
Investigating the Interaction of Cyclic RGD Peptidomimetics with αVβ6 Integrin by Biochemical and Molecular Docking Studies

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
2.1. Integrin Receptor Competitive Binding Assays
2.2. Docking Model of αVβ6 Integrin and X-ray Structure Analysis
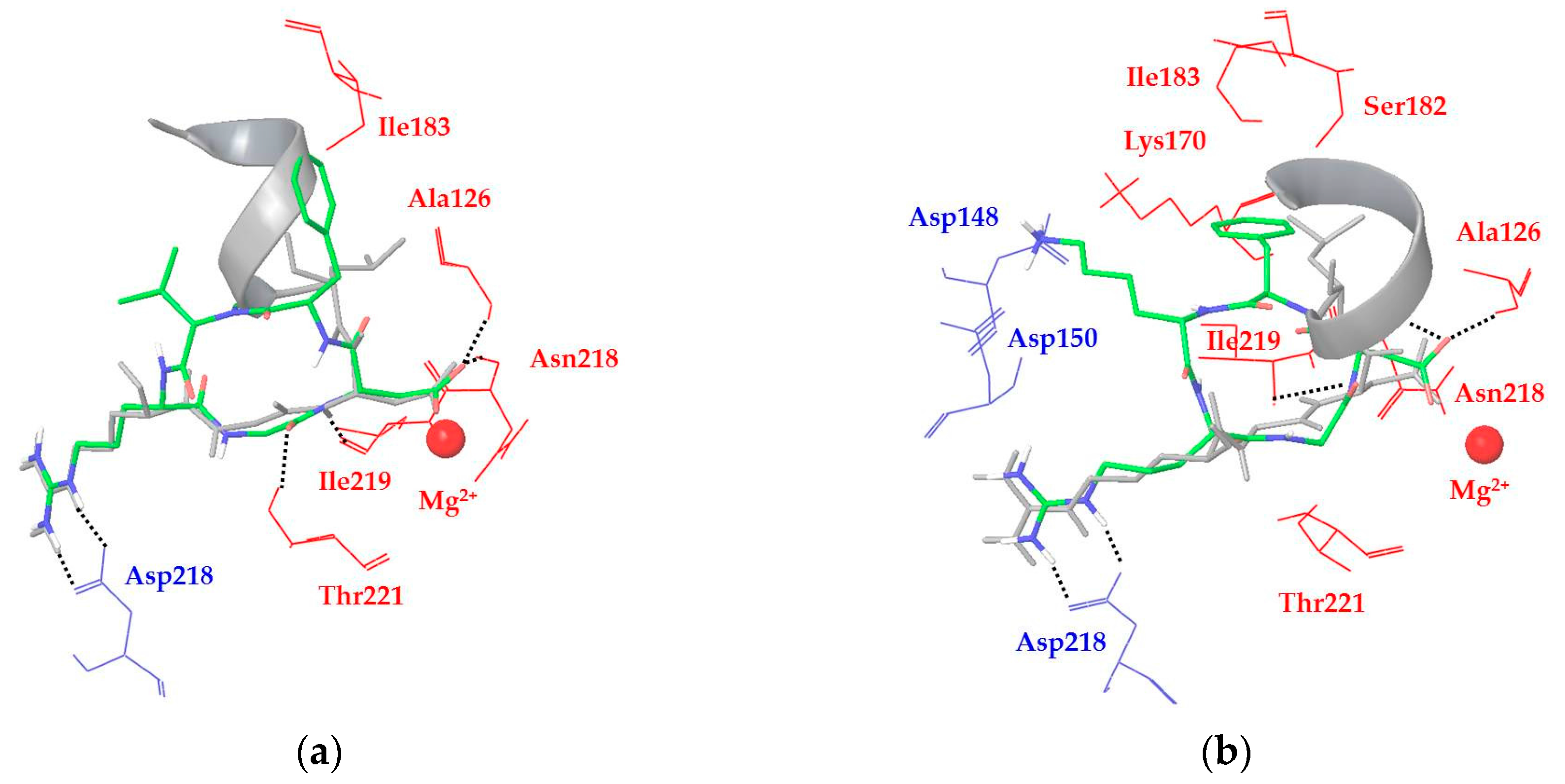
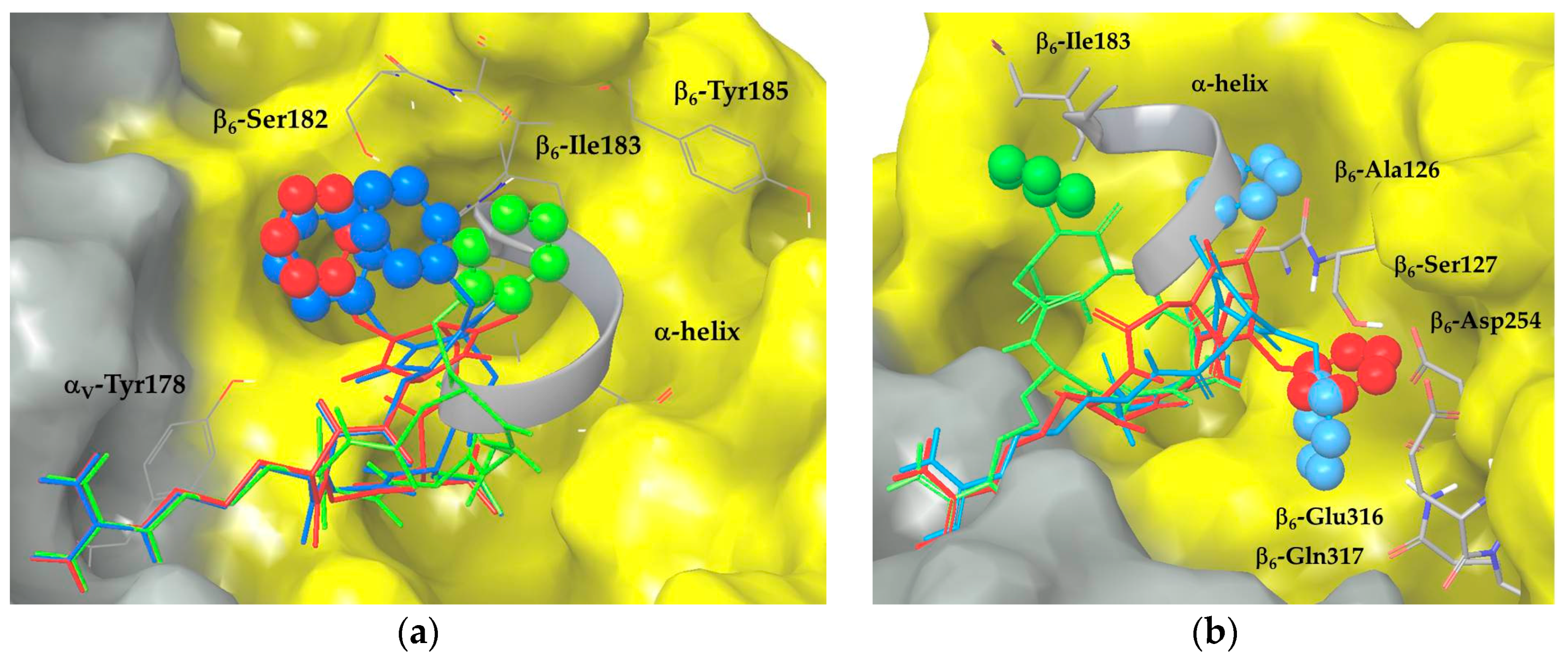
2.3. Docking of Cyclic DKP-RGD Peptidomimetics into αVβ6 Integrin
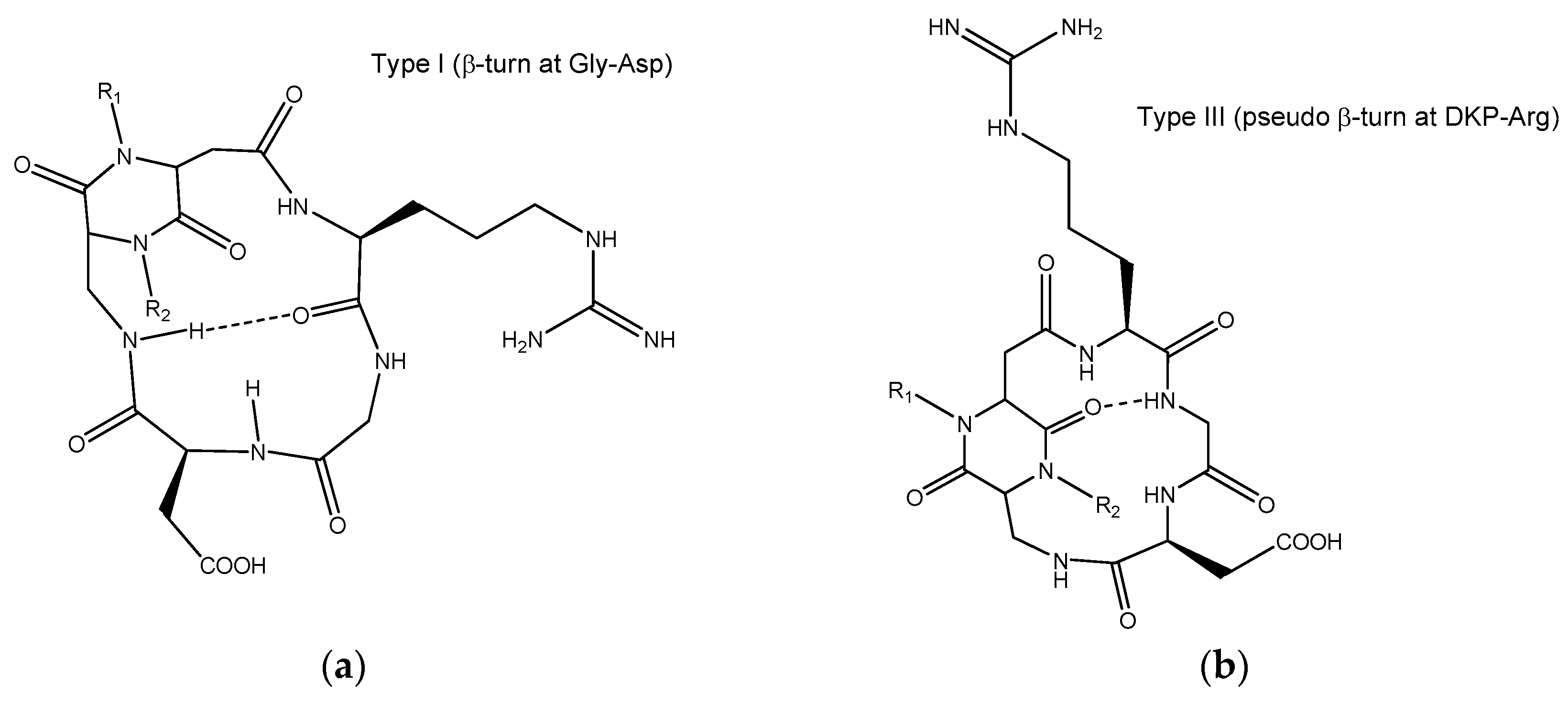
3. Discussion
4. Materials and Methods
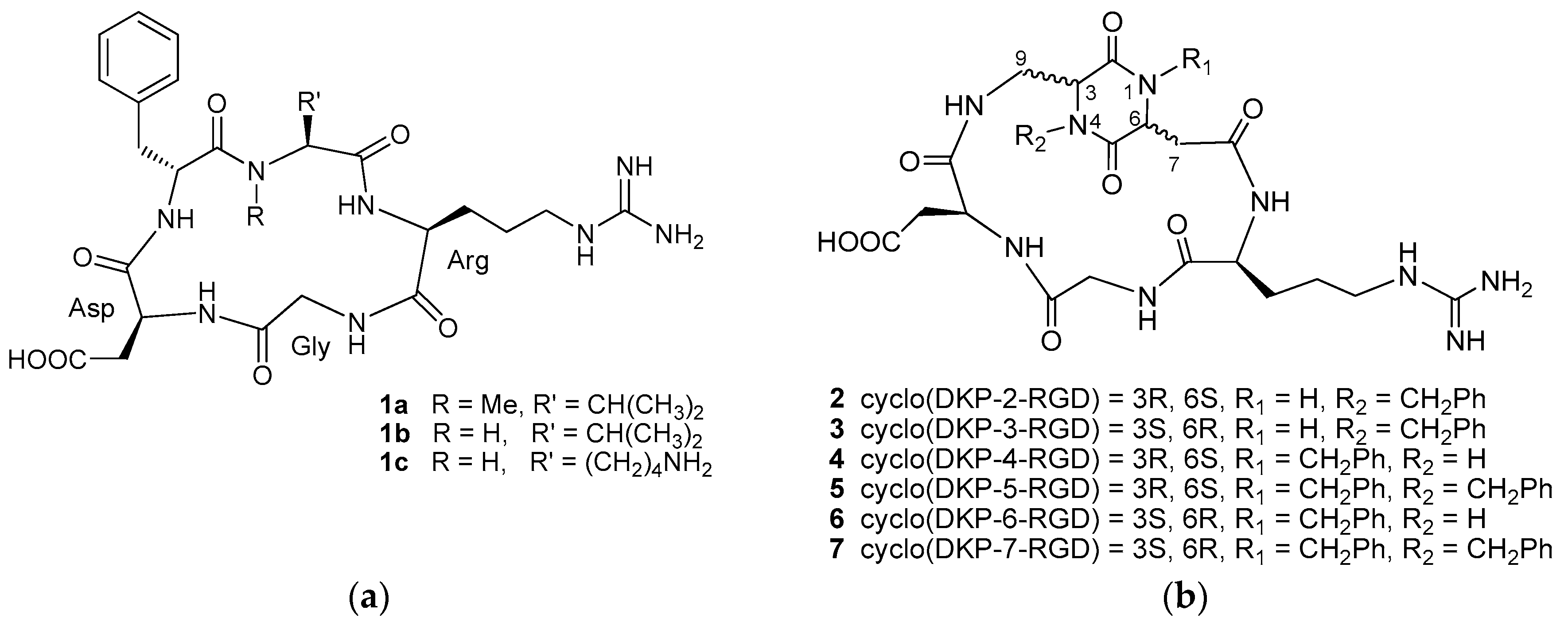
4.1. Integrin Ligands
4.2. Solid-Phase Receptor Binding Assay
4.3. Computational Studies
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

References and Notes
- Takada, Y.; Ye, X.; Simon, S. The integrins. Genome Biol. 2007, 8, 215.1–215.9. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Shimaoka, M.; Springer, T.A. Therapeutic antagonists and conformational regulation of integrin function. Nat. Rev. Drug Discov. 2003, 2, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Rivera-Nieves, J.; Sandborn, W.J.; Shattil, S. Integrin-based therapeutics: Biological basis, clinical use and new drugs. Nat. Rev. Drug Discov. 2016, 15, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.L.; Picard, M. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Brennan, M.; Moran, N. Integrins as therapeutic targets: Lessons and opportunities. Nat. Rev. Drug Discov. 2010, 9, 804–820. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Sheldrake, H.M.; Patterson, L.H. Strategies to inhibit tumor associated integrin receptors: Rationale for dual and multi-antagonists. J. Med. Chem. 2014, 57, 6301–6315. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Fraioli, R.; Rechenmacher, F.; Neubauer, S.; Kapp, T.G.; Kessler, H. αVβ3- or α5β1-integrin-selective peptidomimetics for surface coating. Angew. Chem. Int. Ed. Engl. 2016, 55, 7048–7068. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, M.; Russo, M.A.; Serra, M.; Colombo, L.; Schinelli, S. Small molecule integrin antagonists in cancer therapy. Mini-Rev. Med. Chem. 2009, 9, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Auzzas, L.; Zanardi, F.; Battistini, L.; Burreddu, P.; Carta, P.; Rassu, G.; Curti, C.; Casiraghi, G. Targeting αVβ3 integrin: Design and applications of mono- and multifunctional RGD-based peptides and semipeptides. Curr. Med. Chem. 2010, 17, 1255–1299. [Google Scholar] [CrossRef] [PubMed]
- Marelli, U.K.; Rechenmacher, F.; Sobahi, T.R.A.; Mas-Moruno, C.; Kessler, H. Tumor targeting via integrin ligands. Front. Oncol. 2013, 3, 222. [Google Scholar] [CrossRef] [PubMed]
- Arosio, D.; Casagrande, C. Advancement in integrin facilitated drug delivery. Adv. Drug Deliv. Rev. 2016, 97, 111–143. [Google Scholar] [CrossRef] [PubMed]
- Arosio, D.; Manzoni, L.; Corno, C.; Perego, P. Integrin-targeted Peptide- and peptidomimetic-drug conjugates for the treatment of tumors. Recent Pat. Anticancer Drug Discov. 2017, 12, 148–168. [Google Scholar] [CrossRef] [PubMed]
- Dal Corso, A.; Pignataro, L.; Belvisi, L.; Gennari, C. αVβ3 Integrin-targeted peptide/peptidomimetic-drug conjugates: In-depth analysis of the linker technology. Curr. Top. Med. Chem. 2016, 16, 314–329. [Google Scholar] [CrossRef] [PubMed]
