The Role of Platelet-Derived ADP and ATP in Promoting Pancreatic Cancer Cell Survival and Gemcitabine Resistance



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
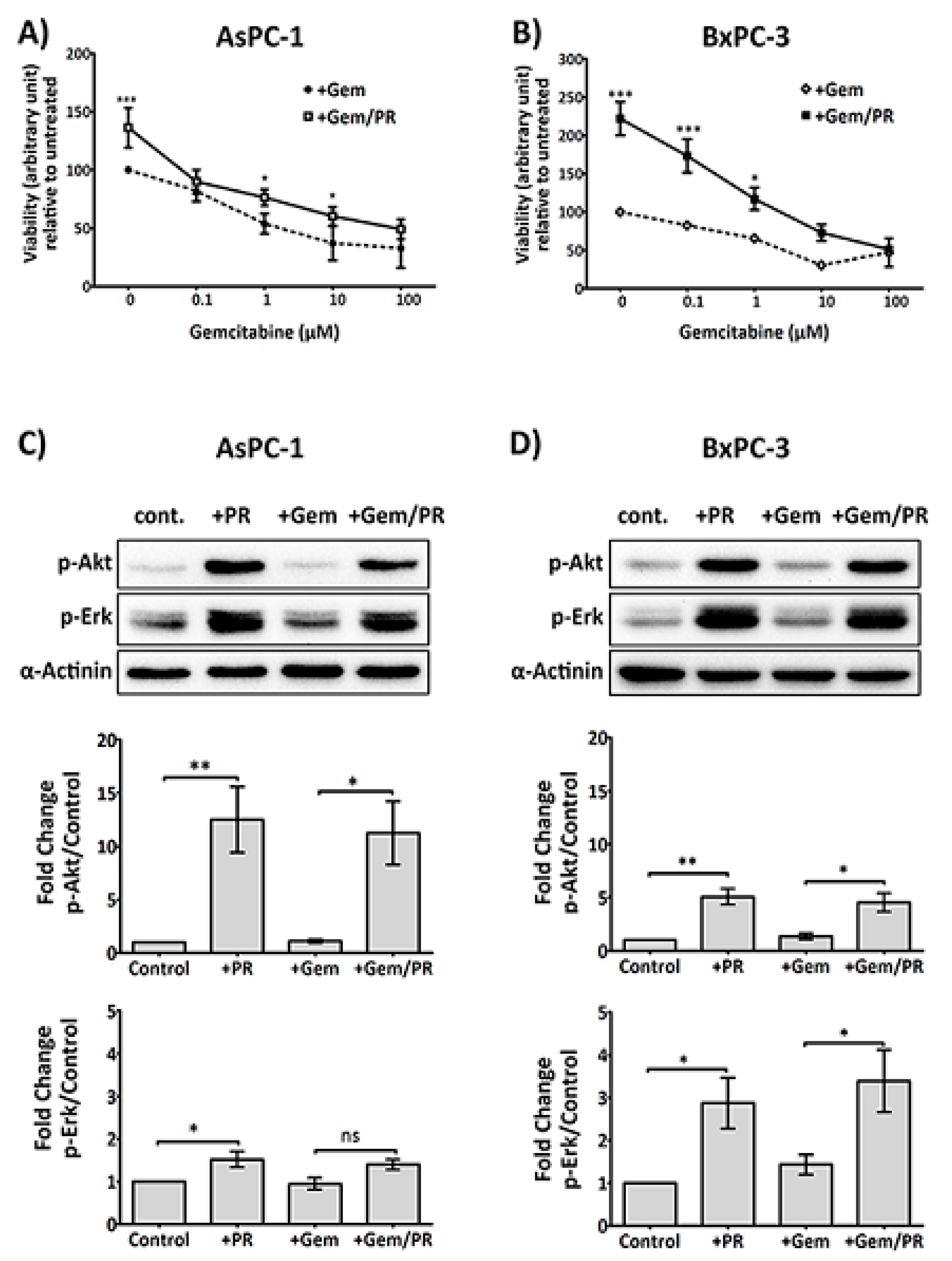
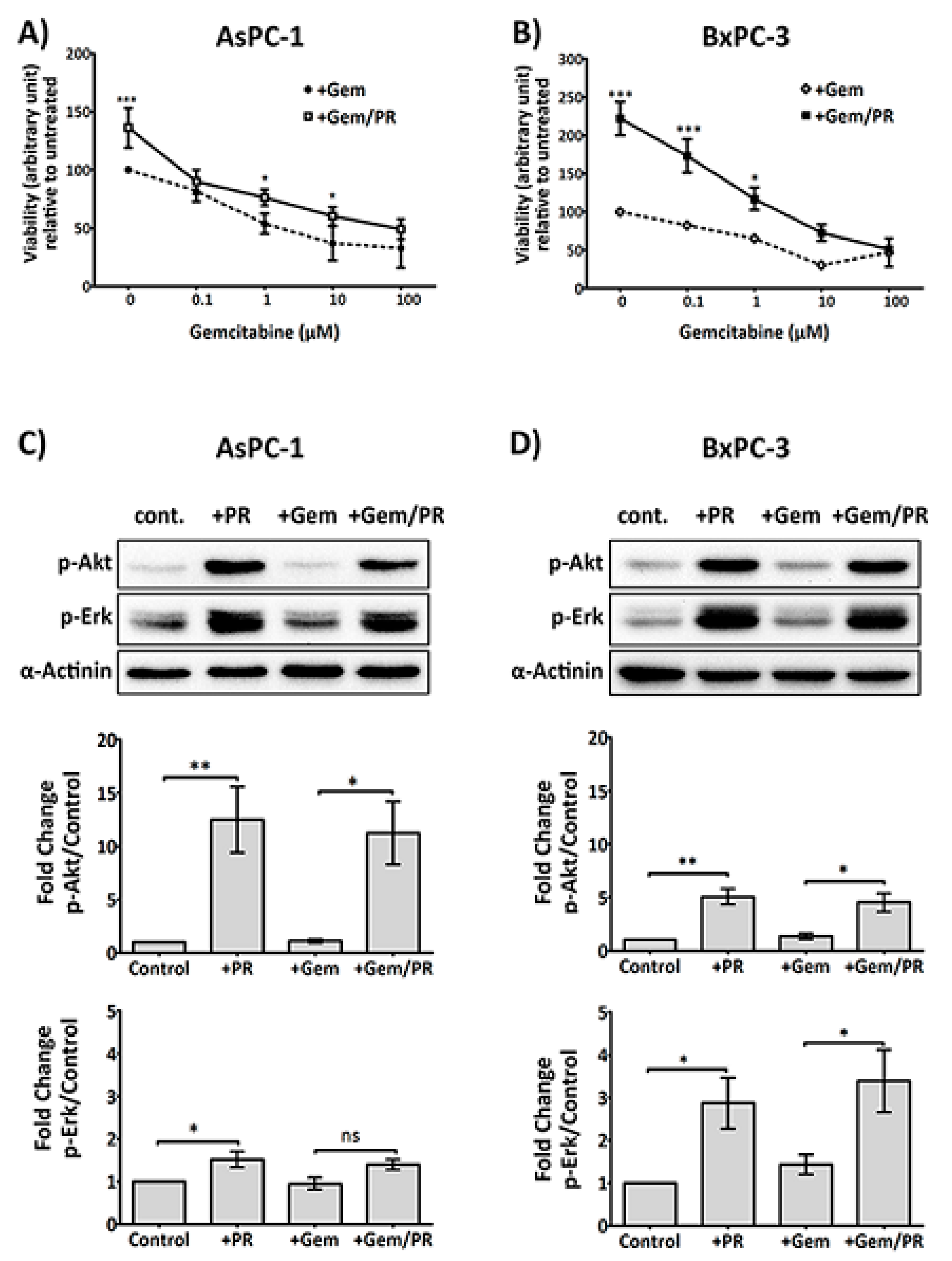
2.1. Platelet Releasate Promotes Proliferation and Survival Signals in PDAC Cells Challenged with Gemcitabine
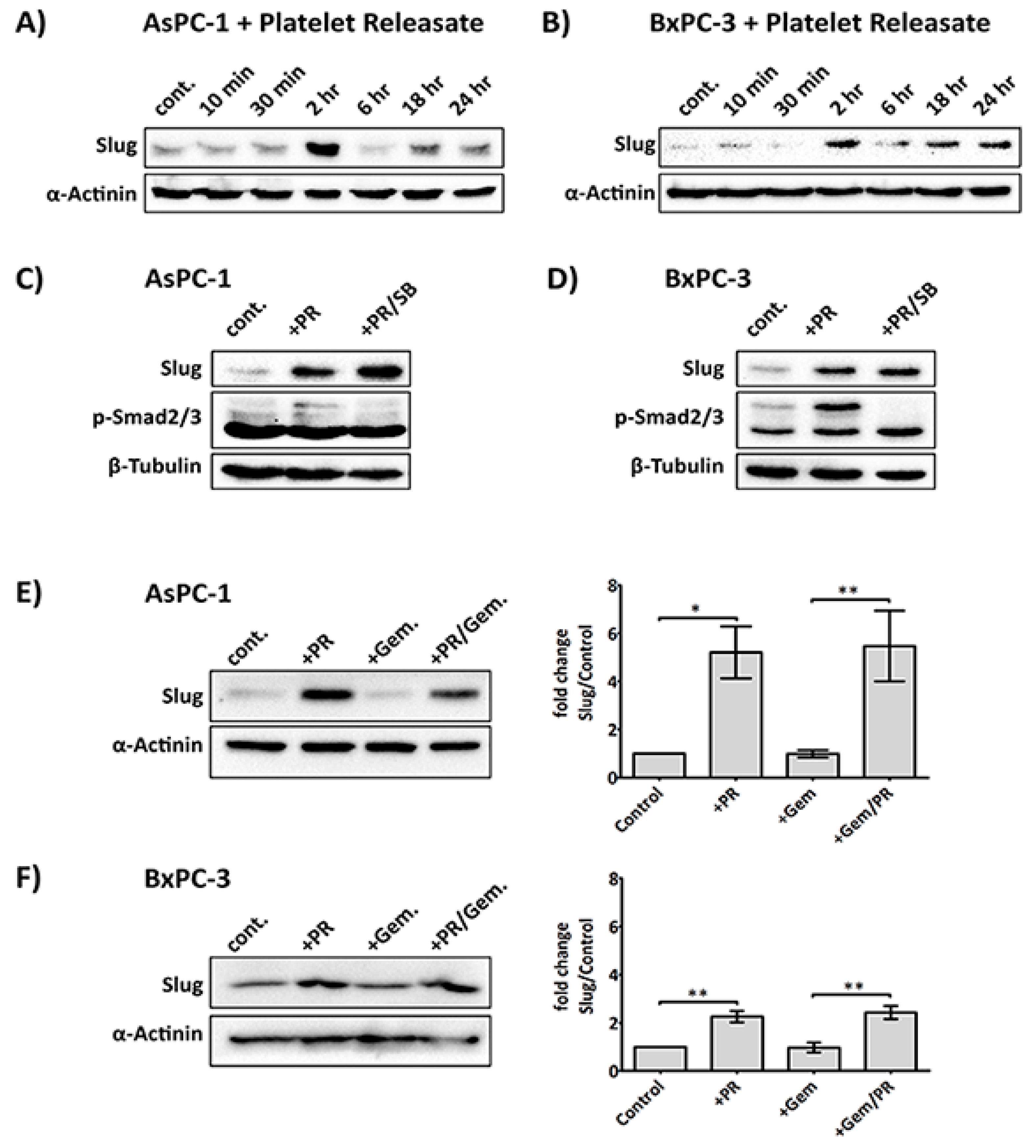
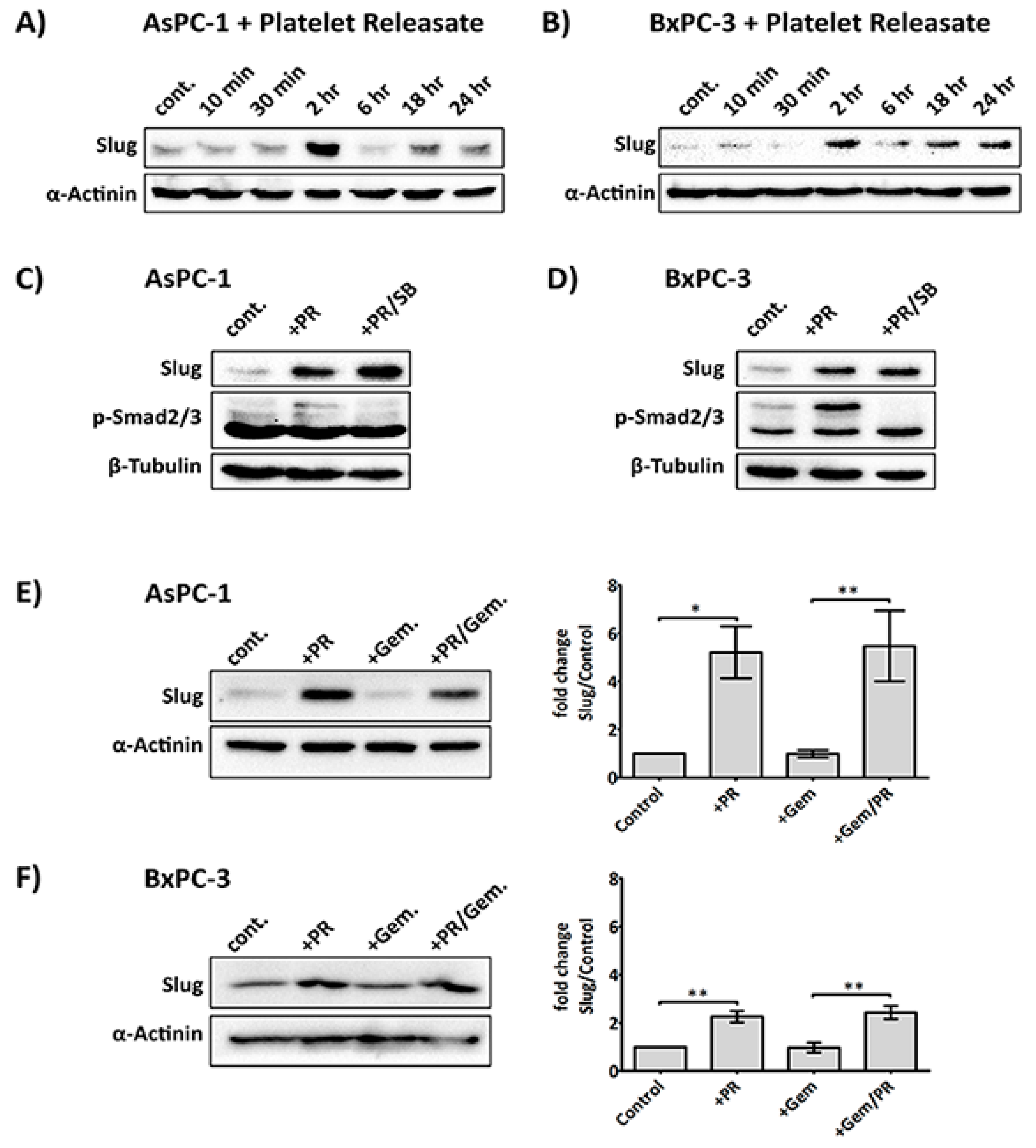
2.2. Platelet Releasate Induces A Rapid Upregulation of the EMT Transcription Factor, Slug, Independent of the TGFβ/Smad Pathway
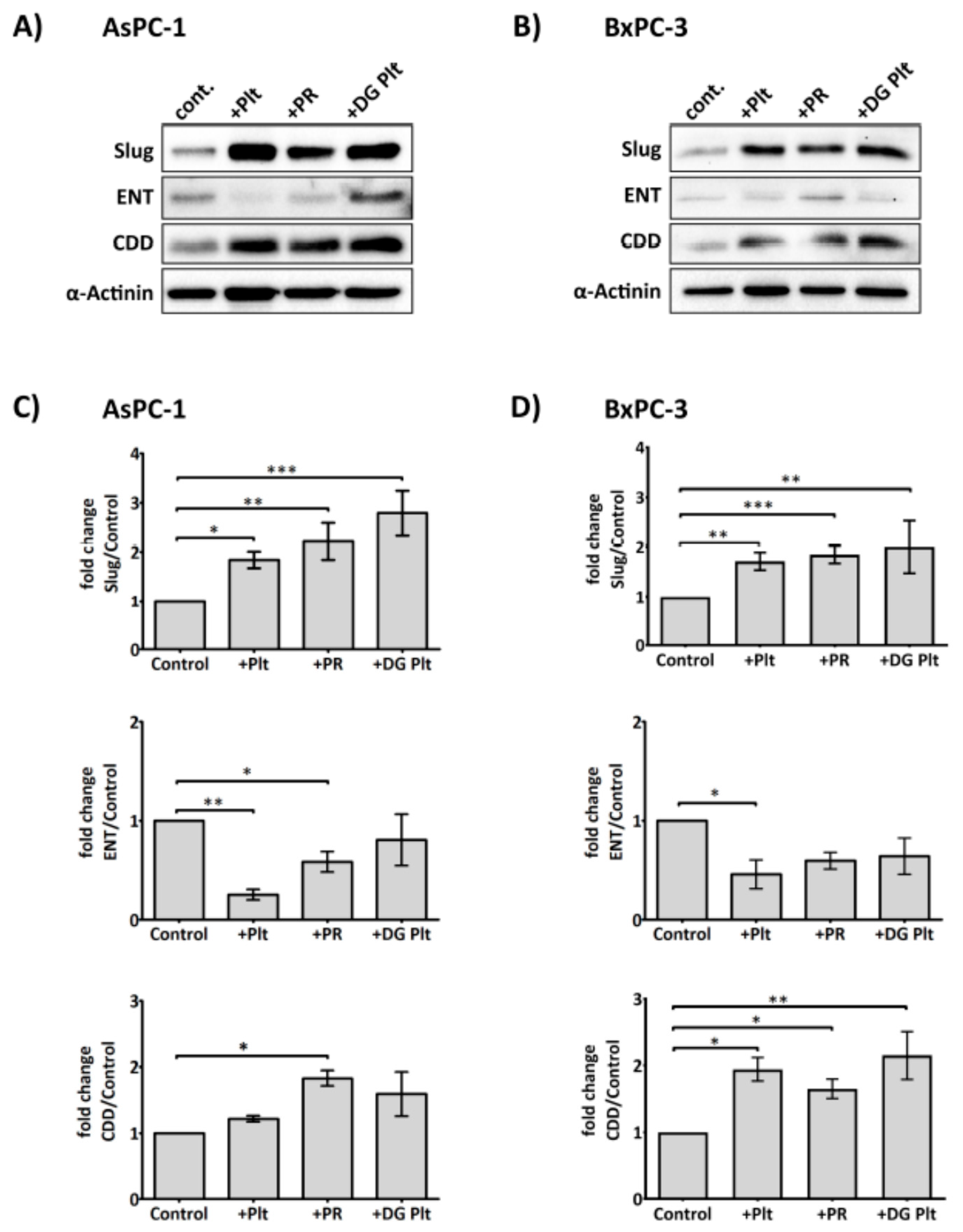
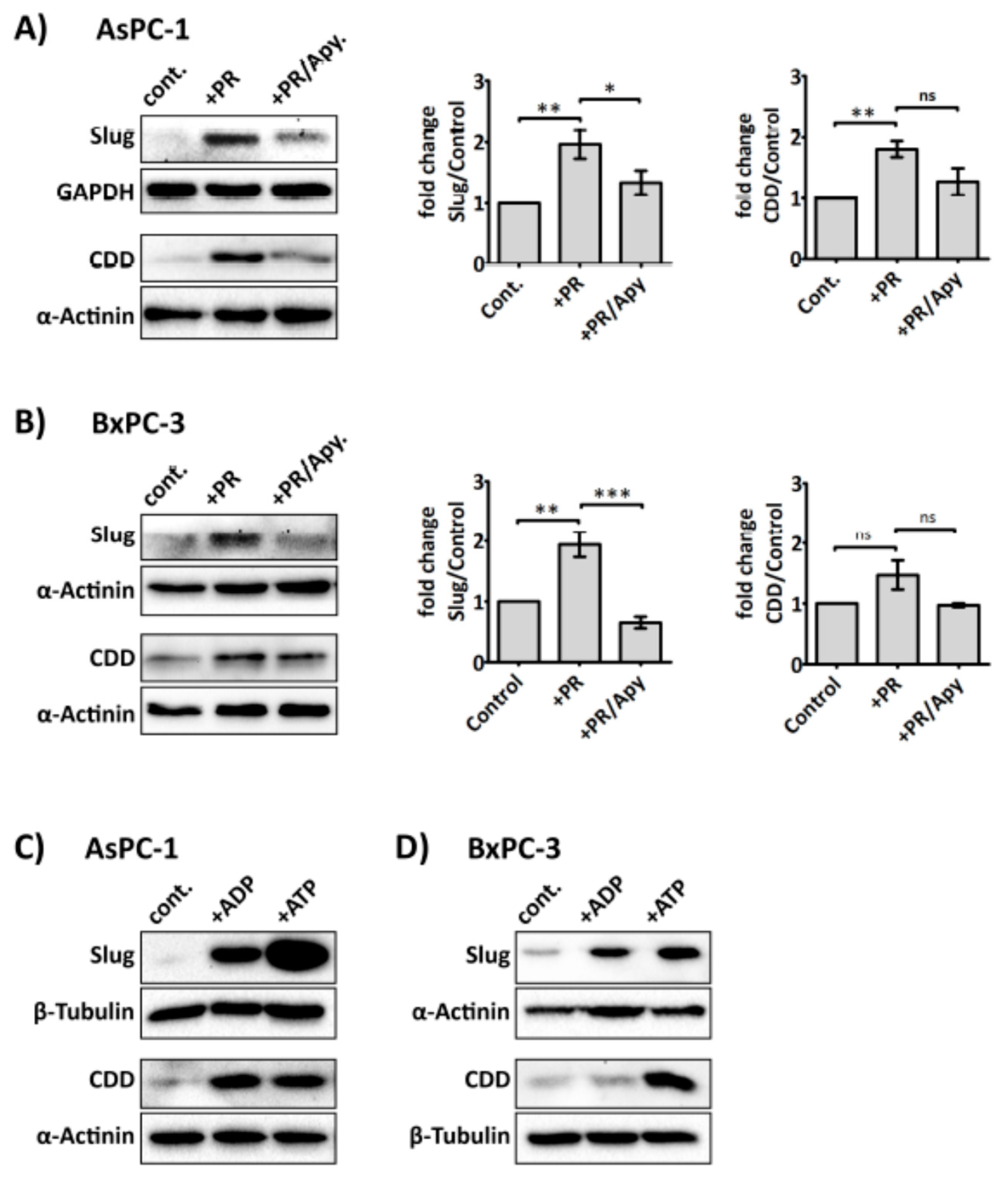
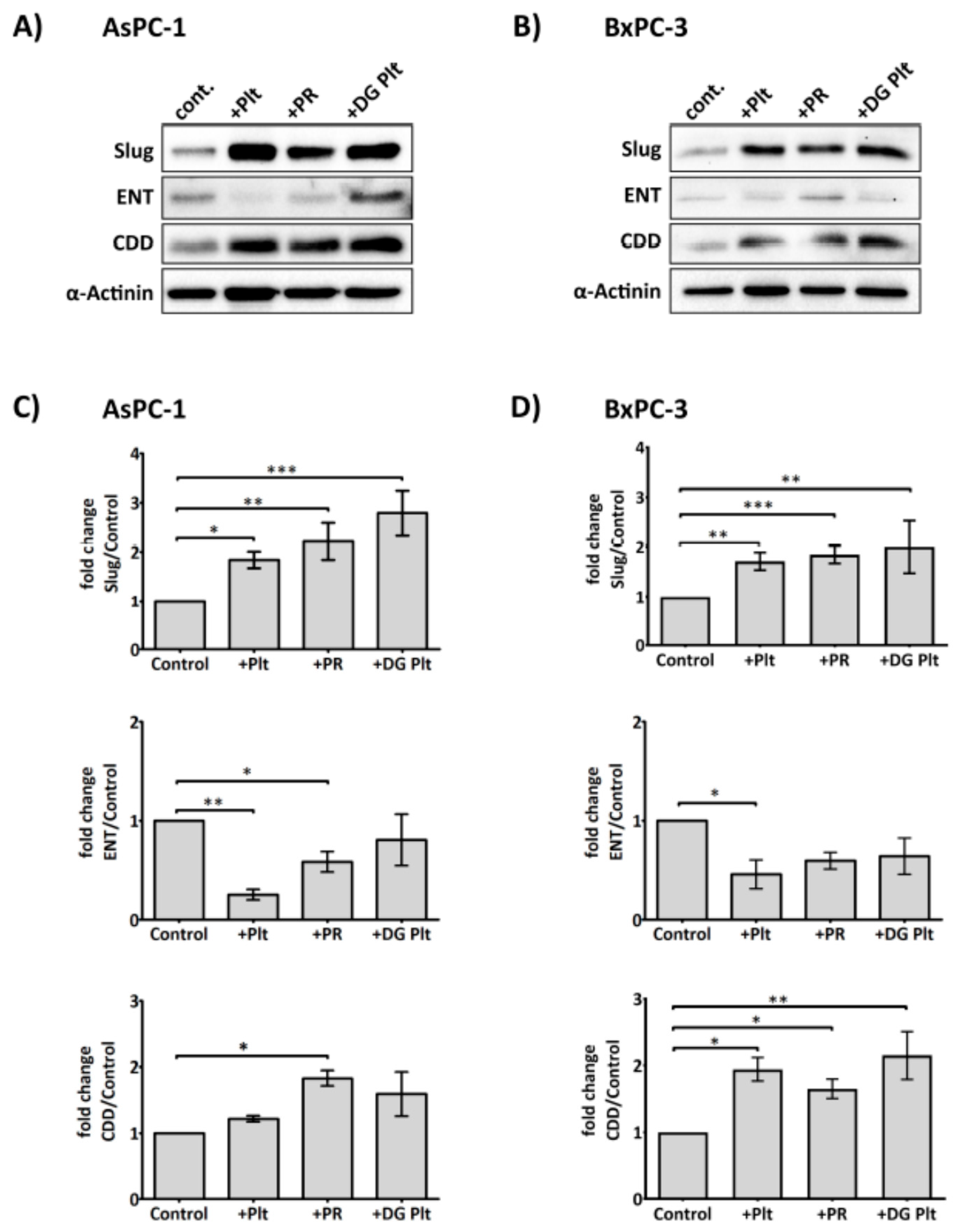
2.3. Platelets Modulate hENT1 and CDD in PDAC Cells
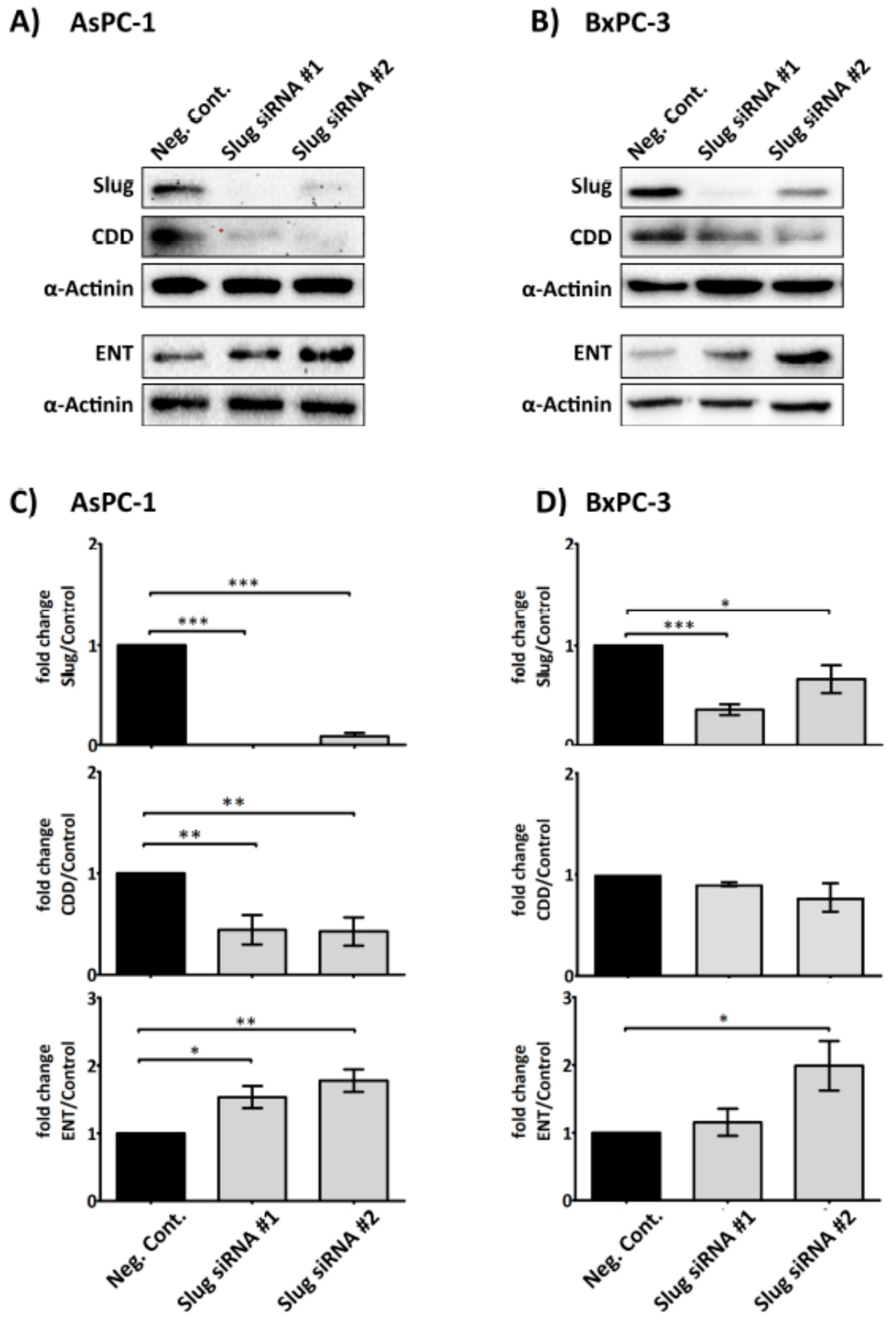
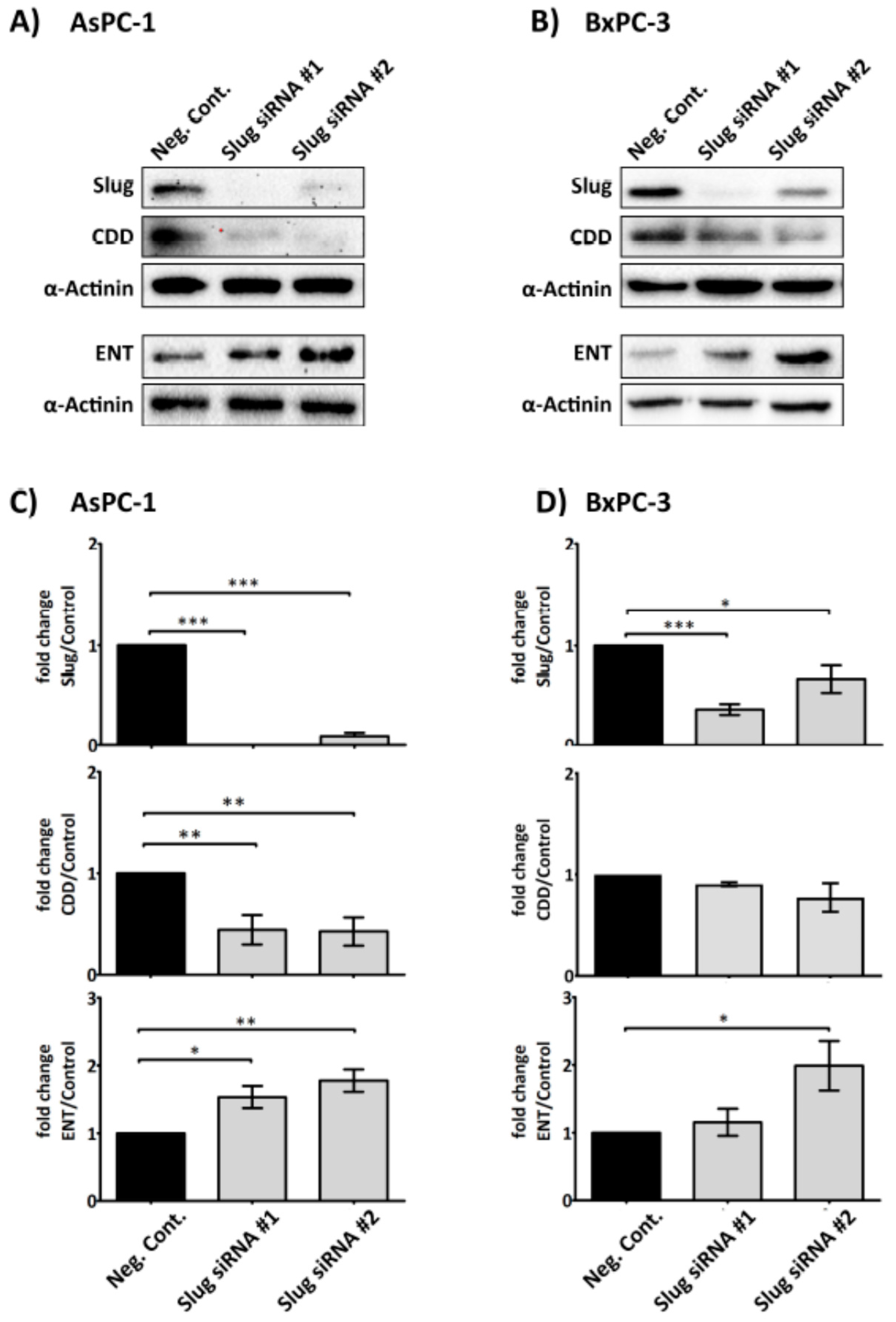
2.4. Slug Modulates Expression of CDD and hENT1 in PDAC Cells
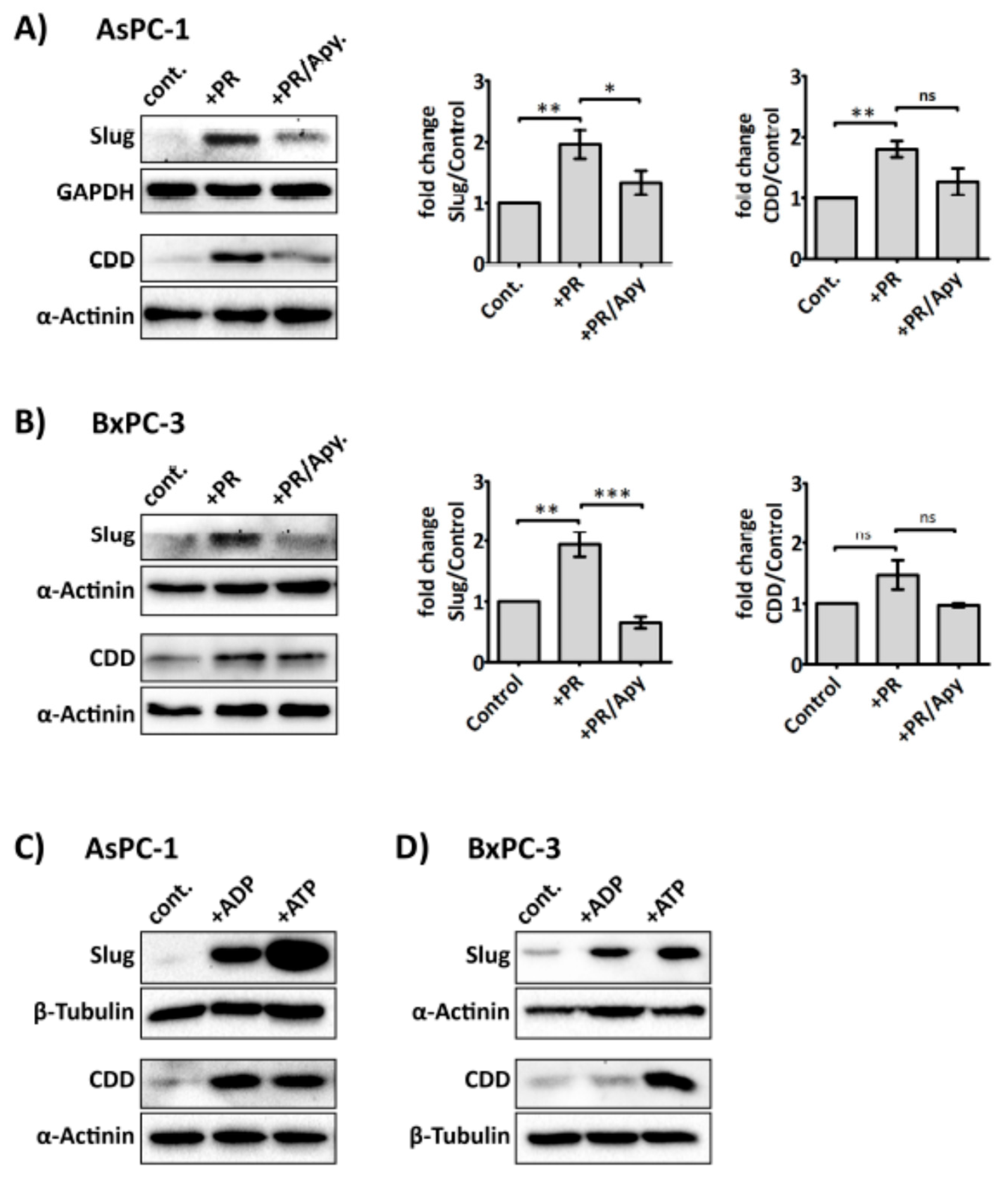
2.5. Platelet-Derived ADP and ATP Promote Slug and CDD Expression Levels in PDAC Cells
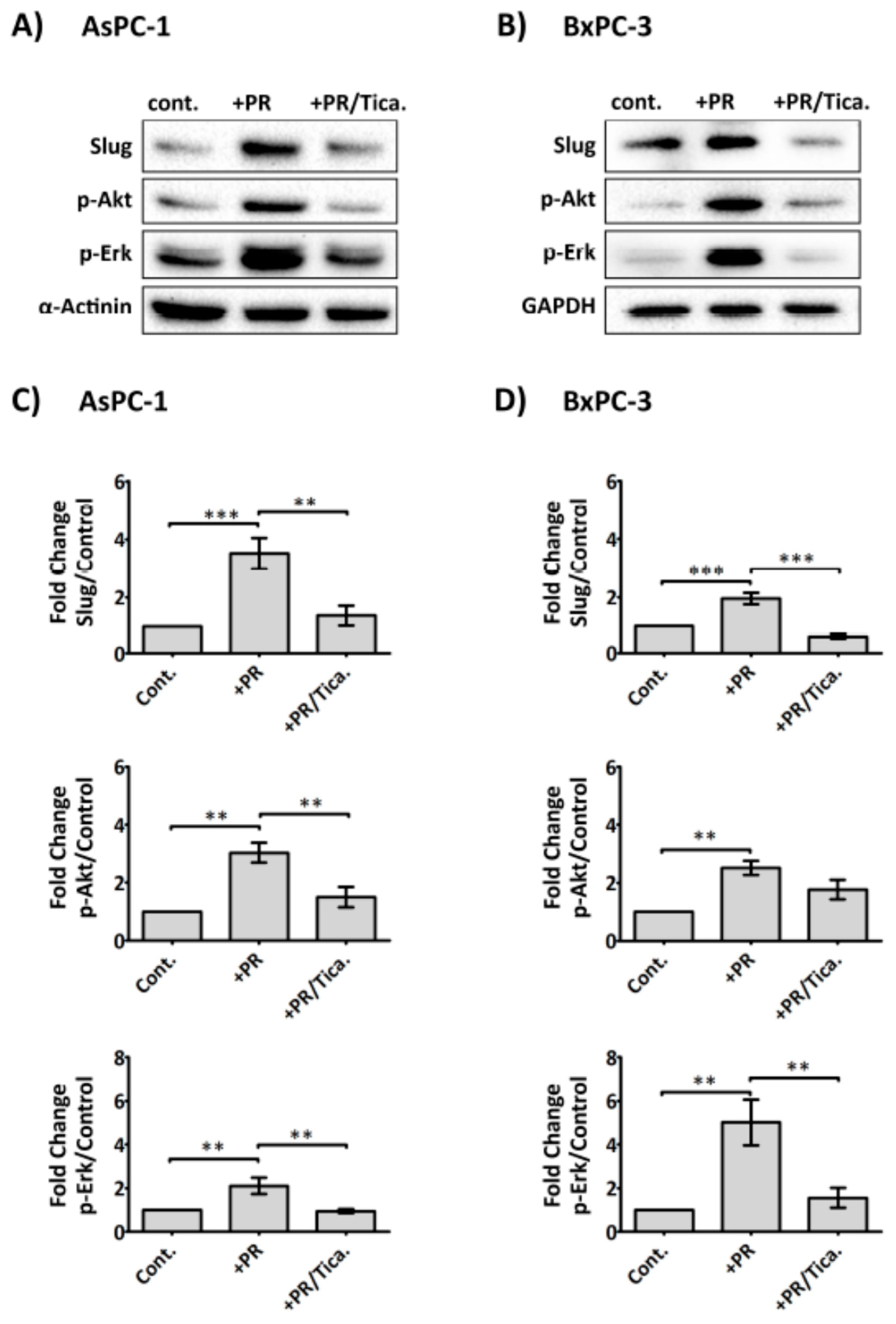
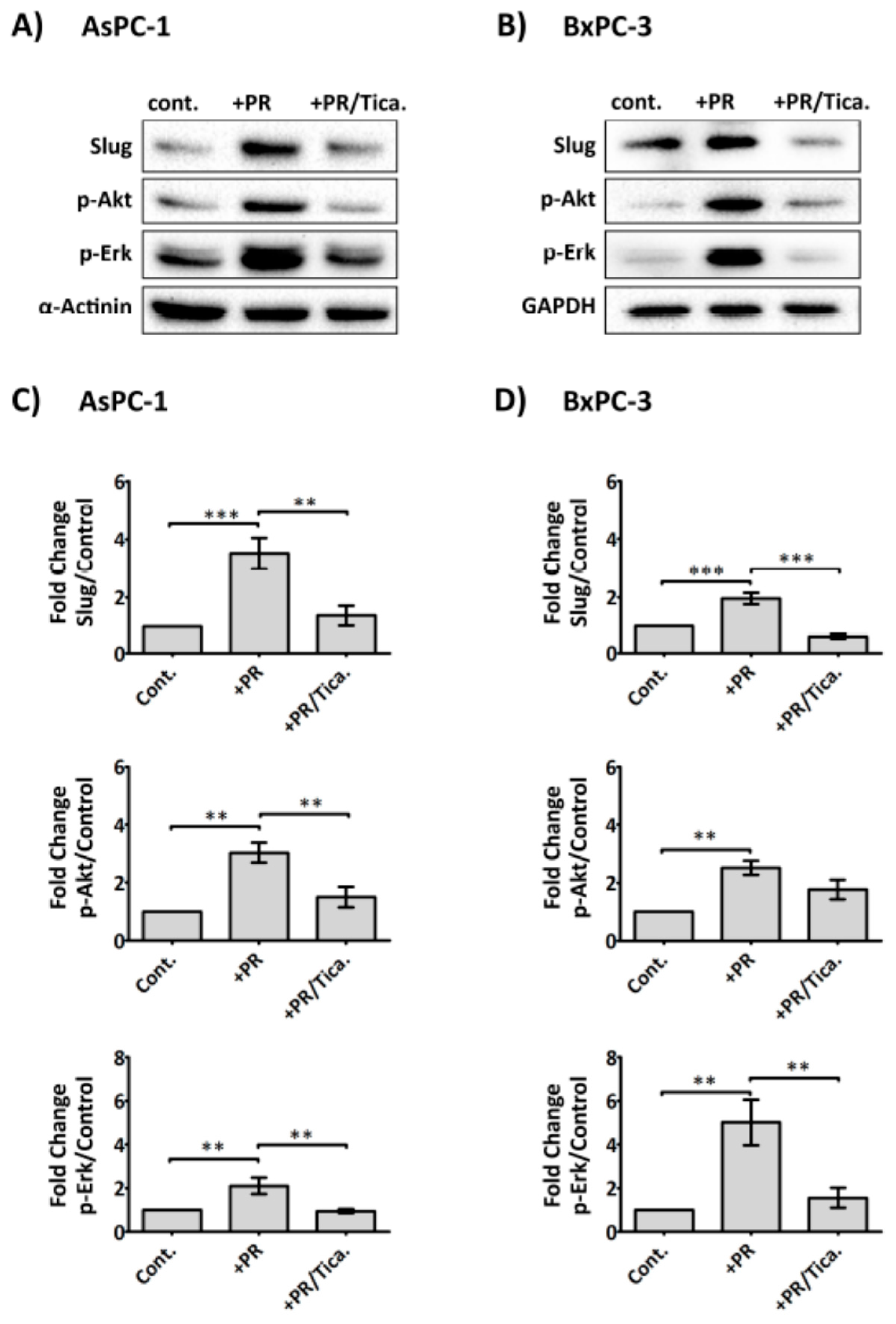
2.6. The Antiplatelet Drug, Ticagrelor, Reduces PR-Induced Akt, Erk Activation and Slug Upregulation in Cancer Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Cell Lines
4.2. Preparation of Human Washed Platelets
4.3. Cell Viability
4.4. Western Blot:
4.5. Slug Knockdown
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Falasca, M.; Kim, M.; Casari, I. Pancreatic cancer: Current research and future directions. Biochim. Biophys. Acta 2016, 1865, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Delluc, A.; Rousseau, A.; Delluc, C.; Le Moigne, E.; Le Gal, G.; Mottier, D.; van Dreden, P.; Lacut, K. Venous thromboembolism in patients with pancreatic cancer: Implications of circulating tissue factor. Blood Coagul. Fibrinolysis 2011, 22, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Weiler, H. A platelet cloak for tumor cells. Blood 2005, 105, 5–6. [Google Scholar] [CrossRef]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirouskova, M.; Degen, J.L. Platelets and fibrin (ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Kopp, H.-G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-β down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [PubMed]
- Geddings, J.E.; Hisada, Y.; Boulaftali, Y.; Getz, T.M.; Whelihan, M.; Fuentes, R.; Dee, R.; Cooley, B.C.; Key, N.S.; Wolberg, A.S.; et al. Tissue factor-positive tumor microvesicles activate platelets and enhance thrombosis in mice. J. Thromb. Haemost. 