
Alcohol and Cancer Stem Cells


{kind=link}
Abstract
:1. Introduction
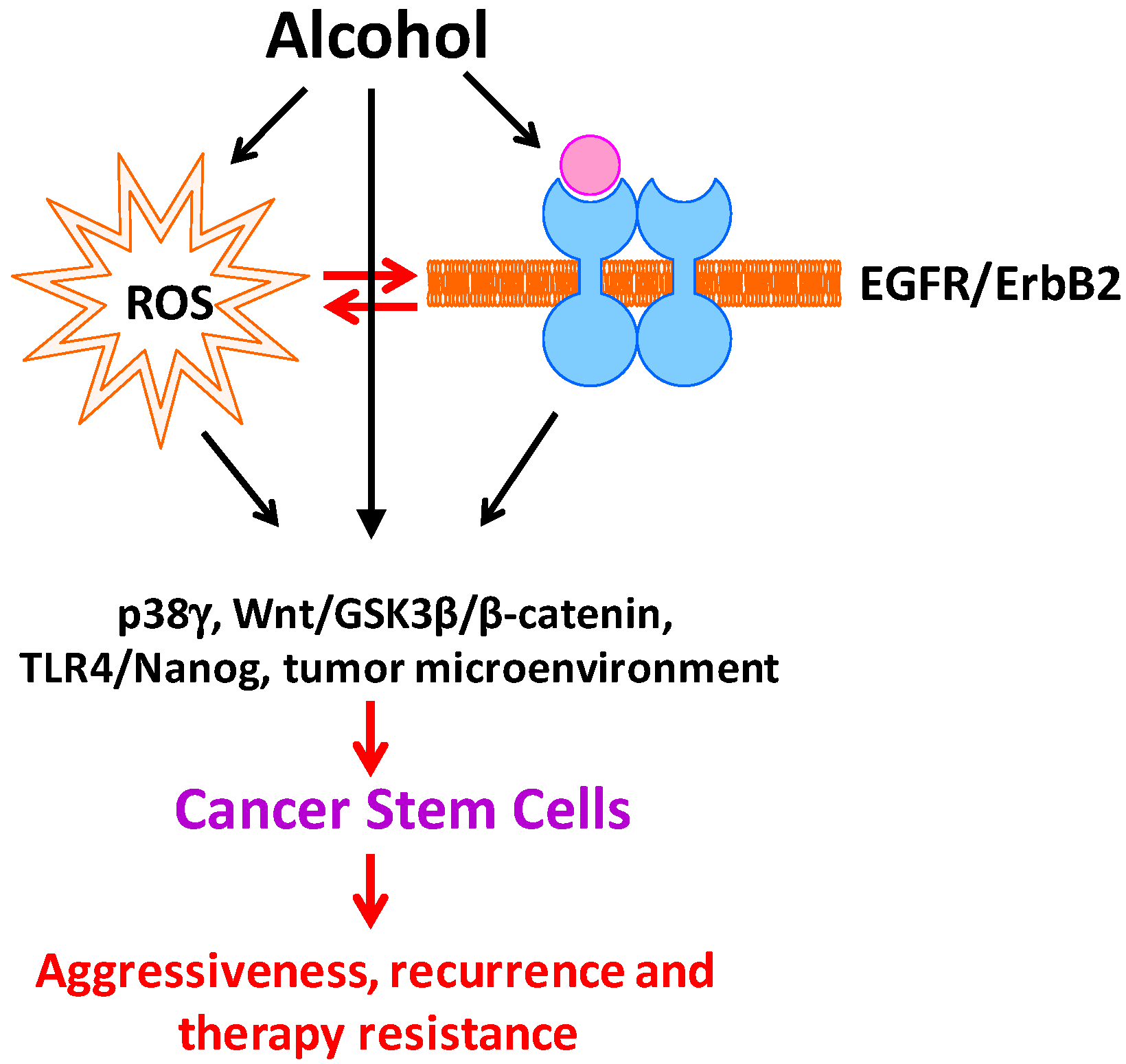
2. Alcohol Alters CSC Population
3. Cellular and Molecular Mechanisms Underlying Alcohol Stimulation of CSCs
3.1. Oxidative Stress and Tumor Microenvironment in Alcohol Promotion of CSCs
3.2. Epidermal Growth Factor Receptor (EGFR) Family in Alcohol Promotion of CSCs
3.3. p38γ MAPK in Alcohol-Induced Increase in CSC Population
3.4. Other Signaling Proteins Regulating Stemness in Alcohol Promotion of CSCs
4. Summary, Future Studies, and Potential Therapeutic Approaches
Acknowledgments
Conflicts of Interest
Abbreviations
| ADH | alcohol dehydrogenase |
| ALDH2 | aldehyde dehydrogenase |
| CSC | cancer stem cells |
| CYP2E1 | cytochrome P450 2E1 |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| HCC | hepatocellular carcinoma |
| HNSCC | head and neck squamous cell carcinoma |
| MMP | matrix metalloproteinase |
| ROS | reactive oxygen species |
References
- Pflaum, T.; Hausler, T.; Baumung, C.; Ackermann, S.; Kuballa, T.; Rehm, J.; Lachenmeier, D.W. Carcinogenic compounds in alcoholic beverages: An update. Arch. Toxicol. 2016, 90, 2349–2367. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Giovannucci, E.L. Alcohol as a Risk Factor for Cancer. Semin. Oncol. Nurs. 2016, 32, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.E.; Jarman, D.W.; Rehm, J.; Greenfield, T.K.; Rey, G.; Kerr, W.C.; Miller, P.; Shield, K.D.; Ye, Y.; Naimi, T.S. Alcohol-attributable cancer deaths and years of potential life lost in the United States. Am. J. Public Health 2013, 103, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, M.; Ke, Z.-J.; Luo, J. Cellular and molecular mechanisms underlying alcohol-induced aggressiveness of breast cancer. Pharmacol. Res. 2017, 115, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Hall, R.R., III; Ahmed, A.U. Cancer Stem Cells: Cellular Plasticity, Niche, and its Clinical Relevance. J. Stem Cell Res. Ther. 2016, 6, 363. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Garcia-Mayea, Y.; Mir, C.; Lorente, J.; Rubio, I.; LLeonart, M. The cancer stem-cell signaling network and resistance to therapy. Cancer Treat. Rev. 2016, 49, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; Song, S.; Hochster, H.S.; Steinberg, I.B. Cancer stem cells: The promise and the potential. Semin. Oncol. 2015, 42, S3–S17. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, S.; Ren, Z.; Frank, J.A.; Yang, X.H.; Zhang, Z.; Ke, Z.J.; Shi, X.; Luo, J. Chronic ethanol exposure enhances the aggressiveness of breast cancer: The role of p38gamma. Oncotarget 2016, 7, 3489–3505. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Ren, Z.; Wang, X.; Comer, A.; Frank, J.A.; Ke, Z.-J.; Huang, Y.; Zhang, Z.; Shi, X.; Wang, S. ErbB2 and p38γ MAPK mediate alcohol-induced increase in breast cancer stem cells and metastasis. Mol. Cancer 2016, 15, 52. [Google Scholar] [CrossRef] [PubMed]