- Dechantsreiter, M.A.; Planker, E.; Mathä, B.; Lohof, E.; Hölzemann, G.; Jonczyk, A.; Goodman, S.L.; Kessler, H. N-methylated cyclic RGD peptides as highly active and selective αVβ3 integrin antagonists. J. Med. Chem. 1999, 42, 3033–3040. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate. Design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicenter, randomized, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; Barlesi, F.; Waller, C.F.; Bennouna, J.; Gridelli, C.; Goekkurt, E.; Verhoeven, D.; Szczesna, A.; Feurer, M.; Milanowski, J.; et al. Cilengitide combined with cetuximab and platinum-based chemotherapy as first-line treatment in advanced non-small-cell lung cancer (NSCLC) patients: Results of an open-label, randomized, controlled phase II study (CERTO). Ann. Oncol. 2015, 26, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Kapp, T.G.; Rechenmacher, F.; Neubauer, S.; Maltsev, O.V.; Cavalcanti-Adam, E.A.; Zarka, R.; Reuning, U.; Notni, J.; Wester, H.-J.; Mas-Moruno, C.; et al. A comprehensive evaluation of the activity and selectivity profile of ligands for RGD-binding integrins. Sci. Rep. 2017, 7, 39805. [Google Scholar] [CrossRef] [PubMed]
- Ressurreição, A.S.M.; Bordessa, A.; Civera, M.; Belvisi, L.; Gennari, C.; Piarulli, U. Synthesis and conformational studies of peptidomimetics containing a new bifunctional diketopiperazine scaffold acting as a β-hairpin inducer. J. Org. Chem. 2008, 73, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Ressurreição, A.S.M.; Vidu, A.; Civera, M.; Belvisi, L.; Potenza, D.; Manzoni, L.; Ongeri, S.; Gennari, C.; Piarulli, U. Cyclic RGD-peptidomimetics containing bifunctional diketopiperazine scaffolds as new potent integrin ligands. Chem. Eur. J. 2009, 15, 12184–12188. [Google Scholar] [CrossRef] [PubMed]
- Marchini, M.; Mingozzi, M.; Colombo, R.; Guzzetti, I.; Belvisi, L.; Vasile, F.; Potenza, D.; Piarulli, U.; Arosio, D.; Gennari, C. Cyclic RGD peptidomimetics containing bifunctional diketopiperazine scaffolds as new potent integrin ligands. Chem. Eur. J. 2012, 18, 6195–6207. [Google Scholar] [CrossRef] [PubMed]