2016, 14, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Mitrugno, A.; Williams, D.; Kerrigan, S.W.; Moran, N. A novel and essential role for FCγRIIa in cancer cell-induced platelet activation. Blood 2014, 123, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Battinelli, E.M.; Markens, B.A.; Italiano, J.E., Jr. Release of angiogenesis regulatory proteins from platelet alpha granules: Modulation of physiologic and pathologic angiogenesis. Blood 2011, 118, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Pucci, F.; Rickelt, S.; Newton, A.P.; Garris, C.; Nunes, E.; Evavold, C.; Pfirschke, C.; Engblom, C.; Mino-Kenudson, M.; Hynes, R.O.; et al. PF4 promotes platelet production and lung cancer growth. Cell Rep. 2016, 17, 1764–1772. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Guillem-Llobat, P.; Dovizio, M.; Bruno, A.; Ricciotti, E.; Cufino, V.; Sacco, A.; Grande, R.; Alberti, S.; Arena, V.; Cirillo, M.; et al. Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget 2016, 7, 32462–32477. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Tillo, E.; Liu, Y.; de Barrios, O.; Siles, L.; Fanlo, L.; Cuatrecasas, M.; Darling, D.S.; Dean, D.C.; Castells, A.; Postigo, A. EMT-activating transcription factors in cancer: Beyond EMT and tumor invasiveness. Cell. Mol. Life Sci. 2012, 69, 3429–3456. [Google Scholar] [CrossRef] [PubMed]
- Haslehurst, A.M.; Koti, M.; Dharsee, M.; Nuin, P.; Evans, K.; Geraci, J.; Childs, T.; Chen, J.; Li, J.; Weberpals, J.; et al. EMT transcription factors snail and slug directly contribute to cisplatin resistance in ovarian cancer. BMC Cancer 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Tsukasa, K.; Ding, Q.; Yoshimitsu, M.; Miyazaki, Y.; Matsubara, S.; Takao, S. Slug contributes to gemcitabine resistance through epithelial-mesenchymal transition in CD133(+) pancreatic cancer cells. Hum. Cell 2015, 28, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Plunkett, W.; Huang, P.; Xu, Y.Z.; Heinemann, V.; Grunewald, R.; Gandhi, V. Gemcitabine: Metabolism, mechanisms of action, and self-potentiation. Semin. Oncol. 1995, 22, 3–10. [Google Scholar] [PubMed]
- Huang, P.; Plunkett, W. Induction of apoptosis by gemcitabine. Semin. Oncol. 1995, 22, 19–25. [Google Scholar] [PubMed]
- Mini, E.; Nobili, S.; Caciagli, B.; Landini, I.; Mazzei, T. Cellular pharmacology of gemcitabine. Ann. Oncol. 2006, 17 (Suppl. 5), v7–v12. [Google Scholar] [CrossRef] [PubMed]
- Perez-Torras, S.; Garcia-Manteiga, J.; Mercade, E.; Casado, F.J.; Carbo, N.; Pastor-Anglada, M.; Mazo, A. Adenoviral-mediated overexpression of human equilibrative nucleoside transporter 1 (hENT1) enhances gemcitabine response in human pancreatic cancer. Biochem. Pharmacol. 2008, 76, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Frese, K.K.; Neesse, A.; Cook, N.; Bapiro, T.E.; Lolkema, M.P.; Jodrell, D.I.; Tuveson, D.A. nab-Paclitaxel Potentiates Gemcitabine Activity by Reducing Cytidine Deaminase Levels in a Mouse Model of Pancreatic Cancer. Cancer Discov. 