- De Beça, F.F.; Caetano, P.; Gerhard, R.; Alvarenga, C.A.; Gomes, M.; Paredes, J.; Schmitt, F. Cancer stem cells markers CD44, CD24 and ALDH1 in breast cancer special histological types. J. Clin. Pathol. 2013, 66, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Tsukamoto, H.; Mkrtchyan, H.; Duan, L.; Dynnyk, A.; Liu, H.M.; Asahina, K.; Govindarajan, S.; Ray, R.; Ou, J.-H.J. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc. Natl. Acad. Sci. USA 2009, 106, 1548–1553. [Google Scholar] [CrossRef] [PubMed]
- Ambade, A.; Satishchandran, A.; Szabo, G. Alcoholic hepatitis accelerates early hepatobiliary cancer by increasing stemness and miR-122-mediated HIF-1alpha activation. Sci. Rep. 2016, 6, 21340. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Chen, C.L.; Liu, J.C.; Kashiwabara, C.; Feldman, D.; French, S.W.; Sher, L.; Hyeongnam, J.J.; Tsukamoto, H. Cancer stem cells generated by alcohol, diabetes, and hepatitis C virus. J. Gastroenterol. Hepatol. 2012, 27, 19–22. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M. A local mechanism by which alcohol consumption causes cancer. Oral Oncol. 2016, 62, 149–152. [Google Scholar] [CrossRef] [PubMed]
- Osei-Sarfo, K.; Tang, X.-H.; Urvalek, A.M.; Scognamiglio, T.; Gudas, L.J. The molecular features of tongue epithelium treated with the carcinogen 4-nitroquinoline-1-oxide and alcohol as a model for HNSCC. Carcinogenesis 2013, 34, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Khalid, O.; Kim, J.J.; Kim, H.-S.; Hoang, M.; Tu, T.G.; Elie, O.; Lee, C.; Vu, C.; Horvath, S.; Spigelman, I. Gene expression signatures affected by alcohol-induced DNA methylomic deregulation in human embryonic stem cells. Stem Cell Res. 2014, 12, 791–806. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.; Kim, J.J.; Kim, Y.; Tong, E.; Trammell, B.; Liu, Y.; Shi, S.; Lee, C.-R.; Hong, C.; Wang, C.-Y. Alcohol-induced suppression of KDM6B dysregulates the mineralization potential in dental pulp stem cells. Stem Cell Res. 2016, 17, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Azmi, A.S.; Li, Y.; Ahmad, A.; Ali, S.; Banerjee, S.; Kong, D.; H Sarkar, F. Targeting CSCs in tumor microenvironment: The potential role of ROS-associated miRNAs in tumor aggressiveness. Curr. Stem Cell Res. Ther. 2014, 9, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Hoek, J.B.; Cahill, A.; Pastorino, J.G. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ke, Z.; Chen, G.; Xu, M.; Bower, K.A.; Frank, J.A.; Zhang, Z.; Shi, X.; Luo, J. Cdc42-dependent activation of NADPH oxidase is involved in ethanol-induced neuronal oxidative stress. PLoS ONE 2012, 7, e38075. [Google Scholar] [CrossRef] [PubMed]
