Eukaryotic Elongation Factor 2 Kinase (eEF2K) in Cancer


{kind=link}
{kind=link}
Abstract
1. Introduction to eEF2K
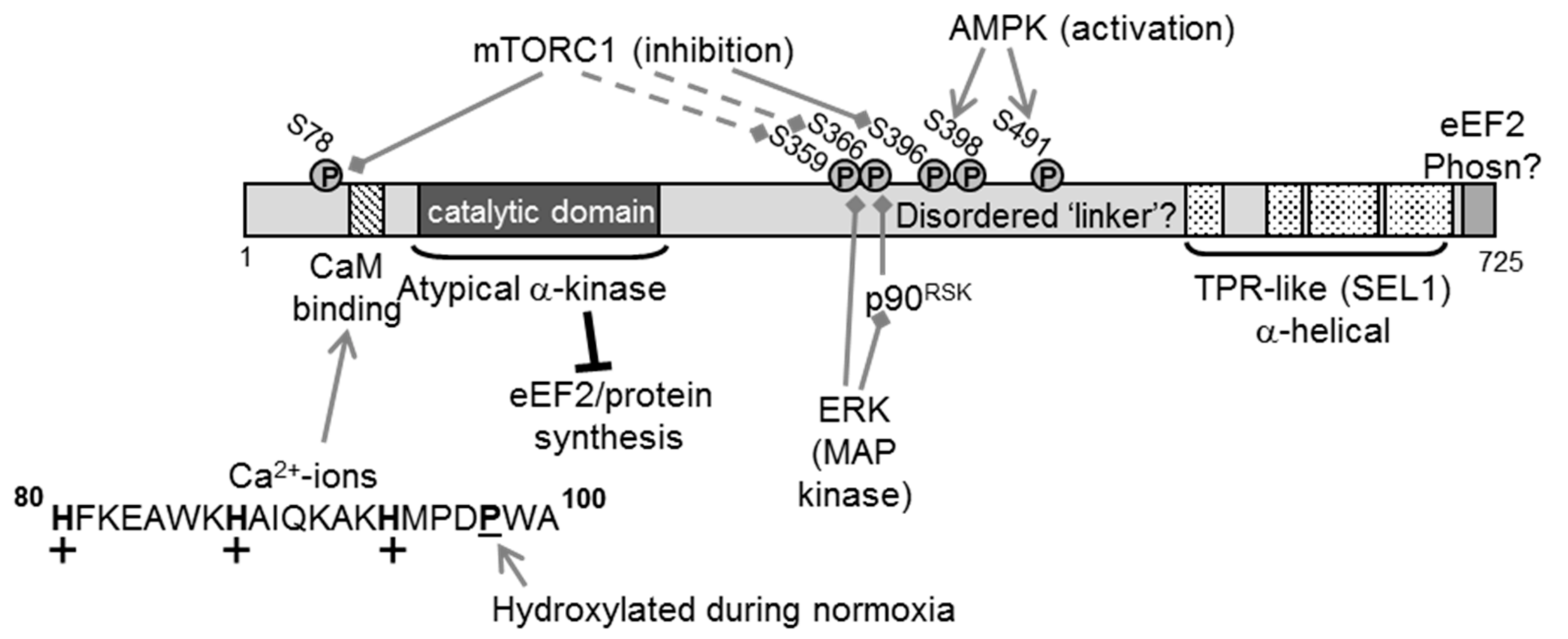
2. The eEF2K Protein