2012, 2, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Ebrahem, Q.; Mahfouz, R.; Ng, K.P.; Saunthararajah, Y. High cytidine deaminase expression in the liver provides sanctuary for cancer cells from decitabine treatment effects. Oncotarget 2012, 3, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Kizer, N.T.; Hatem, H.; Nugent, E.K.; Zhou, G.F.; Moore, K.; Heller, P.; Mutch, D.G.; Thaker, P.H. Chemotherapy response rates among patients with endometrial cancer who have elevated serum platelets. Int. J. Gynecol. Cancer 2015, 25, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Ho-Tin-Noé, B.; Goerge, T.; Cifuni, S.M.; Duerschmied, D.; Wagner, D.D. Platelet granule secretion continuously prevents intratumor hemorrhage. Cancer Res. 2008, 68, 6851–6858. [Google Scholar] [CrossRef] [PubMed]
- Golebiewska, E.M.; Poole, A.W. Platelet secretion: From haemostasis to wound healing and beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Erpenbeck, L.; Schon, M.P. Deadly allies: The fatal interplay between platelets and metastasizing cancer cells. Blood 2010, 115, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Maier, T.J.; Alberti, S.; Di Francesco, L.; Marcantoni, E.; Münch, G.; John, C.M.; Suess, B.; Sgambato, A.; Steinhilber, D.; et al. Pharmacological inhibition of platelet-tumor cell cross-talk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Mol. Pharmacol. 2013, 84, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.T.; Zhang, S.Q.; Miao, L.L.; Wang, J.B.; Jin, Z.J.; Gu, B.; Duan, Z.H.; Zhao, Z.L.; Ma, S.M.; Zhang, W.J.; et al. Activation of platelet protease-activated receptor-1 induces epithelial-mesenchymal transition and chemotaxis of colon cancer cell line SW620. Oncol. Rep. 2015, 33, 2681–2688. [Google Scholar] [CrossRef] [PubMed]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M. Transforming growth factor-β and breast cancer: Transforming growth factor-β/SMAD signaling defects and cancer. Breast Cancer Res. 2000, 2, 107. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, A.; Moepert, K.; Dames, S.; Sternberger, M.; Kaufmann, J.; Klippel, A. Differential regulation of TGF-β signaling through Smad2, Smad3 and Smad4. Oncogene 2003, 22, 6748–6763. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Wilkes, M.C.; Penheiter, S.G.; Gupta, S.K.; Edens, M.; Leof, E.B. Non-Smad transforming growth factor-β signaling regulated by focal adhesion kinase binding the p85 subunit of phosphatidylinositol 3-kinase. J. Biol. Chem. 2011, 286, 17841–17850. [Google Scholar] [CrossRef] [PubMed]
- Wijten, P.; van Holten, T.; Woo, L.L.; Bleijerveld, O.B.; Roest, M.; Heck, A.J.; Scholten, A. High precision platelet releasate definition by quantitative reversed protein profiling—Brief report. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Mackey, J.R.; Mani, R.S.; Selner, M.; Mowles, D.; Young, J.D.; Belt, J.A.; Crawford, C.R.; Cass, C.E. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998, 58, 4349–4357. [Google Scholar] [PubMed]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812. [Google Scholar] [CrossRef] [PubMed]