- Guzzetti, I.; Civera, M.; Vasile, F.; Arosio, D.; Tringali, C.; Piarulli, U.; Gennari, C.; Pignataro, L.; Belvisi, L.; Potenza, D. Insights into the binding of cyclic RGD peptidomimetics to α5β1 integrin by using live-cell NMR and computational studies. ChemistryOpen 2017, 6, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Guzzetti, I.; Civera, M.; Vasile, F.; Araldi, E.M.V.; Belvisi, L.; Gennari, C.; Potenza, D.; Fanelli, R.; Piarulli, U. Determination of the binding epitope of RGD-peptidomimetics to αVβ3 and αIIbβ3 integrin-rich intact cells by NMR and computational studies. Org. Biomol. Chem. 2013, 11, 3886–3893. [Google Scholar] [CrossRef] [PubMed]
- Panzeri, S.; Zanella, S.; Arosio, D.; Vahdati, L.; Dal Corso, A.; Pignataro, L.; Paolillo, M.; Schinelli, S.; Belvisi, L.; Gennari, C.; et al. Cyclic isoDGR and RGD peptidomimetics containing bifunctional diketopiperazine scaffolds are integrin antagonists. Chem. Eur. J. 2015, 21, 6265–6271. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, R.; Schembri, L.; Piarulli, U.; Pinoli, M.; Rasini, E.; Paolillo, M.; Galiazzo, M.C.; Cosentino, M.; Marino, F. Effects of a novel cyclic RGD peptidomimetic on cell proliferation, migration and angiogenic activity in human endothelial cells. Vasc. Cell 2014, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Colombo, R.; Mingozzi, M.; Belvisi, L.; Arosio, D.; Piarulli, U.; Carenini, N.; Perego, P.; Zaffaroni, N.; De Cesare, M.; Castiglioni, V.; et al. Synthesis and biological evaluation (in vitro and in vivo) of cyclic arginine-glycine-aspartate (RGD) peptidomimetic-paclitaxel conjugates targeting integrin αVβ3. J. Med. Chem. 2012, 55, 10460–10474. [Google Scholar] [CrossRef] [PubMed]