- Aye, M.M.; Ma, C.; Lin, H.; Bower, K.A.; Wiggins, R.C.; Luo, J. Ethanol-induced in vitro invasion of breast cancer cells: The contribution of MMP-2 by fibroblasts. Int. J. Cancer 2004, 112, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Etique, N.; Grillier-Vuissoz, I.; Flament, S. Ethanol stimulates the secretion of matrix metalloproteinases 2 and 9 in MCF-7 human breast cancer cells. Oncol. Rep. 2006, 15, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Lin, H.; Fan, Z.; Cai, T.Q.; Kaplan, R.A.; Ma, C.; Bower, K.A.; Shi, X.; Luo, J. MMP-2 mediates ethanol-induced invasion of mammary epithelial cells over-expressing ErbB2. Int. J. Cancer 2006, 119, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Bower, K.A.; Chen, G.; Shi, X.; Dong, Z.; Ke, Z.; Luo, J. Ethanol enhances the interaction of breast cancer cells over-expressing ErbB2 with fibronectin. Alcohol. Clin. Exp. Res. 2010, 34, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, M.; Li, F.; Wang, X.; Bower, K.A.; Frank, J.A.; Lu, Y.; Chen, G.; Zhang, Z.; Ke, Z.; et al. Ethanol promotes mammary tumor growth and angiogenesis: The involvement of chemoattractant factor MCP-1. Breast Cancer Res. Treat. 2012, 133, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Ni, F.; Xu, M.; Yang, J.; Chen, J.; Chen, Z.; Wang, X.; Luo, J.; Wang, S. Alcohol promotes mammary tumor growth through activation of VEGF-dependent tumor angiogenesis. Oncol. Lett. 2014, 8, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhang, F.; Ren, H.; Luo, J.; Wang, S. Role of cytokines and chemokines in alcohol-induced tumor promotion. OncoTargets Ther. 2017, 10, 1665. [Google Scholar] [CrossRef] [PubMed]
- Davis, N.M.; Sokolosky, M.; Stadelman, K.; Abrams, S.L.; Libra, M.; Candido, S.; Nicoletti, F.; Polesel, J.; Maestro, R.; D’Assoro, A. Deregulation of the EGFR/PI3K/PTEN/Akt/mTORC1 pathway in breast cancer: Possibilities for therapeutic intervention. Oncotarget 2014, 5, 4603. [Google Scholar] [CrossRef] [PubMed]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Miller, L.; Metheny-Barlow, L.; Lo, H.-W. EGFR and HER2 signaling in breast cancer brain metastasis. Front. Biosci. 2016, 8, 245. [Google Scholar]
- Lohrisch, C.; Piccart, M. An overview of HER2. Semin. Oncol. 2001, 28, 3–11. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; Van De Rijn, M. Molecular portraits of human breast tumours. Nature 2000, 406, 747. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Hu, Z.; He, X.; Livasy, C.; Carey, L.A.; Ewend, M.G.; Glas, A.M.; Perou, C.M.; Van’t Veer, L.J. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res. 2005, 65, 9155–9158. [Google Scholar] [CrossRef] [PubMed]