3. Regulation of eEF2K
4. eEF2K in Cytoprotection
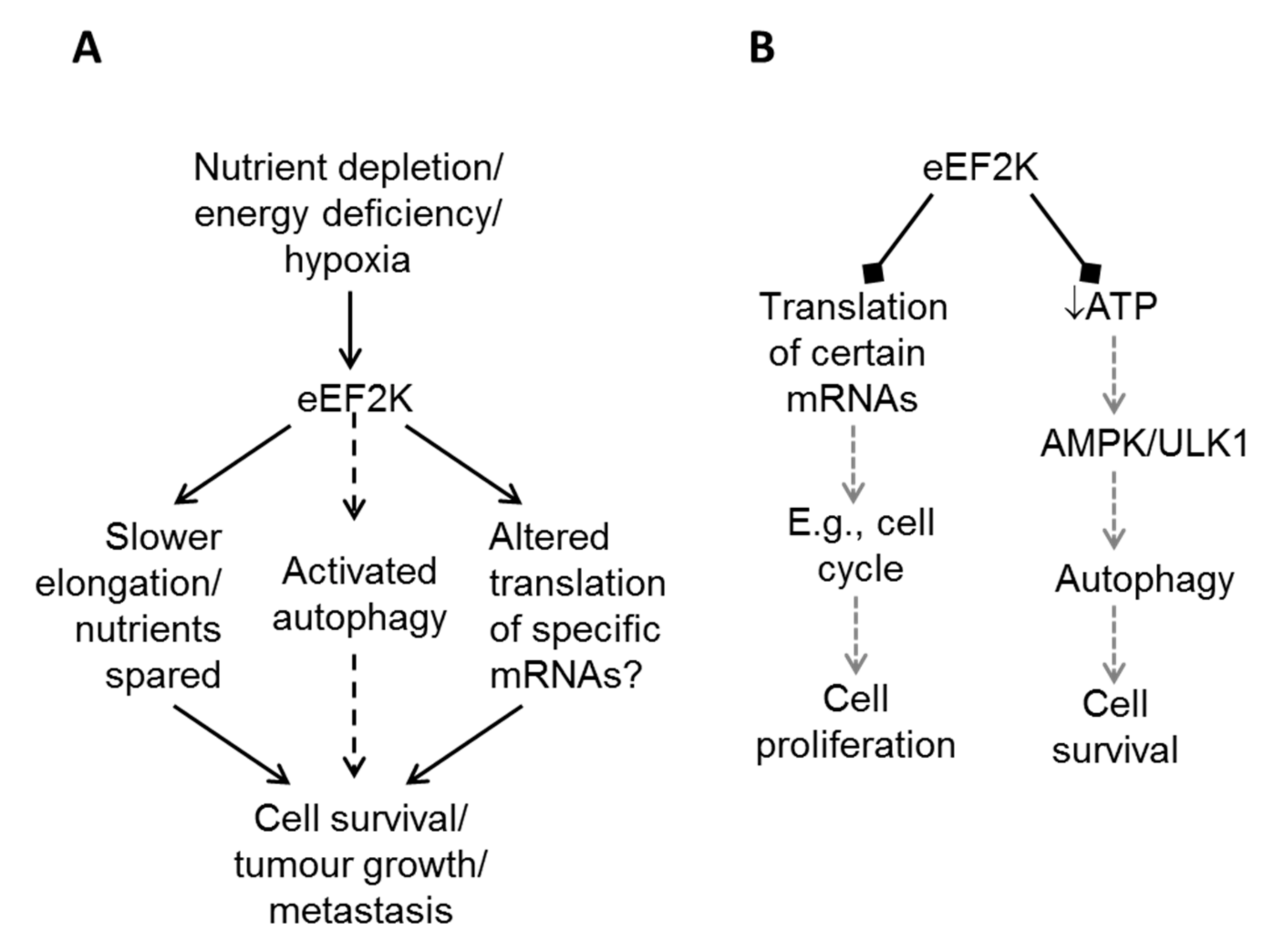
5. eEF2K in Cancer Cell Survival: How Does eEF2K Promote Survival?
