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Giam, M.; Rancati, G. Aneuploidy and chromosomal instability in cancer: A jackpot to chaos. Cell Div. 2015, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Burrell, R.A.; Endesfelder, D.; Novelli, M.R.; Swanton, C. Cancer chromosomal instability: Therapeutic and diagnostic challenges. EMBO Rep. 2012, 13, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Cimini, D.; Mattiuzzo, M.; Torosantuci, L.; Degrassi, F. Histone hyperacetylation in mitosis prevents sister chromatid separation and produces chromosome segregation defects. Mol. Biol. Cell 2003, 14, 3821–3833. [Google Scholar] [CrossRef] [PubMed]
- Vagnarelli, P.; Hudson, D.F.; Ribeiro, S.A.; Trinkle-Mulcahy, L.; Spence, J.M.; Lai, F.; Farr, C.J.; Lamond, A.I.; Earnshaw, W.C. Condensin and Repo-Man–PP1 co-operate in the regulation of chromosome architecture during mitosis. Nat. Cell Biol. 2006, 8, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.; Jeusset, L.; Lepage, C.; McManus, K. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers 2017, 9, 151. [Google Scholar] [CrossRef] [PubMed]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.-Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-Dependent Monoubiquitylation of Histone H2B for Timely Repair of DNA Double-Strand Breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Kato, A.; Kobayashi, J.; Yanagihara, H.; Sakamoto, S.; Oliveira, D.V.N.P.; Shimada, M.; Tauchi, H.; Suzuki, H.; Tashiro, S.; et al. Regulation of Homologous Recombination by RNF20-Dependent H2B Ubiquitination. Mol. Cell 2011, 41, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Meas, R.; Dorgan, K.M.; Smerdon, M.J. UV damage-induced RNA polymerase II stalling stimulates H2B deubiquitylation. Proc. Natl. Acad. Sci. USA 2014, 111, 12811–12816. [Google Scholar] [CrossRef] [PubMed]
- Guppy, B.J.; McManus, K.J. Mitotic accumulation of dimethylated lysine 79 of histone H3 is important for maintaining genome integrity during mitosis in human cells. Genetics 2015, 199, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Vagnarelli, P. Mitotic chromosome condensation in vertebrates. Exp. Cell Res. 2012, 318, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Stirling, P.C.; Bloom, M.S.; Solanki-patil, T.; Smith, S.; Sipahimalani, P.; Li, Z.; Kofoed, M.; Ben-Aroya, S.; Myung, K.; Hieter, P. The Complete Spectrum of Yeast Chromosome Instability Genes Identifies Candidate CIN Cancer Genes and Functional Roles for ASTRA Complex Components. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.W.Y.; Warren, C.D.; Chen, O.; Kwok, T.; Hieter, P.; Spencer, F.A. Systematic genome instability screens in yeast and their potential relevance to cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3925–3930. [Google Scholar] [CrossRef] [PubMed]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining “chromosomal instability”. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A.; Cleveland, D.W. Does aneuploidy cause cancer? Curr. Opin. Cell Biol. 2006, 18, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, H.; Nowak, M.A.; Vogelstein, B.; Lengauer, C. The significance of unstable chromosomes in colorectal cancer. Nat. Rev. Cancer 2003, 3, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Birkbak, N.J.; Eklund, A.C.; Li, Q.; McClelland, S.E.; Endesfelder, D.; Tan, P.; Tan, I.B.; Richardson, A.L.; Szallasi, Z.; Swanton, C. Paradoxical Relationship between Chromosomal Instability and Survival Outcome in Cancer. Cancer Res. 2011, 71, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.M.; Murnane, J.P. Telomeres, chromosome instability and cancer. Nucleic Acids Res. 2006, 34, 2408–2417. [Google Scholar] [CrossRef] [PubMed]
- Van Steensel, B.; de Lange, T. Control of telomere length by the human telomeric protein TRF1. Nature 1997, 385, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Atanassov, B.S.; Evrard, Y.A.; Multani, A.S.; Zhang, Z.; Tora, L.; Devys, D.; Chang, S.; Dent, S.Y.R. Gcn5 and SAGA Regulate Shelterin Protein Turnover and Telomere Maintenance. Mol. Cell 2009, 35, 352–364. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Accession Number | Species | Length A | Identity B (%) | Similarity C (%) | E-Value D |
|---|---|---|---|---|---|---|
| USP22 | NP_056091.1 | Homo sapiens | 525 | N/A | N/A | N/A |
| USP22 | NP_001004143.2 | Mus musculus | 525 | 98 | 99 | 0.0 |
| USP22 | XP_021335835.1 | Danio rerio | 506 | 93 | 97 | 0.0 |
| USP22 | NP_001085912.1 | Xenopus laevis | 523 | 92 | 95 | 0.0 |
| Nonstop | NP_524140.2 | Drosophila melanogaster | 496 | 51 | 65 | 0.0 |
| Ubp8 | NP_592992.1 | Schizosaccharomyces pombe | 449 | 32 | 46 | 5e-70 |
| Ubp8 | NP_013950.1 | Saccharomyces cerevisiae | 471 | 31 | 46 | 8e-63 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeusset, L.M.-P.; McManus, K.J. Ubiquitin Specific Peptidase 22 Regulates Histone H2B Mono-Ubiquitination and Exhibits Both Oncogenic and Tumor Suppressor Roles in Cancer. Cancers 2017, 9, 167. https://doi.org/10.3390/cancers9120167
Jeusset LM-P, McManus KJ. Ubiquitin Specific Peptidase 22 Regulates Histone H2B Mono-Ubiquitination and Exhibits Both Oncogenic and Tumor Suppressor Roles in Cancer. Cancers. 2017; 9(12):167. https://doi.org/10.3390/cancers9120167
Chicago/Turabian StyleJeusset, Lucile M-P., and Kirk J. McManus. 2017. "Ubiquitin Specific Peptidase 22 Regulates Histone H2B Mono-Ubiquitination and Exhibits Both Oncogenic and Tumor Suppressor Roles in Cancer" Cancers 9, no. 12: 167. https://doi.org/10.3390/cancers9120167
APA StyleJeusset, L. M.-P., & McManus, K. J. (2017). Ubiquitin Specific Peptidase 22 Regulates Histone H2B Mono-Ubiquitination and Exhibits Both Oncogenic and Tumor Suppressor Roles in Cancer. Cancers, 9(12), 167. https://doi.org/10.3390/cancers9120167