- Dal Corso, A.; Caruso, M.; Belvisi, L.; Arosio, D.; Piarulli, U.; Albanese, C.; Gasparri, F.; Marsiglio, A.; Sola, F.; Troiani, S.; et al. Synthesis and biological evaluation of RGD peptidomimetic-paclitaxel conjugates bearing lysosomally cleavable linkers. Chem. Eur. J. 2015, 21, 6921–6929. [Google Scholar] [CrossRef] [PubMed]
- Pina, A.; Dal Corso, A.; Caruso, M.; Belvisi, L.; Arosio, D.; Zanella, S.; Gasparri, F.; Albanese, C.; Cucchi, U.; Fraietta, I.; et al. Targeting integrin αVβ3 with theranostic RGD-camptothecin conjugates bearing a disulfide linker: Biological evaluation reveals a complex scenario. ChemistrySelect 2017, 2, 4759–4766. [Google Scholar] [CrossRef]
- Dong, X.; Hudson, N.E.; Lu, C.; Springer, T.A. Structural determinants of integrin β-subunit specificity for latent TGF-β. Nat. Struct. Mol. Biol. 2014, 21, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, M.; Serra, M.; Schinelli, S. Integrins in glioblastoma: Still an attractive target? Pharmacol. Res. 2016, 113, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.L.; Hölzemann, G.; Sulyok, G.A.; Kessler, H. Nanomolar small molecule inhibitors for αVβ6, αVβ5, and αVβ3 integrins. J. Med. Chem. 2002, 45, 1045–1051. [Google Scholar] [CrossRef] [PubMed]
- Glide, version 5.7; Schrödinger, LLC: New York, NY, USA, 2011.
- For the sake of precision, in [32] the ligand-free αVβ6 structure is defined similar to the state 1 and the ligand-bound αVβ6 similar to the state 2 of the eight intermediate states identified for integrin αIIbβ3 opening in Zhu, J.; Zhu, J.; Springer, T.A. Complete integrin headpiece opening in eight steps. J. Cell Biol. 2013, 201, 1053–1068. [Google Scholar] [CrossRef]
