
The Role of Histone Protein Modifications and Mutations in Histone Modifiers in Pediatric B-Cell Progenitor Acute Lymphoblastic Leukemia

Abstract
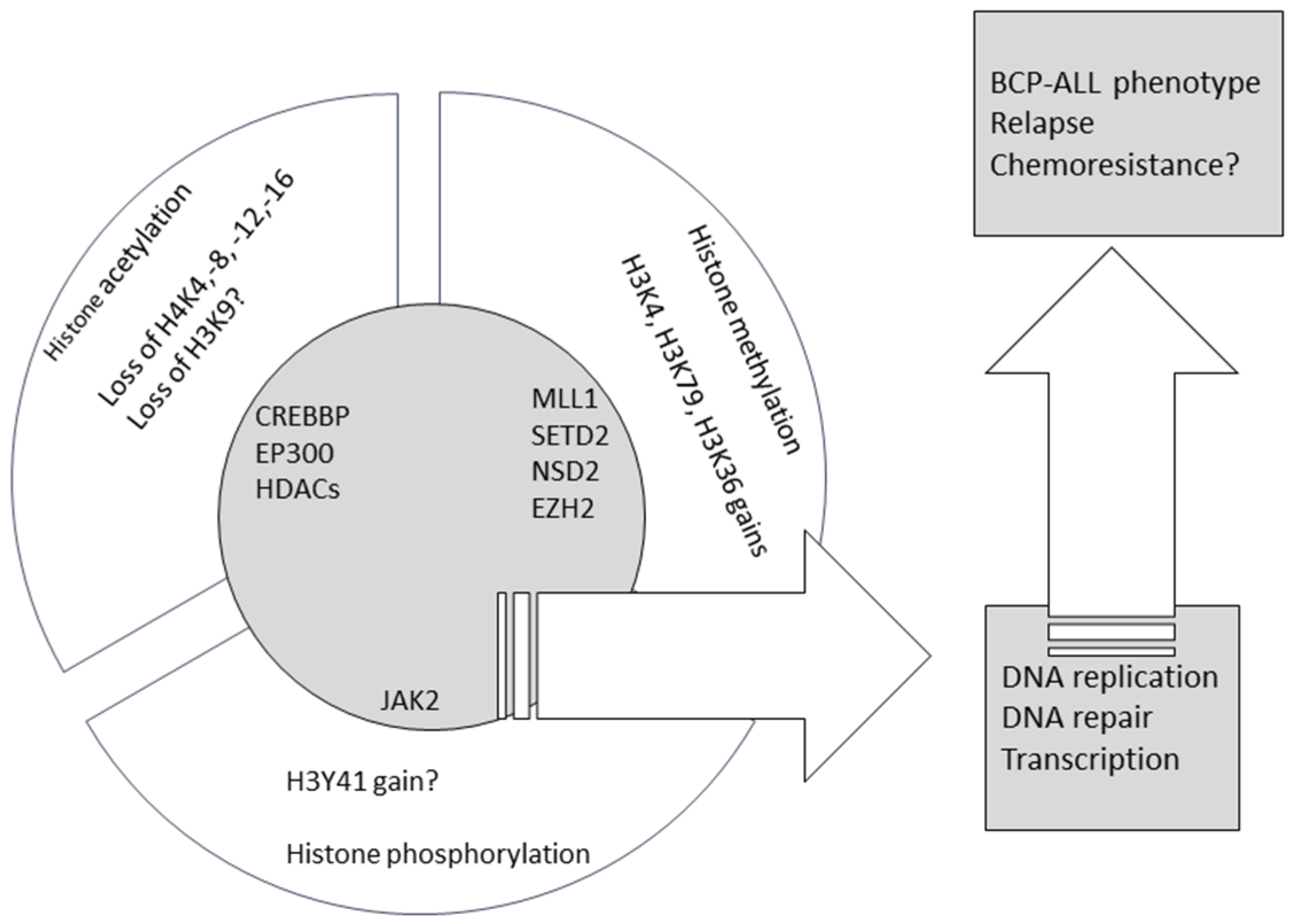
:1. Introduction
1.1. Acute Lymphoblastic Leukemia in Children
1.2. Histone Modifications
1.3. Histone Lysine Acetylation
1.3.1. Mutations/Rearrangements in Genes Involved in Histone Lysine Acetylation
1.3.2. Differential Expression of Genes Involved in Histone Lysine Deacetylation
1.3.3. The Association of Global and Loci-Specific Levels of Histone Acetylation
1.4. Histone Lysine Methylation
Mutations/Rearrangements in Genes Involved in Histone Lysine Methylation
1.5. Histone Phosphorylation
Histone Phosphorylation in BCP-ALL
1.6. Histone Gene Disruption in BCP-ALL
1.7. The Evidence for the Role of Histone Modification in B-Cell Differentiation
2. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hunger, S.P.; Raetz, E.A.; Loh, M.L.; Mullighan, C.G. Improving outcomes for high-risk ALL: Translating new discoveries into clinical care. Pediatr. Blood Cancer 2011, 56, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.V.; Enshaei, A.; Schwab, C.; Wade, R.; Chilton, L.; Elliott, A.; Richardson, S.; Hancock, J.; Kinsey, S.E.; Mitchell, C.D.; et al. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood 2014, 124, 1434–1444. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. Genomic analysis of acute leukemia. Int. J. Lab. Hematol. 2009, 31, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Downing, J.R. Global genomic characterization of acute lymphoblastic leukemia. Semin. Hematol. 2009, 46, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. Genomic profiling of B-progenitor acute lymphoblastic leukemia. Best Pract. Res. Clin. Haematol. 2011, 24, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. New strategies in acute lymphoblastic leukemia: Translating advances in genomics into clinical practice. Clin. Cancer Res. 2011, 17, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Mullighan, C.G.; Evans, W.E.; Relling, M.V. Pediatric acute lymphoblastic leukemia: Where are we going and how do we get there? Blood 2012, 120, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Chi, P.; Allis, C.D.; Wang, G.G. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 10, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T.; Huntly, B.J. Targeting epigenetic readers in cancer. N. Engl. J. Med. 2012, 367, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Wouters, B.J.; Delwel, R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 2016, 127, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Plass, C.; Oakes, C.; Blum, W.; Marcucci, G. Epigenetics in acute myeloid leukemia. Semin. Oncol. 2008, 35, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Pastore, F.; Levine, R.L. Epigenetic regulators and their impact on therapy in acute myeloid leukemia. Haematologica 2016, 101, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.J.; Bhatla, T. Epigenetic modifications in pediatric acute lymphoblastic leukemia. Front. Pediatr. 2014, 2, 42. [Google Scholar] [CrossRef] [PubMed]
- Busche, S.; Ge, B.; Vidal, R.; Spinella, J.F.; Saillour, V.; Richer, C.; Healy, J.; Chen, S.H.; Droit, A.; Sinnett, D.; Pastinen, T. Integration of high-resolution methylome and transcriptome analyses to dissect epigenomic changes in childhood acute lymphoblastic leukemia. Cancer Res. 2013, 73, 4323–4336. [Google Scholar] [CrossRef] [PubMed]