- White, N.; Burnstock, G. P2 receptors and cancer. Trends Pharmacol. Sci. 2006, 27, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Giannuzzo, A.; Saccomano, M.; Napp, J.; Ellegaard, M.; Alves, F.; Novak, I. Targeting of the P2X7 receptor in pancreatic cancer and stellate cells. Int. J. Cancer 2016, 139, 2540–2552. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Qiu, Y.; Zhang, H.Q.; Liu, Y.; You, J.F.; Tian, X.X.; Fang, W.G. P2Y2 receptor promotes cell invasion and metastasis in prostate cancer cells. Br. J. Cancer 2013, 109, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Li, W.-H.; Zhang, H.-Q.; Liu, Y.; Tian, X.-X.; Fang, W.-G. P2X7 mediates ATP-driven invasiveness in prostate cancer cells. PLoS ONE 2014, 9, e114371. [Google Scholar] [CrossRef] [PubMed]
- Cedervall, J.; Zhang, Y.; Ringvall, M.; Thulin, A.; Moustakas, A.; Jahnen-Dechent, W.; Siegbahn, A.; Olsson, A.K. Hrg regulates tumor progression, epithelial to mesenchymal transition and metastasis via platelet-induced signaling in the pre-tumorigenic microenvironment. Angiogenesis 2013, 16, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Mitrugno, A.; Sylman, J.L.; Ngo, A.T.; Pang, J.; Sears, R.C.; Williams, C.; McCarty, O.J. Aspirin therapy reduces the ability of platelets to promote colon and pancreatic cancer cell proliferation: Implications for the oncoprotein c-MYC. Am. J. Physiol. Cell Physiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Radziwon-Balicka, A.; Medina, C.; O’Driscoll, L.; Treumann, A.; Bazou, D.; Inkielewicz-Stepniak, I.; Radomski, A.; Jow, H.; Radomski, M.W. Platelets increase survival of adenocarcinoma cells challenged with anticancer drugs: Mechanisms and implications for chemoresistance. Br. J. Pharmacol. 2012, 167, 787–804. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Fushida, S.; Miyashita, T.; Oyama, K.; Yamaguchi, T.; Tsukada, T.; Kinoshita, J.; Tajima, H.; Ninomiya, I.; Ohta, T. Potential of extravasated platelet aggregation as a surrogate marker for overall survival in patients with advanced gastric cancer treated with preoperative docetaxel, cisplatin and S-1: A retrospective observational study. BMC Cancer 2017, 17, 294. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Bottsford-Miller, J.; Vasquez, H.G.; Stone, R.; Zand, B.; Kroll, M.H.; Sood, A.K.; Afshar-Kharghan, V. Platelets increase the proliferation of ovarian cancer cells. Blood 2012, 120, 4869–4872. [Google Scholar] [CrossRef] [PubMed]