5.1. eEF2K and Cellular Nutrient Levels
5.2. eEF2K and the Control of the Synthesis of Specific Proteins
5.3. eEF2K and Autophagy
5.4. Other Roles of eEF2K in Cancer Cells
6. eEF2K in Tumour Growth
7. eEF2K Promotes the Efficacy of Other Anticancer Agents
8. eEF2K in Migration and Metastasis
9. Can eEF2K Inhibition Promote Cancer?
10. Inhibitors of eEF2K
11. Conclusions and Perspective
Acknowledgments
Conflicts of Interest
References
- Liu, R.; Proud, C.G. Eukaryotic elongation factor 2 kinase as a drug target in cancer, and in cardiovascular and neurodegenerative diseases. Acta Pharmacol. Sin. 2016, 37, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Kenney, J.W.; Moore, C.E.; Wang, X.; Proud, C.G. Eukaryotic elongation factor 2 kinase, an unusual enzyme with multiple roles. Adv. Biol. Regul. 2014, 55, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, U.; Nilsson, A.; Nygard, O. Functional properties of phosphorylated elongation factor 2. Eur. J. Biochem. 1990, 191, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Redpath, N.T.; Price, N.T.; Proud, C.G. Cloning and expression of cDNA encoding protein synthesis elongation factor-2 kinase. J. Biol. Chem. 1996, 271, 17547–17554. [Google Scholar] [CrossRef] [PubMed]
- Ryazanov, A.G.; Pavur, K.S.; Dorovkov, M.V. Alpha kinases: A new class of protein kinases with a novel catalytic domain. Curr. Biol. 1999, 9, R43–R45. [Google Scholar] [CrossRef]
- Ryazanov, A.G.; Ward, M.D.; Mendola, C.E.; Pavur, K.S.; Dorovkov, M.V.; Wiedmann, M.; Erdjument-Bromage, H.; Tempst, P.; Parmer, T.G.; Prostko, C.R.; et al. Identification of a new class of protein kinases represented by eukaryotic elongation factor-2 kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 4884–4889. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Crawley, S.W.; Yang, Y.; Cote, G.P.; Jia, Z. Crystal structure of the alpha-kinase domain of Dictyostelium myosin heavy chain kinase A. Sci. Signal. 2010, 3, ra17. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Matsushita, M.; Nairn, A.C.; Kuriyan, J. Crystal structure of the atypical protein kinase domain of a TRP channel with phosphotransferase activity. Mol. Cell 2001, 7, 1047–1057. [Google Scholar] [CrossRef]
- Fawaz, M.V.; Topper, M.E.; Firestine, S.M. The ATP-grasp enzymes. Bioorg. Chem. 2011, 39, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Pavur, K.S.; Petrov, A.N.; Ryazanov, A.G. Mapping the functional domains of elongation factor-2 kinase. Biochemistry 2000, 39, 12216–12224. [Google Scholar] [CrossRef] [PubMed]
- Diggle, T.A.; Seehra, C.K.; Hase, S.; Redpath, N.T. Analysis of the domain structure of elongation factor-2 kinase by mutagenesis. FEBS Lett. 1999, 457, 189–192. [Google Scholar] [CrossRef]
- Ryazanov, A.G.; Natapov, P.G.; Shestakova, E.A.; Severin, F.F.; Spirin, A.S. Phosphorylation of the elongation factor 2: The fifth Ca2+/calmodulin-dependent system of protein phosphorylation. Biochimie 1988, 70, 619–626. [Google Scholar] [CrossRef]
- Nairn, A.C.; Palfrey, H.C. Phosphorylation of elongation factor-II by Ca2+ calmodulin-depedent kinase-III. FASEB J. 1988, 2, A777. [Google Scholar]
- Mittl, P.R.; Schneider-Brachert, W. Sel1-like repeat proteins in signal transduction. Cell. Signal. 2007, 19, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Will, N.; Piserchio, A.; Snyder, I.; Ferguson, S.B.; Giles, D.H.; Dalby, K.N.; Ghose, R. Structure of the C-Terminal helical repeat domain of eukaryotic elongation factor 2 Kinase. Biochemistry 2016, 55, 5377–5386. [Google Scholar] [CrossRef] [PubMed]