- Chatterton, Z.; Morenos, L.; Saffery, R.; Craig, J.M.; Ashley, D.; Wong, N.C. DNA methylation and miRNA expression profiling in childhood B-cell acute lymphoblastic leukemia. Epigenomics 2010, 2, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Chatterton, Z.; Morenos, L.; Mechinaud, F.; Ashley, D.M.; Craig, J.M.; Sexton-Oates, A.; Halemba, M.S.; Parkinson-Bates, M.; Ng, J.; Morrison, D.; et al. Epigenetic deregulation in pediatric acute lymphoblastic leukemia. Epigenetics 2014, 9, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, L.; Aifantis, I. Fingerprinting acute leukemia: DNA methylation profiling of B-acute lymphoblastic leukemia. Cancer Discov. 2012, 2, 976–978. [Google Scholar] [CrossRef] [PubMed]
- Davidsson, J.; Lilljebjörn, H.; Andersson, A.; Veerla, S.; Heldrup, J.; Behrendtz, M.; Fioretos, T.; Johansson, B. The DNA methylome of pediatric acute lymphoblastic leukemia. Hum. Mol. Genet. 2009, 18, 4054–4065. [Google Scholar] [CrossRef] [PubMed]
- Milani, L.; Lundmark, A.; Kiialainen, A.; Nordlund, J.; Flaegstad, T.; Forestier, E.; Heyman, M.; Jonmundsson, G.; Kanerva, J.; Schmiegelow, K.; et al. DNA methylation for subtype classification and prediction of treatment outcome in patients with childhood acute lymphoblastic leukemia. Blood 2010, 115, 1214–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordlund, J.; Bäcklin, C.L.; Wahlberg, P.; Busche, S.; Berglund, E.C.; Eloranta, M.L.; Flaegstad, T.; Forestier, E.; Frost, B.M.; Harila-Saari, A.; et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Nordlund, J.; Bäcklin, C.L.; Zachariadis, V.; Cavelier, L.; Dahlberg, J.; Öfverholm, I.; Barbany, G.; Nordgren, A.; Övernäs, E.; Abrahamsson, J.; et al. DNA methylation-based subtype prediction for pediatric acute lymphoblastic leukemia. Clin. Epigenet. 2015, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval, J.; Heyn, H.; Méndez-González, J.; Gomez, A.; Moran, S.; Baiget, M.; Melo, M.; Badell, I.; Nomdedéu, J.F.; Esteller, M. Genome-wide DNA methylation profiling predicts relapse in childhood B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2013, 160, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Peirs, S.; Van der Meulen, J.; Van de Walle, I.; Taghon, T.; Speleman, F.; Poppe, B.; Van Vlierberghe, P. Epigenetics in T-cell acute lymphoblastic leukemia. Immunol. Rev. 2015, 263, 50–67. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, D.; Nakagawa, S.; Hori, M.; Kurokawa, N.; Soejima, A.; Nakano, K.; Yamochi, T.; Nakashima, M.; Kobayashi, S.; Tanaka, Y.; et al. Polycomb-dependent epigenetic landscape in adult T-cell leukemia. Blood 2016, 127, 1790–1802. [Google Scholar] [CrossRef] [PubMed]
- Knoechel, B.; Roderick, J.E.; Williamson, K.E.; Zhu, J.; Lohr, J.G.; Cotton, M.J.; Gillespie, S.M.; Fernandez, D.; Ku, M.; Wang, H.; et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Benyoucef, A.; Palii, C.G.; Wang, C.; Porter, C.J.; Chu, A.; Dai, F.; Tremblay, V.; Rakopoulos, P.; Singh, K.; Huang, S.; et al. UTX inhibition as selective epigenetic therapy against TAL1-driven T-cell acute lymphoblastic leukemia. Genes Dev. 2016, 30, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Bhatla, T.; Wang, J.; Morrison, D.J.; Raetz, E.A.; Burke, M.J.; Brown, P.; Carroll, W.L. Epigenetic reprogramming reverses the relapse-specific gene expression signature and restores chemosensitivity in childhood B-lymphoblastic leukemia. Blood 2012, 119, 5201–5210. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P.; Tsirigos, A.; Welstead, G.G.; Trimarchi, T.; Bakogianni, S.; Xu, L.; Loizou, E.; Holmfeldt, L.; Strikoudis, A.; King, B.; et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature 2014, 514, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.F.; Lim, H.W.; Liu, S.; et al. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New nomenclature for chromatin-modifying enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Bartke, T.; Kouzarides, T. Decoding the chromatin modification landscape. Cell Cycle 2011, 10, 182. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. SnapShot: Histone-modifying enzymes. Cell 2007, 131, 822. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.B.; Chen, Z.L.; Li, J.G.; Hu, X.D.; Shi, X.J.; Sun, Z.M.; Zhang, F.; Zhao, Z.R.; Li, Z.T.; Liu, Z.Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Nair, D.; Guan, X.; Rastogi, N.; Freitas, M.A.; Parthun, M.R. Sites of acetylation on newly synthesized histone H4 are required for chromatin assembly and DNA damage response signaling. Mol. Cell. Biol. 2013, 33, 3286–3298. [Google Scholar] [CrossRef] [PubMed]
- Huether, R.; Dong, L.; Chen, X.; Wu, G.; Parker, M.; Wei, L.; Ma, J.; Edmonson, M.N.; Hedlund, E.K.; Rusch, M.C.; et al. The landscape of somatic mutations in epigenetic regulators across 1000 paediatric cancer genomes. Nat. Commun. 2014, 5, 3630. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Dang, W.; Steffen, K.K.; Perry, R.; Dorsey, J.A.; Johnson, F.B.; Shilatifard, A.; Kaeberlein, M.; Kennedy, B.K.; Berger, S.L. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009, 459, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.S.; Kannan, K.; Roy, D.M.; Morris, L.G.; Ganly, I.; Katabi, N.; Ramaswami, D.; Walsh, L.A.; Eng, S.; Huse, J.T.; et al. The mutational landscape of adenoid cystic carcinoma. Nat. Genet. 2013, 45, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Fernández-Cuesta, L.; Sos, M.L.; George, J.; Seidel, D.; Kasper, L.H.; Plenker, D.; Leenders, F.; Sun, R.; Zander, T.; et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 2012, 44, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Zhang, J.; Kasper, L.H.; Lerach, S.; Payne-Turner, D.; Phillips, L.A.; Heatley, S.L.; Holmfeldt, L.; Collins-Underwood, J.R.; Ma, J.; et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011, 471, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. The genomic landscape of acute lymphoblastic leukemia in children and young adults. Hematol. Am. Soc. Hematol. Educ. Program 2014, 2014, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mullighan, C.G.; Harvey, R.C.; Wu, G.; Chen, X.; Edmonson, M.; Buetow, K.H.; Carroll, W.L.; Chen, I.M.; Devidas, M.; et al. Key pathways are frequently mutated in high-risk childhood acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Blood 2011, 118, 3080–3087. [Google Scholar] [CrossRef] [PubMed]
- Bouska, A.; Zhang, W.; Gong, Q.; Iqbal, J.; Scuto, A.; Vose, J.; Ludvigsen, M.; Fu, K.; Weisenburger, D.D.; Greiner, T.C.; et al. Combined copy number and mutation analysis identifies oncogenic pathways associated with transformation of follicular lymphoma. Leukemia 2016. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Bartenhagen, C.; Gombert, M.; Okpanyi, V.; Binder, V.; Röttgers, S.; Bradtke, J.; Teigler-Schlegel, A.; Harbott, J.; Ginzel, S.; et al. Next-generation-sequencing of recurrent childhood high hyperdiploid acute lymphoblastic leukemia reveals mutations typically associated with high risk patients. Leuk. Res. 2015, 39, 990–1001. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Almeida, A.C.; Abate, F.; Khiabanian, H.; Martinez-Escala, E.; Guitart, J.; Tensen, C.P.; Vermeer, M.H.; Rabadan, R.; Ferrando, A.; Palomero, T. The mutational landscape of cutaneous T cell lymphoma and Sézary syndrome. Nat. Genet. 2015, 47, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Inthal, A.; Zeitlhofer, P.; Zeginigg, M.; Morak, M.; Grausenburger, R.; Fronkova, E.; Fahrner, B.; Mann, G.; Haas, O.A.; Panzer-Grümayer, R. CREBBP HAT domain mutations prevail in relapse cases of high hyperdiploid childhood acute lymphoblastic leukemia. Leukemia 2012, 26, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Lunning, M.A.; Green, M.R. Mutation of chromatin modifiers; an emerging hallmark of germinal center B-cell lymphomas. Blood Cancer J. 2015, 5, e361. [Google Scholar] [CrossRef] [PubMed]
- Malinowska-Ozdowy, K.; Frech, C.; Schönegger, A.; Eckert, C.; Cazzaniga, G.; Stanulla, M.; zur Stadt, U.; Mecklenbräuker, A.; Schuster, M.; Kneidinger, D.; et al. KRAS and CREBBP mutations: A relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia 2015, 29, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Mar, B.G.; Bullinger, L.B.; McLean, K.M.; Grauman, P.V.; Harris, M.H.; Stevenson, K.; Neuberg, D.S.; Sinha, A.U.; Sallan, S.E.; Silverman, L.B.; et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat. Commun. 2014, 5, 3469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Okosun, J.; Bödör, C.; Wang, J.; Araf, S.; Yang, C.Y.; Pan, C.; Boller, S.; Cittaro, D.; Bozek, M.; Iqbal, S.; et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat. Genet. 2014, 46, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, S.N.; Zhou, Q.; Zhou, T.; Cheng, Z.; Abboud-Werner, S.L.; Horn, D.; Lecocke, M.; White, R.; Krivtsov, A.V.; Armstrong, S.A.; et al. Crebbp haploinsufficiency in mice alters the bone marrow microenvironment, leading to loss of stem cells and excessive myelopoiesis. Blood 2011, 118, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Gocho, Y.; Kiyokawa, N.; Ichikawa, H.; Nakabayashi, K.; Osumi, T.; Ishibashi, T.; Ueno, H.; Terada, K.; Oboki, K.; Sakamoto, H.; et al. A novel recurrent EP300-ZNF384 gene fusion in B-cell precursor acute lymphoblastic leukemia. Leukemia 2015, 29, 2445–2448. [Google Scholar] [CrossRef] [PubMed]
- Josling, G.A.; Selvarajah, S.A.; Petter, M.; Duffy, M.F. The role of bromodomain proteins in regulating gene expression. Genes 2012, 3, 320–343. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, P.; Müller, S.; O’Mahony, A.; Fedorov, O.; Filippakopoulos, P.; Hunt, J.P.; Lasater, E.A.; Pallares, G.; Picaud, S.; Wells, C.; et al. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat. Chem. Biol. 2014, 10, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Fedorov, O.; Thanasopoulou, A.; Leonards, K.; Jones, K.; Meier, J.; Olzscha, H.; Monteiro, O.; Martin, S.; Philpott, M.; et al. Generation of a Selective Small Molecule Inhibitor of the CBP/p300 Bromodomain for Leukemia Therapy. Cancer Res. 2015, 75, 5106–5119. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.J.; Kopp, N.; Bird, L.; Paranal, R.M.; Qi, J.; Bowman, T.; Rodig, S.J.; Kung, A.L.; Bradner, J.E.; Weinstock, D.M. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 2012, 120, 2843–2852. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, D.; Agathanggelou, A.; Perry, T.; Weston, V.; Petermann, E.; Zlatanou, A.; Oldreive, C.; Wei, W.; Stewart, G.; Longman, J.; Smith, E.; Kearns, P.; Knapp, S.; Stankovic, T. BET inhibition as a single or combined therapeutic approach in primary paediatric B-precursor acute lymphoblastic leukaemia. Blood Cancer J. 2013, 3, e126. [Google Scholar] [CrossRef] [PubMed]