- Carr, B.I.; Cavallini, A.; D’Alessandro, R.; Refolo, M.G.; Lippolis, C.; Mazzocca, A.; Messa, C. Platelet extracts induce growth, migration and invasion in human hepatocellular carcinoma in vitro. BMC Cancer 2014, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- Fahy, B.N.; Schlieman, M.; Virudachalam, S.; Bold, R.J. AKT inhibition is associated with chemosensitisation in the pancreatic cancer cell line MIA-PaCa-2. Br. J. Cancer 2003, 89, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.S.W.; Tsao, M.S.; Nicklee, T.; Hedley, D.W. Wortmannin Inhibits PKB/Akt Phosphorylation and Promotes Gemeitabine Antitumor Activity in Orthotopic Human Pancreatic Cancer Xenografts in Immunodeficient Mice. Clin. Cancer Res. 2001, 7, 3269–3275. [Google Scholar] [PubMed]
- Wang, M.; Lu, X.; Dong, X.; Hao, F.; Liu, Z.; Ni, G.; Chen, D. pERK1/2 silencing sensitizes pancreatic cancer BXPC-3 cell to gemcitabine-induced apoptosis via regulating Bax and Bcl-2 expression. World J. Surg. Oncol. 2015, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Jiao, X.; Jiang, Y.; Sun, S. ERK1/2 activity contributes to gemcitabine resistance in pancreatic cancer cells. J. Int. Med. Res. 2013, 41, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, A.; Okitaka, M.; Takagi, S.; Takami, M.; Sato, S.; Nishio, M.; Okumura, S.; Fujita, N. A critical role of platelet TGF-β release in podoplanin-mediated tumour invasion and metastasis. Sci. Rep. 2017, 7, 42186. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- De Caestecker, M.P.; Piek, E.; Roberts, A.B. Role of transforming growth factor-β signaling in cancer. J. Natl. Cancer Inst. 2000, 92, 1388–1402. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Imamura, T.; Souchelnytskyi, S.; Kawabata, M.; Ishisaki, A.; Oeda, E.; Tamaki, K.; Hanai, J.; Heldin, C.H.; Miyazono, K.; et al. TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 1997, 16, 5353–5362. [Google Scholar] [CrossRef] [PubMed]
- Truty, M.J.; Urrutia, R. Basics of TGF-β and pancreatic cancer. Pancreatology 2007, 7, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Joseph, M.J.; Dangi-Garimella, S.; Shields, M.A.; Diamond, M.E.; Sun, L.; Koblinski, J.E.; Munshi, H.G. Slug is a downstream mediator of transforming growth factor-β1-induced matrix metalloproteinase-9 expression and invasion of oral cancer cells. J. Cell. Biochem. 2009, 108, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Hino, M.; Kamo, M.; Saito, D.; Kyakumoto, S.; Shibata, T.; Mizuki, H.; Ishisaki, A. Transforming growth factor-β1 induces invasion ability of HSC-4 human oral squamous cell carcinoma cells through the Slug/Wnt-5b/MMP-10 signalling axis. J. Biochem. 2016, 159, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.M.; Wang, L.; Shen, H.; Jiang, C.F.; Ge, X.; Li, D.M.; Wen, Y.Y.; Sun, H.R.; Pan, M.H.; Li, W.; et al. Downregulation of miR-218 contributes to epithelial-mesenchymal transition and tumor metastasis in lung cancer by targeting Slug/Zeb2 signaling. Oncogene 2017, 36, 2577–2588. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, G.; Schwarz, R.E.; Higgins, L.; McEnroe, G.; Chakravarty, S.; Dugar, S.; Reiss, M. Targeting endogenous transforming growth factor β receptor signaling in SMAD4-deficient human pancreatic carcinoma cells inhibits their invasive phenotype1. Cancer Res. 2004, 64, 5200–5211. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Ammanamanchi, S.; Brattain, M.; Cao, L.; Thangasamy, A.; Wang, J.; Freeman, J.W. Smad4-dependent TGF-β signaling suppresses RON receptor tyrosine kinase-dependent motility and invasion of pancreatic cancer cells. J. Biol. Chem. 2008, 283, 11293–11301. [Google Scholar] [CrossRef] [PubMed]
- Su, H.T.; Weng, C.C.; Hsiao, P.J.; Chen, L.H.; Kuo, T.L.; Chen, Y.W.; Kuo, K.K.; Cheng, K.H. Stem cell marker nestin is critical for TGF-β 1-mediated tumor progression in pancreatic cancer. Mol. Cancer Res. 2013, 11, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Cao, J.X.; Pei, Y.J.; Zhang, J.Y.; Wang, Q.Y. Smad4 inhibits cell migration via suppression of JNK activity in human pancreatic carcinoma PANC-1 cells. Oncol. Lett. 2016, 11, 3465–3470. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Hsiao, P.J.; Weng, C.C.; Kuo, K.K.; Kuo, T.L.; Wu, D.C.; Hung, W.C.; Cheng, K.H. SMAD4 loss triggers the phenotypic changes of pancreatic ductal adenocarcinoma cells. BMC Cancer 2014, 14, 181. [Google Scholar] [CrossRef] [PubMed]
- Marechal, R.; Mackey, J.R.; Lai, R.; Demetter, P.; Peeters, M.; Polus, M.; Cass, C.E.; Salmon, I.; Deviere, J.; Van Laethem, J.L. Deoxycitidine kinase is associated with prolonged survival after adjuvant gemcitabine for resected pancreatic adenocarcinoma. Cancer 2010, 116, 5200–5206. [Google Scholar] [CrossRef] [PubMed]
- Pires, B.R.; Mencalha, A.L.; Ferreira, G.M.; de Souza, W.F.; Morgado-Diaz, J.A.; Maia, A.M.; Correa, S.; Abdelhay, E.S. NF-κB is involved in the regulation of EMT genes in breast cancer cells. PLoS ONE 2017, 12, e0169622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lapidus, R.G.; Liu, P.; Choi, E.Y.; Adediran, S.; Hussain, A.; Wang, X.; Liu, X.; Dan, H.C. Targeting IκB kinase β/NF-κB signaling in human prostate cancer by a novel IκB kinase β inhibitor CmpdA. Mol. Cancer Ther. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tozzi, F.; Chen, J.; Fan, F.; Xia, L.; Wang, J.; Gao, G.; Zhang, A.; Xia, X.; Brasher, H.; et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res. 2012, 72, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Shen, Z.; Huang, S.; Zhao, L.; Chen, S.; Mak, T.W.; Wang, X. The ER UDPase ENTPD5 promotes protein N-glycosylation, the Warburg effect, and proliferation in the PTEN pathway. Cell 2010, 143, 711–724. [Google Scholar] [CrossRef] [PubMed]
