
Regulation of mTOR, Metabolic Fitness, and Effector Functions by Cytokines in Natural Killer Cells

 ,
,
Abstract
1. Introduction
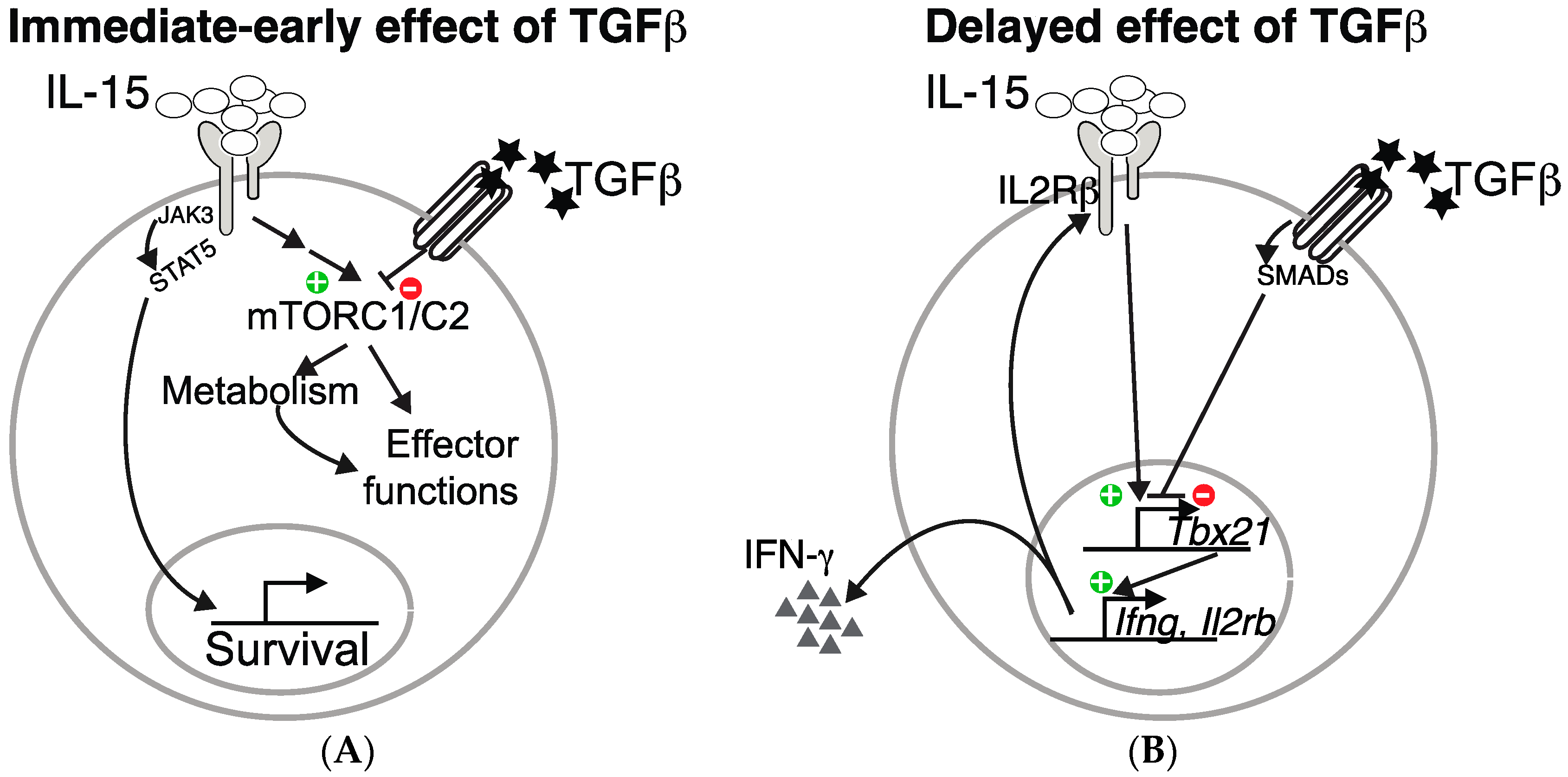
2. IL-15 Activates mTOR in NK Cells and Boosts Cellular Metabolism
3. TGF-β Represses the mTOR Pathway in NK Cells
4. Strategies to Boost NK Cell Metabolism
5. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Vivier, E.; Tomasello, E.; Baratin, M.; Walzer, T.; Ugolini, S. Functions of natural killer cells. Nat. Immunol. 2008, 9, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, C.M.; Finlay, D.K. What Fuels Natural Killers? Metabolism and NK Cell Responses. Front. Immunol. 2017, 8, 367. [Google Scholar] [CrossRef] [PubMed]
- Stojanovic, A.; Correia, M.P.; Cerwenka, A. Shaping of NK Cell Responses by the Tumor Microenvironment. Cancer Microenviron. 2013, 6, 135. [Google Scholar] [CrossRef] [PubMed]
- Ardolino, M.; Azimi, C.S.; Iannello, A.; Trevino, T.N.; Horan, L.; Zhang, L.; Deng, W.; Ring, A.M.; Fischer, S.; Garcia, K.C.; et al. Cytokine therapy reverses NK cell anergy in MHC-deficient tumors. J. Clin. Investig. 2014, 124, 4781–4794. [Google Scholar] [CrossRef] [PubMed]
- Mamessier, E.; Sylvain, A.; Thibult, M.-L.; Houvenaeghel, G.; Jacquemier, J.; Castellano, R.; Gonçalves, A.; André, P.; Romagné, F.; Thibault, G.; et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Invest. 2011, 121, 3609–3622. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.K.; et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Schurich, A.; Pallett, L.J.; Jajbhay, D.; Wijngaarden, J.; Otano, I.; Gill, U.S.; Hansi, N.; Kennedy, P.T.; Nastouli, E.; Gilson, R.; et al. Distinct Metabolic Requirements of Exhausted and Functional Virus-Specific CD8 T Cells in the Same Host. Cell Rep. 2016, 16, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8+ T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Wang, C.; Cong, L.; Marjanovic, N.D.; Kowalczyk, M.S.; Zhang, H.; Nyman, J.; Sakuishi, K.; Kurtulus, S.; Gennert, D.; et al. A Distinct Gene Module for Dysfunction Uncoupled from Activation in Tumor-Infiltrating T Cells. Cell 2016, 166, 1500–1511.e9. [Google Scholar] [CrossRef] [PubMed]
- Marçais, A.; Cherfils-Vicini, J.; Viant, C.; Degouve, S.; Viel, S.; Fenis, A.; Rabilloud, J.; Mayol, K.; Tavares, A.; Bienvenu, J.; et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 2014, 15, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Keppel, M.P.; Saucier, N.; Mah, A.Y.; Vogel, T.P.; Cooper, M.A. Activation-specific metabolic requirements for NK cell IFN-γ production. J. Immunol. 2015, 194, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.P.; Loftus, R.M.; Keating, S.E.; Liou, K.T.; Biron, C.A.; Gardiner, C.M.; Finlay, D.K. mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. 2014, 193, 4477–4484. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.E.; Zaiatz-Bittencourt, V.; Loftus, R.M.; Keane, C.; Brennan, K.; Finlay, D.K.; Gardiner, C.M. Metabolic reprogramming supports IFN-γ production by CD56bright NK cells. J. Immunol. 2016, 196, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Viel, S.; Marçais, A.; Guimaraes, F.S.-F.; Loftus, R.; Rabilloud, J.; Grau, M.; Degouve, S.; Djebali, S.; Sanlaville, A.; Charrier, E.; et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal. 2016, 9, ra19. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.; Schachterle, W.; Oberle, K.; Aichele, P.; Diefenbach, A. Dendritic Cells Prime Natural Killer Cells by trans-Presenting Interleukin 15. Immunity 2007, 26, 503–517. [Google Scholar] [CrossRef] [PubMed]