- Moreno, D.A.; Scrideli, C.A.; Cortez, M.A.; de Paula Queiroz, R.; Valera, E.T.; da Silva Silveira, V.; Yunes, J.A.; Brandalise, S.R.; Tone, L.G. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Gruhn, B.; Naumann, T.; Gruner, D.; Walther, M.; Wittig, S.; Becker, S.; Beck, J.F.; Sonnemann, J. The expression of histone deacetylase 4 is associated with prednisone poor-response in childhood acute lymphoblastic leukemia. Leuk. Res. 2013, 37, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Sonnemann, J.; Gruhn, B.; Wittig, S.; Becker, S.; Beck, J.F. Increased activity of histone deacetylases in childhood acute lymphoblastic leukaemia and acute myeloid leukaemia: Support for histone deacetylase inhibitors as antileukaemic agents. Br. J. Haematol. 2012, 158, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Mummery, A.; Narendran, A.; Lee, K.Y. Targeting epigenetics through histone deacetylase inhibitors in acute lymphoblastic leukemia. Curr. Cancer Drug Targets 2011, 11, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Vilas-Zornoza, A.; Agirre, X.; Abizanda, G.; Moreno, C.; Segura, V.; De Martino Rodriguez, A.; José-Eneriz, E.S.; Miranda, E.; Martín-Subero, J.I.; Garate, L.; et al. Preclinical activity of LBH589 alone or in combination with chemotherapy in a xenogeneic mouse model of human acute lymphoblastic leukemia. Leukemia 2012, 26, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Janczar, K.; Janczar, S.; Pastorczak, A.; Mycko, K.; Paige, A.J.; Zalewska-Szewczyk, B.; Wagrowska-Danilewicz, M.; Danilewicz, M.; Mlynarski, W. Preserved global histone H4 acetylation linked to ETV6-RUNX1 fusion and PAX5 deletions is associated with favorable outcome in pediatric B-cell progenitor acute lymphoblastic leukemia. Leuk. Res. 2015, 39, 1455–1461. [Google Scholar] [CrossRef] [PubMed]
- Advani, A.S.; Gibson, S.E.; Douglas, E.; Jin, T.; Zhao, X.; Kalaycio, M.; Copelan, E.; Sobecks, R.; Sekeres, M.; Sungren, S.; Hsi, E.D. Histone H4 acetylation by immunohistochemistry and prognosis in newly diagnosed adult acute lymphoblastic leukemia (ALL) patients. BMC Cancer 2010, 10, 387. [Google Scholar] [CrossRef] [PubMed]
- Advani, A.S.; Gibson, S.; Douglas, E.; Diacovo, J.; Elson, P.; Kalaycio, M.; Copelan, E.; Sekeres, M.; Sobecks, R.; Sungren, S.; et al. Histone H4 acetylation by immunohistochemistry and prognosis in relapsed acute lymphocytic leukaemia (ALL). Br. J. Haematol. 2011, 153, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, P.S.; Piazza, R.G.; Janes, M.E.; Wong, N.C.; Davies, C.; Mogavero, A.; Bhadri, V.A.; Szymanska, B.; Geninson, G.; Magistroni, V.; et al. Epigenetic silencing of BIM in glucocorticoid poor-responsive pediatric acute lymphoblastic leukemia, and its reversal by histone deacetylase inhibition. Blood 2010, 116, 3013–3022. [Google Scholar] [CrossRef] [PubMed]
- Vu, L.P.; Luciani, L.; Nimer, S.D. Histone-modifying enzymes: Their role in the pathogenesis of acute leukemia and their therapeutic potential. Int. J. Hematol. 2013, 97, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Armstrong, S.A. Targeting DOT1L and HOX gene expression in MLL-rearranged leukemia and beyond. Exp. Hematol. 2015, 43, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Bernt, K.M.; Armstrong, S.A. Targeting epigenetic programs in MLL-rearranged leukemias. Hematol. Am. Soc. Hematol. Educ. Program 2011, 2011, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. Molecular genetics of B-precursor acute lymphoblastic leukemia. J. Clin. Investig. 