- Pigott, C.R.; Mikolajek, H.; Moore, C.E.; Finn, S.J.; Phippen, C.W.; Werner, J.M.; Proud, C.G. Insights into the regulation of eukaryotic elongation factor 2 kinase and the interplay between its domains. Biochem. J. 2011, 442, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Ruys, S.P.D.; Wang, X.; Smith, E.M.; Herinckx, G.; Hussain, N.; Rider, M.H.; Vertommen, D.; Proud, C.G. Identification of autophosphorylation sites in eukaryotic elongation factor-2 kinase. Biochem. J. 2012, 442, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Crawley, S.W.; Gharaei, M.S.; Ye, Q.; Yang, Y.; Raveh, B.; London, N.; Schueler-Furman, O.; Jia, Z.; Cote, G.P. Autophosphorylation activates Dictyostelium myosin II heavy chain kinase A by providing a ligand for an allosteric binding site in the alpha-kinase domain. J. Biol. Chem. 2011, 286, 2607–2616. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.E.; Regufe da Mota, S.; Mikolajek, H.; Proud, C.G. A conserved loop in the catalytic domain of eukaryotic elongation factor 2 kinase plays a key role in its substrate specificity. Mol. Cell. Biol. 2014, 34, 2294–2307. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.M.; Liu, X.Y.; Sham, K.W.; Lai, J.M.; Cheng, C.H. Silencing of EEF2K (eukaryotic elongation factor-2 kinase) reveals AMPK-ULK1-dependent autophagy in colon cancer cells. Autophagy 2014, 10, 1495–1508. [Google Scholar] [CrossRef] [PubMed]
- Dorovkov, M.V.; Pavur, K.S.; Petrov, A.N.; Ryazanov, A.G. Regulation of elongation factor-2 kinase by Ph. Biochemistry 2002, 41, 13444–13450. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Mikolajek, H.; Pigott, C.R.; Hooper, K.J.; Mellows, T.; Moore, C.E.; Mohammed, H.; Werner, J.M.; Thomas, G.J.; Proud, C.G. Molecular mechanism for the control of eukaryotic elongation factor 2 Kinase by pH: Role in cancer cell survival. Mol. Cell. Biol. 2015, 35, 1805–1824. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.E.; Mikolajek, H.; Regufe da Mota, S.; Wang, X.; Kenney, J.W.; Werner, J.M.; Proud, C.G. Elongation factor 2 kinase is regulated by proline hydroxylation and protects cells during hypoxia. Mol. Cell. Biol. 2015, 35, 1788–1804. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in growth, metabolism, and disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, W.; Williams, M.; Terada, N.; Alessi, D.R.; Proud, C.G. Regulation of elongation factor 2 kinase by p90RSK1 and p70 S6 kinase. EMBO J. 2001, 20, 4370–4379. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Regufe da Mota, S.; Liu, R.; Moore, C.E.; Xie, J.; Lanucara, F.; Agarwala, U.; Dit, R.S.P.; Vertommen, D.; Rider, M.H.; et al. Eukaryotic elongation factor 2 kinase activity is controlled by multiple inputs from oncogenic signaling. Mol. Cell. Biol. 2014, 34, 4088–4103. [Google Scholar] [CrossRef] [PubMed]
- Browne, G.J.; Proud, C.G. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol. Cell Biol. 2004, 24, 2986–2997. [Google Scholar] [CrossRef] [PubMed]
- Knebel, A.; Haydon, C.E.; Morrice, N.; Cohen, P. Stress-induced regulation of eEF2 kinase by SB203580-sensitive and -insensitive pathways. Biochem. J. 2002, 367, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Knebel, A.; Morrice, N.; Cohen, P. A novel method to identify protein kinase substrates: eEF2 kinase is phosphorylated and inhibited by SAPK4/p38delta. EMBO J. 2001, 20, 4360–4369. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- McLeod, L.E.; Proud, C.G. ATP depletion increases phosphorylation of elongation factor eEF2 in adult cardiomyocytes independently of inhibition of mTOR signalling. FEBS Lett. 2002, 531, 448–452. [Google Scholar] [CrossRef]
- Browne, G.J.; Finn, S.G.; Proud, C.G. Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J. Biol. Chem. 2004, 279, 12220–12231. [Google Scholar] [CrossRef] [PubMed]
- Horman, S.; Browne, G.J.; Krause, U.; Patel, J.V.; Vertommen, D.; Bertrand, L.; Lavoinne, A.; Hue, L.; Proud, C.G.; Rider, M.H. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr. Biol. 2002, 12, 1419–1423. [Google Scholar] [CrossRef]
- Johanns, M.; Ruys, S.P.D.; Houddane, A.; Vertommen, D.; Herinckx, G.; Hue, L.; Proud, C.G.; Rider, M.H. Direct and indirect activation of eukaryotic elongation factor 2 kinase by AMP-activated protein kinase. Cell. Signal. 2017, 36, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Leprivier, G.; Sorensen, P.H. How does oncogene transformation render tumor cells hypersensitive to nutrient deprivation? Bioessays 2014, 36, 1082–1090. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D. Adaptation to starvation: Translating a matter of life or death. Cancer Cell 2013, 23, 713–715. [Google Scholar] [CrossRef] [PubMed]