- Ko, T.; An, H.J.; Ji, Y.G.; Kim, O.J.; Lee, D.H. P2Y receptors regulate proliferation of human pancreatic duct epithelial cells. Pancreas 2012, 41, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Giannuzzo, A.; Pedersen, S.F.; Novak, I. The P2X7 receptor regulates cell survival, migration and invasion of pancreatic ductal adenocarcinoma cells. Mol. Cancer 2015, 14, 203. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.T.; Kunapuli, S.P. Central role of the P2Y12 receptor in platelet activation. J. Clin. Investig. 2004, 113, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Hollopeter, G.; Jantzen, H.M.; Vincent, D.; Li, G.; England, L.; Ramakrishnan, V.; Yang, R.B.; Nurden, P.; Nurden, A.; Julius, D.; et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature 2001, 409, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.L.; Luo, L.; Gustafson, E.; Lachowicz, J.; Smith, M.; Qiao, X.; Liu, Y.H.; Chen, G.; Pramanik, B.; Laz, T.M.; et al. ADP is the cognate ligand for the orphan G protein-coupled receptor SP1999. J. Biol. Chem. 2001, 276, 8608–8615. [Google Scholar] [CrossRef] [PubMed]
- Satonaka, H.; Nagata, D.; Takahashi, M.; Kiyosue, A.; Myojo, M.; Fujita, D.; Ishimitsu, T.; Nagano, T.; Nagai, R.; Hirata, Y. Involvement of P2Y12 receptor in vascular smooth muscle inflammatory changes via MCP-1 upregulation and monocyte adhesion. Am. J. Physiol. 2015, 308, H853–H861. [Google Scholar]
- Rauch, B.H.; Rosenkranz, A.C.; Ermler, S.; Bohm, A.; Driessen, J.; Fischer, J.W.; Sugidachi, A.; Jakubowski, J.A.; Schror, K. Regulation of functionally active P2Y12 ADP receptors by thrombin in human smooth muscle cells and the presence of P2Y12 in carotid artery lesions. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Garcia, L.; Nadal, B.; Gomis, R.; Petit, P.; Gross, R.; Lajoix, A.D. Human pancreatic islets express the purinergic P2Y11 and P2Y12 receptors. Horm. Metab. Res. 2008, 40, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.; Summers, C.; Ewart, L.; Nylander, S.; Sidaway, J.E.; van Giezen, J.J. Characterization of the adenosine pharmacology of ticagrelor reveals therapeutically relevant inhibition of equilibrative nucleoside transporter 1. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.; Schulz, R.; Nylander, S. Adenosine-mediated effects of ticagrelor: Evidence and potential clinical relevance. J. Am. Coll. Cardiol. 2014, 63, 2503–2509. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla-Varela, M.; Luddy, K.; Noyes, D.; Khalil, F.K.; Neuger, A.M.; Soliman, H.; Antonia, S.J. Antagonism of adenosine A2A receptor expressed by lung adenocarcinoma tumor cells and cancer associated fibroblasts inhibits their growth. Cancer Biol. Ther. 2013, 14, 860–868. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Pharmacology Review(s). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/022433Orig1s000PharmR.pdf (accessed on 23 October 2017).