2012, 122, 3407–3415. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.C.; Dou, Y. Hijacked in cancer: The KMT2 (MLL) family of methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Basavapathruni, A.; Jin, L.; Boriack-Sjodin, P.A.; Allain, C.J.; Klaus, C.R.; Raimondi, A.; Scott, M.P.; et al. Potent inhibition of DOT1L as treatment of MLL-fusion leukemia. Blood 2013, 122, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.J.; Chen, L.; Fazio, M.; Sinha, A.U.; Bernt, K.M.; Banka, D.; Dias, S.; Chang, J.; Olhava, E.J.; Daigle, S.R.; et al. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood 2013, 121, 2533–2541. [Google Scholar] [CrossRef] [PubMed]
- Bernt, K.M.; Armstrong, S.A. A role for DOT1L in MLL-rearranged leukemias. Epigenomics 2011, 3, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Song, E.J.; Kawasawa, Y.I.; Li, J.; Dovat, S.; Song, C. WDR5 high expression and its effect on tumorigenesis in leukemia. Oncotarget 2016, 7, 37740–37754. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Townsend, E.C.; Karatas, H.; Xu, J.; Li, L.; Lee, S.; Liu, L.; Chen, Y.; Ouillette, P.; Zhu, J.; et al. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol. Cell 2014, 53, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, J.D.; Wang, Y.; Chan, H.M.; Zhang, J.; Huether, R.; Kryukov, G.V.; Bhang, H.E.; Taylor, J.E.; Hu, M.; Englund, N.P.; et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Oyer, J.A.; Huang, X.; Zheng, Y.; Shim, J.; Ezponda, T.; Carpenter, Z.; Allegretta, M.; Okot-Kotber, C.I.; Patel, J.P.; Melnick, A.; et al. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia 2014, 28, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; He, F.; Zeng, H.; Ling, S.; Chen, A.; Wang, Y.; Yan, X.; Wei, W.; Pang, Y.; Cheng, H.; et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat. Genet. 2014, 46, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, V.; Ernst, J.; Rinke, J.; Winkelmann, N.; Beck, J.F.; Hochhaus, A.; Gruhn, B.; Ernst, T. EZH2 mutations and promoter hypermethylation in childhood acute lymphoblastic leukemia. J. Cancer Res. Clin. Oncol. 2016, 142, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Van der Meulen, J.; Sanghvi, V.; Mavrakis, K.; Durinck, K.; Fang, F.; Matthijssens, F.; Rondou, P.; Rosen, M.; Pieters, T.; Vandenberghe, P.; et al. The H3K27me3 demethylase UTX is a gender-specific tumor suppressor in T-cell acute lymphoblastic leukemia. Blood 2015, 125, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, C.; Ding, Y.; Pan, X.; Ge, Z.; Tan, B.H.; Gowda, C.; Sachdev, M.; Muthusami, S.; Ouyang, H.; et al. Transcriptional Regulation of JARID1B/KDM5B Histone Demethylase by Ikaros, Histone Deacetylase 1 (HDAC1), and Casein Kinase 2 (CK2) in B-cell Acute Lymphoblastic Leukemia. J. Biol. Chem. 2016, 291, 4004–4018. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Gowda, C.; Pan, X.; Ding, Y.; Tong, Y.; Tan, B.H.; Wang, H.; Muthusami, S.; Ge, Z.; Sachdev, M.; et al. Targeting casein kinase II restores Ikaros tumor suppressor activity and demonstrates therapeutic efficacy in high-risk leukemia. Blood 2015, 126, 1813–1822. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Zhang, J.; Harvey, R.C.; Collins-Underwood, J.R.; Schulman, B.A.; Phillips, L.A.; Tasian, S.K.; Loh, M.L.; Su, X.; Liu, W.; et al. JAK mutations in high-risk childhood acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2009, 106, 9414–9418. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Tasian, S.K.; Vincent, T.; Hall, J.W.; Sheen, C.; Roberts, K.G.; Seif, A.E.; Barrett, D.M.; Chen, I.M.; Collins, J.R.; et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood 2012, 120, 3510–3518. [Google Scholar] [CrossRef] [PubMed]