- Leprivier, G.; Rotblat, B.; Khan, D.; Jan, E.; Sorensen, P.H. Stress-mediated translational control in cancer cells. Biochim. Biophys. Acta 2015, 1849, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Faller, W.J.; Jackson, T.J.; Knight, J.R.; Ridgway, R.A.; Jamieson, T.; Karim, S.A.; Jones, C.; Radulescu, S.; Huels, D.J.; Myant, K.B.; et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature 2014, 517, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ren, X.; Zhang, Y.; Patel, R.; Sharma, A.; Wu, H.; Robertson, G.P.; Yan, L.; Rubin, E.; Yang, J.M. eEF-2 kinase dictates cross-talk between autophagy and apoptosis induced by Akt Inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK-2206. Cancer Res. 2011, 71, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Li, H.; Ren, X.; Niu, T.; Hait, W.N.; Yang, J. Cytoprotective effect of the elongation factor-2 kinase-mediated autophagy in breast cancer cells subjected to growth factor inhibition. PLoS ONE 2010, 5, e9715. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhu, H.; Liu, D.X.; Niu, T.K.; Ren, X.; Patel, R.; Hait, W.N.; Yang, J.M. Silencing of elongation factor-2 kinase potentiates the effect of 2-deoxy-D-glucose against human glioma cells through blunting of autophagy. Cancer Res. 2009, 69, 2453–2460. [Google Scholar] [CrossRef] [PubMed]
- Py, B.F.; Boyce, M.; Yuan, J. A critical role of eEF-2K in mediating autophagy in response to multiple cellular stresses. Autophagy 2009, 5, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yang, J.M.; Jin, S.; Zhang, H.; Hait, W.N. Elongation factor-2 kinase regulates autophagy in human glioblastoma cells. Cancer Res. 2006, 66, 3015–3023. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.P.; Liao, Y.; Novak, J.S.; Hu, Z.; Merkin, J.J.; Shymkiv, Y.; Braeckman, B.P.; Dorovkov, M.V.; Nguyen, A.; Clifford, P.M.; et al. Germline quality control: eEF2K stands guard to eliminate defective oocytes. Dev. Cell 2014, 28, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.; Braunstein, S.; Formenti, S.; Schneider, R.J. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol. Cell. Biol. 2006, 26, 3955–3965. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cash, T.P.; Jones, R.G.; Keith, B.; Thompson, C.B.; Simon, M.C. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 2006, 21, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Delaidelli, A.; Negri, G.L.; Jan, A.; Jansonius, B.; El-Naggar, A.; Lim, J.K.M.; Khan, D.; Oo, H.Z.; Carnie, C.J.; Remke, M.; et al. MYCN amplified neuroblastoma requires the mRNA translation regulator eEF2 kinase to adapt to nutrient deprivation. Cell Death Differ. 2017, 24, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Leprivier, G.; Remke, M.; Rotblat, B.; Dubuc, A.; Mateo, A.R.; Kool, M.; Agnihotri, S.; El-Naggar, A.; Yu, B.; Prakash, S.S.; et al. The eEF2 kinase confers resistance to nutrient deprivation by blocking translation elongation. Cell 2013, 153, 1064–1079. [Google Scholar] [CrossRef] [PubMed]
- Walden, W.E.; Thach, R.E. Translational control of gene expression in a normal fibroblast. Characterization of a subclass of mRNAs with unusual kinetic properties. Biochemistry 1986, 25, 2033–2041. [Google Scholar] [CrossRef] [PubMed]
- Scheetz, A.J.; Nairn, A.C.; Constantine-Paton, M. NMDA-mediated control of protein synthesis at developing synapses. Nat. Neurosci. 2000, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kenney, J.W.; Genheden, M.; Moon, K.-M.; Foster, L.J.; Proud, C.G. eEF2K regulates the synthesis of microtubule-related proteins in neurons. J. Neurochem. 2015, 136, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Tekedereli, I.; Alpay, S.N.; Tavares, C.D.; Cobanoglu, Z.E.; Kaoud, T.S.; Sahin, I.; Sood, A.K.; Lopez-Berestein, G.; Dalby, K.N.; Ozpolat, B. Targeted silencing of elongation factor 2 kinase suppresses growth and sensitizes tumors to Doxorubicin in an orthotopic model of breast cancer. PLoS ONE 2012, 7, e41171. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, R.; Pichler, M.; Kanlikilicer, P.; Ivan, C.; Bayraktar, E.; Kahraman, N.; Aslan, B.; Ulasli, M.; Arslan, A.; Oguztuzun, S.; et al. MicroRNA 603 acts as a tumor suppressor and inhibits triple-negative breast cancer tumorigenesis by targeting elongation factor 2 kinase. Oncotarget 2017, 8, 11641–11658. [Google Scholar]