- Dubois, S.; Conlon, K.C.; Müller, J.R.; Hsu-Albert, J.; Beltran, N.; Bryant, B.R.; Waldmann, T.A. IL15 infusion of cancer patients expands the subpopulation of cytotoxic CD56bright NK cells and increases NK cell cytokine release capabilities. Cancer Immunol. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; van Hoef, V.; Zhang, X.; Wennerberg, E.; Lorent, J.; Witt, K.; Masvidal, L.; Liang, S.; Murray, S.; Larsson, O.; et al. IL-15 activates mTOR and primes stress-activated gene expression leading to prolonged antitumor capacity of NK cells. Blood 2016, 128, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.-C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.-C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.-R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria Are Required for Antigen-Specific T Cell Activation through Reactive Oxygen Species Signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Travis, M.A.; Sheppard, D. TGF-β Activation and Function in Immunity. Annu. Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.-K.L.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Ménard, C.; Terme, M.; Flament, C.; Taieb, J.; Chaput, N.; Puig, P.E.; Novault, S.; Escudier, B.; Vivier, E.; et al. CD4+CD25+ Regulatory T Cells Inhibit Natural Killer Cell Functions in a Transforming Growth Factor–β–dependent Manner. J. Exp. Med. 2005, 202, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Teng, M.W.L.; Swann, J.; Kyparissoudis, K.; Godfrey, D.I.; Hayakawa, Y. CD4+CD25+ T Regulatory Cells Suppress NK Cell-Mediated Immunotherapy of Cancer. J. Immunol. 2006, 176, 1582–1587. [Google Scholar] [CrossRef] [PubMed]
- Ortaldo, J.R.; Mason, A.T.; O’Shea, J.J.; Smyth, M.J.; Falk, L.A.; Kennedy, I.C.; Longo, D.L.; Ruscetti, F.W. Mechanistic studies of transforming growth factor-beta inhibition of IL-2-dependent activation of CD3- large granular lymphocyte functions. Regulation of IL-2R beta (p75) signal transduction. J. Immunol. 1991, 146, 3791–3798. [Google Scholar] [PubMed]
- Malygin, A.M.; Meri, S.; Timonen, T. Regulation of natural killer cell activity by transforming growth factor-beta and prostaglandin E2. Scand. J. Immunol. 1993, 37, 71–76. [Google Scholar]
- Laouar, Y.; Sutterwala, F.S.; Gorelik, L.; Flavell, R.A. Transforming growth factor-β controls T helper type 1 cell development through regulation of natural killer cell interferon-γ. Nat. Immunol. 2005, 6, 600–607. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.A.; McNutt, N.S.; Prieto, V.G.; Albino, A.P. Expression of transforming growth factor-beta 2 in malignant melanoma correlates with the depth of tumor invasion. Implications for tumor progression. Am. J. Pathol. 1994, 145, 97–104. [Google Scholar] [PubMed]
- Urashima, M.; Ogata, A.; Chauhan, D.; Hatziyanni, M.; Vidriales, M.B.; Dedera, D.A.; Schlossman, R.L.; Anderson, K.C. Transforming growth factor-beta1: Differential effects on multiple myeloma versus normal B cells. Blood 1996, 87, 1928–1938. [Google Scholar] [PubMed]
- Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor β1 inhibits expression of NKp30 and NKG2D receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-C.; Lee, K.-M.; Kim, D.-W.; Heo, D.S. Elevated TGF- 1 Secretion and Down-Modulation of NKG2D Underlies Impaired NK Cytotoxicity in Cancer Patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.L.; Soudja, S.M.; Deceneux, C.; Lauvau, G.; Marie, J.C. NK1.1+ CD8+ T cells escape TGF-β control and contribute to early microbial pathogen response. Nat. Commun. 2014, 5, 5150. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.S.; Ulland, T.K.; Cervantes-Barragan, L.; Bando, J.K.; Robinette, M.L.; Wang, Q.; White, A.J.; Gilfillan, S.; Cella, M.; Colonna, M. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-β signaling. Nat. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wei, M.; Becknell, B.; Trotta, R.; Liu, S.; Boyd, Z.; Jaung, M.S.; Blaser, B.W.; Sun, J.; Benson, D.M.; et al. Pro- and Antiinflammatory Cytokine Signaling: Reciprocal Antagonism Regulates Interferon-gamma Production by Human Natural Killer Cells. Immunity 2006, 24, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Trotta, R.; Col, J.D.; Yu, J.; Ciarlariello, D.; Thomas, B.; Zhang, X.; Allard, J.; Wei, M.; Mao, H.; Byrd, J.C.; et al. TGF-β utilizes SMAD3 to inhibit CD16-mediated IFN- production and antibody-dependent cellular cytotoxicity in human NK Cells. J. Immunol. 2008, 181, 3784–3792. [Google Scholar] [CrossRef] [PubMed]
- Sauer, S.; Bruno, L.; Hertweck, A.; Finlay, D.; Leleu, M.; Spivakov, M.; Knight, Z.A.; Cobb, B.S.; Cantrell, D.; O’Connor, E.; et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl. Acad. Sci. USA 2008, 105, 7797–7802. [Google Scholar] [CrossRef] [PubMed]
- Delisle, J.-S.; Giroux, M.; Boucher, G.; Landry, J.-R.; Hardy, M.-P.; Lemieux, S.; Jones, R.G.; Wilhelm, B.T.; Perreault, C. The TGF-β-Smad3 pathway inhibits CD28-dependent cell growth and proliferation of CD4 T cells. Genes Immun. 2013, 14, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Chi, H. mTOR and lymphocyte metabolism. Curr. Opin. Immunol. 2013, 25, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Hukelmann, J.L.; Anderson, K.E.; Sinclair, L.V.; Grzes, K.M.; Murillo, A.B.; Hawkins, P.T.; Stephens, L.R.; Lamond, A.I.; Cantrell, D.A. The cytotoxic T cell proteome and its shaping by the kinase mTOR. Nat. Immunol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, S. Sirolimus: Its discovery, biological properties, and mechanism of action. Transplant. Proc. 2003, 35, S7–S14. [Google Scholar] [CrossRef]
- Wang, T.; Donahoe, P.K. The immunophilin FKBP12: A molecular guardian of the TGF-family type I receptors. Front. Biosci. 2004, 9, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Chiasson, V.L.; Talreja, D.; Young, K.J.; Chatterjee, P.; Banes-Berceli, A.K.; Mitchell, B.M. FK506 binding protein 12 deficiency in endothelial and hematopoietic cells decreases regulatory T cells and causes hypertension. Hypertension 2011, 57, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Segundo, D.S.; Ruiz, J.C.; Izquierdo, M.; Fernández-Fresnedo, G.; Gómez-Alamillo, C.; Merino, R.; Benito, M.J.; Cacho, E.; Rodrigo, E.; Palomar, R.; et al. Calcineurin inhibitors, but not rapamycin, reduce percentages of CD4+CD25+FOXP3+ regulatory T cells in renal transplant recipients. Transplantation 2006, 82, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Serine/threonine phosphatases: Mechanism through structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [PubMed]