- Xiong, J.P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal Structure of the Extracellular Segment of Integrin αVβ3 in Complex with an Arg-Gly-Asp Ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A.; Zhu, J.; Xiao, T. Structural basis for distinctive recognition of fibrinogen γC peptide by the platelet integrin αIIbβ3. J. Cell Biol. 2008, 182, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Springer, T.A. Metal ion and ligand binding of integrin α5β1. Proc. Natl. Acad. Sci. USA 2014, 111, 17863–17868. [Google Scholar] [CrossRef] [PubMed]
- Nagae, M.; Re, S.; Mihara, E.; Nogi, T.; Sugita, Y.; Takagi, J. Crystal structure of α5β1 integrin ectodomain: Atomic details of the fibronectin receptor. J. Cell Biol. 2012, 197, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Raghavan, S. Defining the role of integrin αVβ6 in cancer. Curr. Drug Targets 2009, 10, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, O.V.; Marelli, U.K.; Kapp, T.G.; Di Leva, F.S.; Di Maro, S.; Nieberler, M.; Reuning, U.; Schwaiger, M.; Novellino, E.; Marinelli, L.; et al. Stable peptides instead of stapled peptides: Highly potent αvβ6-selective integrin ligands. Angew. Chem. Int. Ed. Engl. 2016, 55, 1535–1539. [Google Scholar] [CrossRef] [PubMed]
- Data on the role of the post-RGD helix in stabilizing the RGD interaction and increasing affinity and potency of RGDLXX(L/I) peptides are reported in DiCara, D.; Rapisarda, C.; Sutcliffe, J.L.; Violette, S.M.; Weinreb, P.H.; Hart, I.R.; Howard, M.J.; Marshall, J.F. Structure-Function Analysis of Arg-Gly-Asp Helix Motifs in αVβ6 Integrin Ligands. J. Biol. Chem. 2007, 282, 9657–9665. [Google Scholar] [CrossRef]
- Maestro, version 9.2; Schrödinger, LLC: New York, NY, USA, 2011.