- Zhang, Y.; Cheng, Y.; Zhang, L.; Ren, X.; Huber-Keener, K.J.; Lee, S.; Yun, J.; Wang, H.G.; Yang, J.M. Inhibition of eEF-2 kinase sensitizes human glioma cells to TRAIL and down-regulates Bcl-xL expression. Biochem. Biophys. Res. Commun. 2011, 414, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Holcik, M.; Lefebvre, C.; Yeh, C.; Chow, T.; Korneluk, R.G. A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat. Cell Biol. 1999, 1, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriyarachchi, D.; Cerquozzi, S.; Cheung, H.H.; Holcik, M. Translational induction of the inhibitor of apoptosis protein HIAP2 during endoplasmic reticulum stress attenuates cell death and is mediated via an inducible internal ribosome entry site element. J. Biol. Chem. 2004, 279, 17148–17157. [Google Scholar] [CrossRef] [PubMed]
- Sherrill, K.W.; Byrd, M.P.; van Eden, M.E.; Lloyd, R.E. BCL-2 translation is mediated via internal ribosome entry during cell stress. J. Biol. Chem. 2004, 279, 29066–29074. [Google Scholar] [CrossRef] [PubMed]
- Ungureanu, N.H.; Cloutier, M.; Lewis, S.M.; de Silva, N.; Blais, J.D.; Bell, J.C.; Holcik, M. Internal ribosome entry site-mediated translation of Apaf-1, but not XIAP, is regulated during UV-induced cell death. J. Biol. Chem. 2006, 281, 15155–15163. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Zhang, J.; Ma, H.; Sun, L.; Zhang, X.; Yu, G.; Guan, H.; Wang, W.; Li, C. Synthesis and anti-influenza a virus activity of 6′-amino-6′-deoxy-glucoglycerolipids analogs. Mar. Drugs 2016, 14, 116. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ren, X.; Zhang, Y.; Shan, Y.; Huber-Keener, K.J.; Zhang, L.; Kimball, S.R.; Harvey, H.; Jefferson, L.S.; Yang, J.M. Integrated regulation of autophagy and apoptosis by EEF2K controls cellular fate and modulates the efficacy of curcumin and velcade against tumor cells. Autophagy 2012, 9, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Devkota, A.K.; Tavares, C.D.; Warthaka, M.; Abramczyk, O.; Marshall, K.D.; Kaoud, T.S.; Gorgulu, K.; Ozpolat, B.; Dalby, K.N. Investigating the kinetic mechanism of inhibition of elongation factor 2 kinase by NH125: Evidence of a common in vitro artifact. Biochemistry 2012, 51, 2100–2112. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Gopalakrishnan, S.M.; Bui, M.H.; Soni, N.B.; Warrior, U.; Johnson, E.F.; Donnelly, J.B.; Glaser, K.B. 1-Benzyl-3-cetyl-2-methylimidazolium iodide (NH125) induces phosphorylation of eukaryotic elongation factor-2 (eEF2): A cautionary note on the anticancer mechanism of an eEF2 kinase inhibitor. J. Biol. Chem. 2011, 286, 43951–43958. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.E.; Wang, X.; Xie, J.; Pickford, J.; Barron, J.; Regufe da Mota, S.; Versele, M.; Proud, C.G. Elongation factor 2 kinase promotes cell survival by inhibiting protein synthesis without inducing autophagy. Cell. Signal. 2016, 28, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, E.A.; Tee, A.R. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin. Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The metabolism of tumors in the body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Heiden, M.G.V.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Ren, X.; Yuan, Y.; Shan, Y.; Li, L.; Chen, X.; Zhang, L.; Takahashi, Y.; Yang, J.W.; Han, B.; et al. eEF-2 kinase is a critical regulator of Warburg effect through controlling PP2A-A synthesis. Oncogene 2016, 35, 6293–6308. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Chen, X.; Ma, J.; Peng, H.; Wang, F.; Zha, X.; Wang, Y.; Jing, Y.; Yang, H.; Chen, R.; et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc. Natl. Acad. Sci. USA 2011, 108, 4129–4134. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Voisin, V.; Wang, S.; Wang, D.Y.; Jones, R.A.; Datti, A.; Uehling, D.; Al-Awar, R.; Egan, S.E.; Bader, G.D.; et al. Combined deletion of Pten and p53 in mammary epithelium accelerates triple-negative breast cancer with dependency on eEF2K. EMBO Mol. Med. 2014, 6, 1542–1560. [Google Scholar] [CrossRef] [PubMed]