- Apostolidis, S.A.; Rodríguez-Rodríguez, N.; Suárez-Fueyo, A.; Dioufa, N.; Ozcan, E.; Crispín, J.C.; Tsokos, M.G.; Tsokos, G.C. Phosphatase PP2A is requisite for the function of regulatory T cells. Nat. Immunol. 2016, 17, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Guillerey, C.; Huntington, N.D.; Smyth, M.J. Targeting natural killer cells in cancer immunotherapy. Nat. Immunol. 2016, 17, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Sola, C.; André, P.; Lemmers, C.; Fuseri, N.; Bonnafous, C.; Bléry, M.; Wagtmann, N.R.; Romagné, F.; Vivier, E.; Ugolini, S. Genetic and antibody-mediated reprogramming of natural killer cell missing-self recognition in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 12879–12884. [Google Scholar] [CrossRef] [PubMed]
- Kohrt, H.E.; Thielens, A.; Marabelle, A.; Sagiv-Barfi, I.; Sola, C.; Chanuc, F.; Fuseri, N.; Bonnafous, C.; Czerwinski, D.; Rajapaksa, A.; et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood 2014, 123, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, M.K.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 2013, 19, 3844–3855. [Google Scholar] [CrossRef] [PubMed]
- Romanski, A.; Uherek, C.; Bug, G.; Seifried, E.; Klingemann, H.; Wels, W.S.; Ottmann, O.G.; Tonn, T. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J. Cell. Mol. Med. 2016, 20, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S.; et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Rocca, Y.S.; Roberti, M.P.; Juliá, E.P.; Pampena, M.B.; Bruno, L.; Rivero, S.; Huertas, E.; Sánchez Loria, F.; Pairola, A.; Caignard, A.; et al. Phenotypic and functional dysregulated blood NK cells in colorectal cancer patients can be activated by cetuximab plus IL-2 or IL-15. Front. Immunol. 2016, 7, 413. [Google Scholar] [CrossRef] [PubMed]
- Conlon, K.C.; Lugli, E.; Welles, H.C.; Rosenberg, S.A.; Fojo, A.T.; Morris, J.C.; Fleisher, T.A.; Dubois, S.P.; Perera, L.P.; Stewart, D.M.; et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J. Clin. Oncol. 2015, 33, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Kong, L.; Han, K.; Hong, H.; Marcus, W.D.; Chen, X.; Jeng, E.K.; Alter, S.; Zhu, X.; Rubinstein, M.P.; et al. A Novel Fusion of ALT-803 (Interleukin (IL)-15 Superagonist) with an Antibody Demonstrates Antigen-specific Antitumor Responses. J. Biol. Chem. 2016, 291, 23869–23881. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Jeon, I.; Kim, B.-S.; Park, M.; Bae, E.-A.; Song, B.; Koh, C.-H.; Shin, K.S.; Kim, I.K.; Choi, K.; et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat. Commun. 2017, 8, 15776. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Miller, M.; Stojanovic, A.; Garbi, N.; Cerwenka, A. Sustained effector function of IL-12/15/18–preactivated NK cells against established tumors. J. Exp. Med. 2012, 209, 2351–2365. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Delconte, R.B.; Kolesnik, T.B.; Dagley, L.F.; Rautela, J.; Shi, W.; Putz, E.M.; Stannard, K.; Zhang, J.G.; The, C.; Firth, M.; et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 2016, 17, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Morris, J.C. Transforming growth factor-β: A therapeutic target for cancer. Hum. Vaccines Immunother. 2017, 13, 1741–1750. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wrzesinski, S.H.; Stern, E.; Look, M.; Criscione, J.; Ragheb, R.; Jay, S.M.; Demento, S.L.; Agawu, A.; Licona Limon, P.; et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater. 2012, 11, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; Deng, J.; Chinnadurai, R.; Yuan, S.; Pennati, A.; Galipeau, J. Stimulation of Natural Killer Cell–Mediated Tumor Immunity by an IL15/TGFβ–Neutralizing Fusion Protein. Cancer Res. 2016, 76, 5683–5695. [Google Scholar] [CrossRef] [PubMed]