- Moasser, M.M. The oncogene HER2; Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Abrams, S.L.; Fitzgerald, T.L.; Cocco, L.; Martelli, A.M.; Montalto, G.; Cervello, M.; Scalisi, A.; Candido, S.; Libra, M. Roles of signaling pathways in drug resistance, cancer initiating cells and cancer progression and metastasis. Adv. Biol. Regul. 2015, 57, 75–101. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.R.; Yarden, Y. The EGFR-HER2 module: A stem cell approach to understanding a prime target and driver of solid tumors. Oncogene 2016, 35, 2949. [Google Scholar] [CrossRef] [PubMed]
- Nami, B.; Wang, Z. HER2 in Breast Cancer Stemness: A Negative Feedback Loop towards Trastuzumab Resistance. Cancers 2017, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Forsyth, C.B.; Tang, Y.; Shaikh, M.; Zhang, L.; Keshavarzian, A. Alcohol stimulates activation of Snail, epidermal growth factor receptor signaling, and biomarkers of epithelialGÇômesenchymal transition in colon and breast cancer cells. Alcohol. Clin. Exp. Res. 2010, 34, 19–31. [Google Scholar] [CrossRef] [PubMed]
- León-Buitimea, A.; Rodríguez-Fragoso, L.; Lauer, F.T.; Bowles, H.; Thompson, T.A.; Burchiel, S.W. Ethanol-induced oxidative stress is associated with EGF receptor phosphorylation in MCF-10A cells overexpressing CYP2E1. Toxicol. Lett. 2012, 209, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Lin, H.; Leonard, S.S.; Shi, X.; Ye, J.; Luo, J. Overexpression of ErbB2 enhances ethanol-stimulated intracellular signaling and invasion of human mammary epithelial and breast cancer cells in vitro. Oncogene 2003, 22, 5281–5290. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, D.T.; Iyer, H.; Escudero, S.; Bao, L.; Wu, Z.; Ventura, A.C.; Kleer, C.G.; Arruda, E.M.; Garikipati, K.; Merajver, S.D. p38γ promotes breast cancer cell motility and metastasis through regulation of RhoC GTPase, cytoskeletal architecture, and a novel leading edge behavior. Cancer Res. 2011, 71, 6338–6349. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Yin, N.; Ma, S.; Lepp, A.; Tang, J.; Jing, W.; Johnson, B.; Dwinell, M.B.; Chitambar, C.R.; Chen, G. p38γ MAPK Is a Therapeutic Target for Triple-Negative Breast Cancer by Stimulation of Cancer Stem-Like Cell Expansion. Stem Cells 2015, 33, 2738–2747. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zong, Z.-H.; Xu, H.-M. RhoC expression level is correlated with the clinicopathological characteristics of ovarian cancer and the expression levels of ROCK-I, VEGF, and MMP9. Gynecol. Oncol. 2010, 116, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Ramdass, B.; Nagarajan, S.; Rehman, M.; Mukherjee, G.; Krishna, S. Notch1 regulates the functional contribution of RhoC to cervical carcinoma progression. Br. J. Cancer 2010, 102, 196. [Google Scholar] [CrossRef] [PubMed]