- Torres, A.; Vargas, Y.; Uribe, D.; Jaramillo, C.; Gleisner, A.; Salazar-Onfray, F.; Lopez, M.N.; Melo, R.; Oyarzun, C.; San Martin, R.; et al. Adenosine A3 receptor elicits chemoresistance mediated by multiple resistance-associated protein-1 in human glioblastoma stem-like cells. Oncotarget 2016, 7, 67373–67386. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Cruickshanks, N.; Conley, A.; Durrant, D.E.; Das, A.; Fisher, P.B.; Kukreja, R.C.; Grant, S.; Poklepovic, A.; et al. Phosphodiesterase 5 inhibitors enhance chemotherapy killing in gastrointestinal/genitourinary cancer cells. Mol. Pharmacol. 2014, 85, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Pellegatti, P.; Raffaghello, L.; Bianchi, G.; Piccardi, F.; Pistoia, V.; Di Virgilio, F. Increased level of extracellular ATP at tumor sites: In vivo imaging with plasma membrane luciferase. PLoS ONE 2008, 3, e2599. [Google Scholar] [CrossRef] [PubMed]
- Yegutkin, G.G.; Marttila-Ichihara, F.; Karikoski, M.; Niemelä, J.; Laurila, J.P.; Elima, K.; Jalkanen, S.; Salmi, M. Altered purinergic signaling in CD73-deficient mice inhibits tumor progression. Eur. J. Immunol. 2011, 41, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F. Purines, purinergic receptors, and cancer. Cancer Res. 2012, 72, 5441–5447. [Google Scholar] [CrossRef] [PubMed]
- Radomski, M.; Moncada, S. An improved method for washing of human platelets with prostacyclin. Thromb. Res. 1983, 30, 383–389. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Gogal, R.M., Jr.; Walsh, J.E. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: An alternative to [3H] thymidine incorporation assay. J. Immunol. Methods 1994, 170, 211–224. [Google Scholar] [CrossRef]
- Wen, F.; Shen, A.; Choi, A.; Gerner, E.W.; Shi, J. Extracellular DNA in pancreatic cancer promotes cell invasion and metastasis. Cancer Res. 2013, 73, 4256–4266. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elaskalani, O.; Falasca, M.; Moran, N.; Berndt, M.C.; Metharom, P. The Role of Platelet-Derived ADP and ATP in Promoting Pancreatic Cancer Cell Survival and Gemcitabine Resistance. Cancers 2017, 9, 142. https://doi.org/10.3390/cancers9100142
Elaskalani O, Falasca M, Moran N, Berndt MC, Metharom P. The Role of Platelet-Derived ADP and ATP in Promoting Pancreatic Cancer Cell Survival and Gemcitabine Resistance. Cancers. 2017; 9(10):142. https://doi.org/10.3390/cancers9100142
Chicago/Turabian StyleElaskalani, Omar, Marco Falasca, Niamh Moran, Michael C. Berndt, and Pat Metharom. 2017. "The Role of Platelet-Derived ADP and ATP in Promoting Pancreatic Cancer Cell Survival and Gemcitabine Resistance" Cancers 9, no. 10: 142. https://doi.org/10.3390/cancers9100142
APA StyleElaskalani, O., Falasca, M., Moran, N., Berndt, M. C., & Metharom, P. (2017). The Role of Platelet-Derived ADP and ATP in Promoting Pancreatic Cancer Cell Survival and Gemcitabine Resistance. Cancers, 9(10), 142. https://doi.org/10.3390/cancers9100142