- Siska, P.J.; Rathmell, J.C. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015, 36, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Carlsten, M.; Korde, N.; Kotecha, R.; Reger, R.; Bor, S.; Kazandjian, D.; Landgren, O.; Childs, R.W. Checkpoint Inhibition of KIR2D with the Monoclonal Antibody IPH2101 Induces Contraction and Hyporesponsiveness of NK Cells in Patients with Myeloma. Clin. Cancer Res. 2016, 22, 5211–5222. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.D.; Delgoffe, G.M. The mammalian Target of Rapamycin (mTOR) provides a critical link between T cell differentiation, function and metabolism. Immunity 2010, 33, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Marçais, A.; Marotel, M.; Degouve, S.; Koenig, A.; Fauteux-Daniel, S.; Drouillard, A.; Schlums, H.; Viel, S.; Besson, L.; Allatif, O.; et al. High mTOR activity is a hallmark of reactive natural killer cells and amplifies early signaling through activating receptors. eLife 2017, e26423. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, T.E.; Johnson, L.R.; Kang, H.H.; Sun, J.C. BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity 2015, 43, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000Res. 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Lagrue, K.; Carisey, A.; Morgan, D.J.; Chopra, R.; Davis, D.M. Lenalidomide augments actin remodeling and lowers NK-cell activation thresholds. Blood 2015, 126, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M.; Bakan, C.E.; Zhang, S.; Collins, S.M.; Liang, J.; Srivastava, S.; Hofmeister, C.C.; Efebera, Y.; Andre, P.; Romagne, F.; et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood 2011, 118, 6387–6391. [Google Scholar] [CrossRef] [PubMed]
- Borg, C.; Terme, M.; Taïeb, J.; Ménard, C.; Flament, C.; Robert, C.; Maruyama, K.; Wakasugi, H.; Angevin, E.; Thielemans, K.; et al. Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J. Clin. Invest. 2004, 114, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Tian, Z. NK Cell Exhaustion. Front. Immunol. 2017, 8, 760. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Species | NK Cell Metabolic Activity | Reference | |
|---|---|---|---|
| Increased by | Decreased by | ||
| Murine NK cells | IL-2, IL-2/12, poly(I:C) | Rapamycin | [14] |
| IL-15, poly(I:C) | [12] | ||
| IL-15, IL-15+αTGF-β | Rapamycin, TGF-β | [16] | |
| IL-15 | [13] | ||
| Human NK Cells | IL-2, IL-12/15 | Rapamycin | [15] |
| IL-2, IL-15 | Torin | [17] | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viel, S.; Besson, L.; Marotel, M.; Walzer, T.; Marçais, A. Regulation of mTOR, Metabolic Fitness, and Effector Functions by Cytokines in Natural Killer Cells. Cancers 2017, 9, 132. https://doi.org/10.3390/cancers9100132
Viel S, Besson L, Marotel M, Walzer T, Marçais A. Regulation of mTOR, Metabolic Fitness, and Effector Functions by Cytokines in Natural Killer Cells. Cancers. 2017; 9(10):132. https://doi.org/10.3390/cancers9100132
Chicago/Turabian StyleViel, Sébastien, Laurie Besson, Marie Marotel, Thierry Walzer, and Antoine Marçais. 2017. "Regulation of mTOR, Metabolic Fitness, and Effector Functions by Cytokines in Natural Killer Cells" Cancers 9, no. 10: 132. https://doi.org/10.3390/cancers9100132
APA StyleViel, S., Besson, L., Marotel, M., Walzer, T., & Marçais, A. (2017). Regulation of mTOR, Metabolic Fitness, and Effector Functions by Cytokines in Natural Killer Cells. Cancers, 9(10), 132. https://doi.org/10.3390/cancers9100132