- Ikoma, T.; Takahashi, T.; Nagano, S.; Li, Y.-M.; Ohno, Y.; Ando, K.; Fujiwara, T.; Fujiwara, H.; Kosai, K.-I. A definitive role of RhoC in metastasis of orthotopic lung cancer in mice. Clin. Cancer Res. 2004, 10, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.; Sharma, S.; Teknos, T.N. RhoC regulates cancer stem cells in head and neck squamous cell carcinoma by overexpressing IL-6 and phosphorylation of STAT3. PLoS ONE 2014, 9, e88527. [Google Scholar] [CrossRef] [PubMed]
- Luo, J. Glycogen synthase kinase 3β (GSK3β) in tumorigenesis and cancer chemotherapy. Cancer Lett. 2009, 273, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yi, M.; Chen, S.; Li, J.; Zhang, H.; Xiong, W.; Li, G.; Li, X.; Xiang, B. NOR1 Suppresses Cancer Stem-Like Cells Properties of Tumor Cells via the Inhibition of the AKT-GSK-3β-Wnt/β-catenin-ALDH1A1 Signal Circuit. J. Cell. Physiol. 2017, 232, 2829–2840. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Park, S.-Y.; Jun, Y.; Kim, J.-Y.; Nam, J.-S. Roles of Wnt target genes in the journey of cancer stem cells. Int. J. Mol. Sci. 2017, 18, 1604. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, S.; Qi, Y.; Chen, L.; Frank, J.A.; Yang, X.H.; Zhang, Z.; Shi, X.; Luo, J. Role of MCP-1 in alcohol-induced aggressiveness of colorectal cancer cells. Mol. Carcinog. 2016, 55, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Mercer, K.; Hennings, L.; Ronis, M. Alcohol consumption, Wnt/β-catenin signaling, and hepatocarcinogenesis. In Biological Basis of Alcohol-Induced Cancer; Springer: New York, NY, USA, 2015; pp. 185–195. [Google Scholar]
- Lee, S.; Wottrich, S.; Bonavida, B. Crosstalks between Raf-kinase inhibitor protein and cancer stem cell transcription factors (Oct4, KLF4, Sox2, Nanog). Tumor Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, R.; Vernet, D.; Bruhn, K.W.; Sarkissyan, S.; Heber, D.; Vadgama, J.V.; Gonzalez-Cadavid, N.F. Long-term exposure of MCF-7 breast cancer cells to ethanol stimulates oncogenic features. Int. J. Oncol. 2017, 50, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R. Resistance to cell death and its modulation in cancer stems cells. Crit. Rev. Oncog. 2016, 21, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Atkinson, K.; Zhang, T. Combination of chemotherapy and cancer stem cell targeting agents: Preliclinal and clinical studies. Cancer Lett. 2017, 396, 103–109. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, M.; Luo, J. Alcohol and Cancer Stem Cells. Cancers 2017, 9, 158. https://doi.org/10.3390/cancers9110158
Xu M, Luo J. Alcohol and Cancer Stem Cells. Cancers. 2017; 9(11):158. https://doi.org/10.3390/cancers9110158
Chicago/Turabian StyleXu, Mei, and Jia Luo. 2017. "Alcohol and Cancer Stem Cells" Cancers 9, no. 11: 158. https://doi.org/10.3390/cancers9110158
APA StyleXu, M., & Luo, J. (2017). Alcohol and Cancer Stem Cells. Cancers, 9(11), 158. https://doi.org/10.3390/cancers9110158