- Allen, S.E.; Dokholyan, N.V.; Bowers, A.A. Dynamic Docking of Conformationally Constrained Macrocycles: Methods and Applications. ACS Chem. Biol. 2016, 11, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Vasile, F.; Civera, M.; Belvisi, L.; Potenza, D.; Tiana, G. Thermodynamically-weighted conformational ensemble of cyclic RGD peptidomimetics from NOE data. J. Phys. Chem. B 2016, 120, 7098–7107. [Google Scholar] [CrossRef] [PubMed]
- Marelli, U.K.; Frank, A.O.; Wahl, B.; La Pietra, V.; Novellino, E.; Marinelli, L.; Herdtweck, E.; Groll, M.; Kessler, H. Receptor-bound conformation of cilengitide better represented by its solution-state structure than the solid-state structure. Chem. Eur. J. 2014, 20, 14201–14206. [Google Scholar] [CrossRef] [PubMed]
- Guarnieri, F.; Still, W.C. A rapidly convergent simulation method: Mixed monte carlo/stochastic dynamics. J. Comput. Chem. 1994, 15, 1302–1310. [Google Scholar] [CrossRef]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- MacroModel, version 9.9; Schrödinger, LLC: New York, NY, USA, 2011.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | αVβ6 IC50 [nM] 1 | αVβ3 IC50 [nM] 2 | IC50 (αVβ6)/IC50 (αVβ3) |
|---|---|---|---|
| 1a c[RGDf(N-Me)V] | 82.8 ± 4.9 | 0.71 ± 0.06 | 117 |
| 1b c[RGDfV] | 104.7 ± 18.9 | 3.2 ± 1.3 | 33 |
| 1c c[RGDfK] | 52.0 ± 23.8 | 1.4 ± 0.2 | 37 |
| 2 | 345.0 ± 1.0 | 3.2 ± 2.7 | 108 |
| 3 | 95.6 ± 24.6 | 4.5 ± 1.1 | 21 |
| 4 | 95.3 ± 4.9 | 7.6 ± 4.3 | 13 |
| 5 | 173.5 ± 52.5 | 12.6 ± 5.0 | 14 |
| 6 | 76.6 ± 4.2 | 2.1 ± 0.6 | 37 |
| 7 | 2.3 ± 0.8 | 0.2 ± 0.09 | 12 |
| 8 c[DKP-3-RAD] | 4095 ± 1425 | 1500 ± 540 | 3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Civera, M.; Arosio, D.; Bonato, F.; Manzoni, L.; Pignataro, L.; Zanella, S.; Gennari, C.; Piarulli, U.; Belvisi, L. Investigating the Interaction of Cyclic RGD Peptidomimetics with αVβ6 Integrin by Biochemical and Molecular Docking Studies. Cancers 2017, 9, 128. https://doi.org/10.3390/cancers9100128
Civera M, Arosio D, Bonato F, Manzoni L, Pignataro L, Zanella S, Gennari C, Piarulli U, Belvisi L. Investigating the Interaction of Cyclic RGD Peptidomimetics with αVβ6 Integrin by Biochemical and Molecular Docking Studies. Cancers. 2017; 9(10):128. https://doi.org/10.3390/cancers9100128
Chicago/Turabian StyleCivera, Monica, Daniela Arosio, Francesca Bonato, Leonardo Manzoni, Luca Pignataro, Simone Zanella, Cesare Gennari, Umberto Piarulli, and Laura Belvisi. 2017. "Investigating the Interaction of Cyclic RGD Peptidomimetics with αVβ6 Integrin by Biochemical and Molecular Docking Studies" Cancers 9, no. 10: 128. https://doi.org/10.3390/cancers9100128
APA StyleCivera, M., Arosio, D., Bonato, F., Manzoni, L., Pignataro, L., Zanella, S., Gennari, C., Piarulli, U., & Belvisi, L. (2017). Investigating the Interaction of Cyclic RGD Peptidomimetics with αVβ6 Integrin by Biochemical and Molecular Docking Studies. Cancers, 9(10), 128. https://doi.org/10.3390/cancers9100128