- Kucine, N.; Marubayashi, S.; Bhagwat, N.; Papalexi, E.; Koppikar, P.; Sanchez Martin, M.; Dong, L.; Tallman, M.S.; Paietta, E.; Wang, K.; et al. Tumor-specific HSP90 inhibition as a therapeutic approach in JAK-mutant acute lymphoblastic leukemias. Blood 2015, 126, 2479–2483. [Google Scholar] [CrossRef] [PubMed]
- Loudin, M.G.; Wang, J.; Leung, H.C.; Gurusiddappa, S.; Meyer, J.; Condos, G.; Morrison, D.; Tsimelzon, A.; Devidas, M.; Heerema, N.A.; et al. Genomic profiling in Down syndrome acute lymphoblastic leukemia identifies histone gene deletions associated with altered methylation profiles. Leukemia 2011, 25, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Holmfeldt, L.; Wei, L.; Diaz-Flores, E.; Walsh, M.; Zhang, J.; Ding, L.; Payne-Turner, D.; Churchman, M.; Andersson, A.; Chen, S.C.; et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Bergman, Y. Epigenetics of haematopoietic cell development. Nat. Rev. Immunol. 2011, 11, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Lotem, J.; Sachs, L. Epigenetics and the plasticity of differentiation in normal and cancer stem cells. Oncogene 2006, 25, 7663–7672. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Haery, L.; Thompson, R.C.; Gilmore, T.D. Histone acetyltransferases and histone deacetylases in B- and T-cell development, physiology and malignancy. Genes Cancer 2015, 6, 184–213. [Google Scholar] [PubMed]
- Johnson, K.; Shapiro-Shelef, M.; Tunyaplin, C.; Calame, K. Regulatory events in early and late B-cell differentiation. Mol. Immunol. 2005, 42, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Subrahmanyam, R.; Chakraborty, T.; Sen, R.; Desiderio, S. A plant homeodomain in RAG-2 that binds Hypermethylated lysine 4 of histone H3 is necessary for efficient antigen-receptor-gene rearrangement. Immunity 2007, 27, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Matthews, A.G.; Kuo, A.J.; Ramón-Maiques, S.; Han, S.; Champagne, K.S.; Ivanov, D.; Gallardo, M.; Carney, D.; Cheung, P.; Ciccone, D.N.; et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature 2007, 450, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Ramón-Maiques, S.; Kuo, A.J.; Carney, D.; Matthews, A.G.; Oettinger, M.A.; Gozani, O.; Yang, W. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc. Natl. Acad. Sci. USA 2007, 104, 18993–18998. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, N.; Tsai, A.G.; Lieber, M.R. H3K4me3 stimulates the V(D)J RAG complex for both nicking and hairpinning in trans in addition to tethering in CIS: Implications for translocations. Mol. Cell 2009, 34, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, N.; Lieber, M.R. Histone methylation and V(D)J recombination. Int. J. Hematol. 2014, 100, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Angelin-Duclos, C.; Park, S.; Calame, K.L. Changes in histone acetylation are associated with differences in accessibility of V(H) gene segments to V-DJ recombination during B-cell ontogeny and development. Mol. Cell. Biol. 2003, 23, 2438–2450. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, C.R.; Feeney, A.J. The extent of histone acetylation correlates with the differential rearrangement frequency of individual VH genes in pro-B cells. J. Immunol. 