- de Gassart, A.; Demaria, O.; Panes, R.; Zaffalon, L.; Ryazanov, A.G.; Gilliet, M.; Martinon, F. Pharmacological eEF2K activation promotes cell death and inhibits cancer progression. EMBO Rep. 2016, 17, 1471–1484. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Py, B.F.; Ryazanov, A.G.; Long, K.; Minden, J.S.; Ma, D.; Yuan, J. A pharmacoproteomic approach implicates eukaryotic elongation factor 2 kinase in ER stress-induced cell death. Cell Death Differ. 2008, 15, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Nijima, R.; Sakatsume, T.; Otani, K.; Kameshima, S.; Okada, M.; Yamawaki, H. Eukaryotic elongation factor 2 kinase controls proliferation and migration of vascular smooth muscle cells. Acta Physiol. 2015, 213, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Hamurcu, Z.; Ashour, A.; Kahraman, N.; Ozpolat, B. FOXM1 regulates expression of eukaryotic elongation factor 2 kinase and promotes proliferation, invasion and tumorgenesis of human triple negative breast cancer cells. Oncotarget 2016, 7, 16619. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Song, H.; Chen, G.; Yang, X.; Liu, J.; Ge, Y.; Lu, J.; Qin, Q.; Zhang, C.; Xu, L.; et al. eEF2K promotes progression and radioresistance of esophageal squamous cell carcinoma. Radiother. Oncol. 2017, 124, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Xu, X.; Liu, Q.; Luo, F.; Shi, J.; He, X. MicroRNA-877 acts as a tumor suppressor by directly targeting eEF2K in renal cell carcinoma. Oncol. Lett. 2016, 11, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Palm, W.; Park, Y.; Wright, K.; Pavlova, N.N.; Tuveson, D.A.; Thompson, C.B. The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 2015, 162, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Gschwendt, M.; Kittstein, W.; Marks, F. Elongation factor-2 kinase: Effective inhibition by the novel protein kinase inhibitor rottlerin and relative insensitivity towards staurosporine. FEBS Lett. 1994, 338, 85–88. [Google Scholar] [CrossRef]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Yang, J.M.; Kinzy, T.G.; Utsumi, R.; Okamoto, T.; Kitayama, T.; Ortiz, P.A.; Hait, W.N. Identification and characterization of an inhibitor of eukaryotic elongation factor 2 kinase against human cancer cell lines. Cancer Res. 2003, 63, 6894–6899. [Google Scholar] [PubMed]
- Edupuganti, R.; Wang, Q.; Tavares, C.D.; Chitjian, C.A.; Bachman, J.L.; Ren, P.; Anslyn, E.V.; Dalby, K.N. Synthesis and biological evaluation of pyrido[2,3-d]pyrimidine-2,4-dione derivatives as eEF-2K inhibitors. Bioorg. Med. Chem. 2014, 22, 4910–4916. [Google Scholar] [CrossRef] [PubMed]
- Hori, H.; Nagasawa, H.; Ishibashi, M.; Uto, Y.; Hirata, A.; Saijo, K.; Ohkura, K.; Kirk, K.L.; Uehara, Y. TX-1123: An antitumor 2-hydroxyarylidene-4-cyclopentene-1,3-dione as a protein tyrosine kinase inhibitor having low mitochondrial toxicity. Bioorg. Med. Chem. 2002, 10, 3257–3265. [Google Scholar] [CrossRef]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Xie, J.; Proud, C.G. Eukaryotic Elongation Factor 2 Kinase (eEF2K) in Cancer. Cancers 2017, 9, 162. https://doi.org/10.3390/cancers9120162
Wang X, Xie J, Proud CG. Eukaryotic Elongation Factor 2 Kinase (eEF2K) in Cancer. Cancers. 2017; 9(12):162. https://doi.org/10.3390/cancers9120162
Chicago/Turabian StyleWang, Xuemin, Jianling Xie, and Christopher G. Proud. 2017. "Eukaryotic Elongation Factor 2 Kinase (eEF2K) in Cancer" Cancers 9, no. 12: 162. https://doi.org/10.3390/cancers9120162
APA StyleWang, X., Xie, J., & Proud, C. G. (2017). Eukaryotic Elongation Factor 2 Kinase (eEF2K) in Cancer. Cancers, 9(12), 162. https://doi.org/10.3390/cancers9120162