2005, 175, 6668–6675. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, K.P.; Baumann, M.; Eberharter, A.; Mamais, A.; Becker, P.B.; Boyes, J. Acetylation increases access of remodelling complexes to their nucleosome targets to enhance initiation of V(D)J recombination. Nucleic Acids Res. 2007, 35, 6311–6321. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Rapado, I.; Li, Y.; Potter, N.E.; Wedge, D.C.; Tubio, J.; Alexandrov, L.B.; Van Loo, P.; Cooke, S.L.; Marshall, J.; et al. RAG-mediated recombination is the predominant driver of oncogenic rearrangement in ETV6-RUNX1 acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Liu, F.; Wu, C.; Li, S.; Zhao, X.; Zhang, P.; Jiao, J.; Yu, X.; Ji, Y.; Zhang, M. Illegitimate RAG-mediated recombination events are involved in IKZF1 Δ3–6 deletion in BCR-ABL1 lymphoblastic leukaemia. Clin. Exp. Immunol. 2016, 185, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Heinäniemi, M.; Vuorenmaa, T.; Teppo, S.; Kaikkonen, M.U.; Bouvy-Liivrand, M.; Mehtonen, J.; Niskanen, H.; Zachariadis, V.; Laukkanen, S.; Liuksiala, T.; et al. Transcription-coupled genetic instability marks acute lymphoblastic leukemia structural variation hotspots. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Chen, S.C.; Andersson, A.K.; Phillips, L.A.; Li, Y.; Sotzen, J.; Kundu, M.; Downing, J.R.; Melnick, A.; Mullighan, C.G. Integrated genetic and epigenetic analysis of childhood acute lymphoblastic leukemia. J. Clin. Investig. 2013, 123, 3099–3111. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G. Genomic characterization of childhood acute lymphoblastic leukemia. Semin. Hematol. 2013, 50, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Cheng, T. Evidences for mutations in the histone modifying gene SETD2 as critical drivers in leukemia development. Sci. China Life Sci. 2014, 57, 944–946. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.C.; Mullighan, C.G.; Chen, I.M.; Wharton, W.; Mikhail, F.M.; Carroll, A.J.; Kang, H.; Liu, W.; Dobbin, K.K.; Smith, M.A.; et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 2010, 115, 5312–5321. [Google Scholar] [CrossRef] [PubMed]
- Loh, M.L.; Zhang, J.; Harvey, R.C.; Roberts, K.; Payne-Turner, D.; Kang, H.; Wu, G.; Chen, X.; Becksfort, J.; Edmonson, M.; et al. Tyrosine kinome sequencing of pediatric acute lymphoblastic leukemia: A report from the Children’s Oncology Group TARGET Project. Blood 2013, 121, 485–488. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Gene (Reference) | Histone-Modifying Function | Frequency of Mutations/Rearrangements in BCP-ALL | Subgroups Enriched |
|---|---|---|---|
| CREBBP [50,51,53,55,57] | H3K18 acetyltransferase (and other H3/H4 residues) | Rare in cases without hyperdiploidy and relapse | Relapse, hyperdiploidy |
| EP300 [50,55] | H3K18 acetyltransferase (and other H3/H4 residues) | <1% | - |
| MLL1 [3,5,6,82,122,123] | H3K4 methyltransferase | 5% | - |
| NSD2 [44,91] | H3K36 methyltransferase | Not documented in unselected patients, up to 14% in subgroups | ETV6-RUNX1-rearranged |
| SETD2 [44,60,93,124] | H3K36 methyltransferase | 12% | MLL- and ETV6-RUNX1- rearrangements, relapse |
| EZH2 [44,55,94] | H3K4 methyltransferase | 1.3% | hypodiploidy |
| JAK2 [99,100,125,126] | H3Y41 phosphorylase | Not determined in unselected patients, up to 10% in high risk disease | BCR-ABL1-like, Down syndrome, high-risk disease |
| Study Identifier | Start Year | Drug Targeting (or Potentially Targeting) Histone Modifications | ALL Population | Phase | Status |
|---|---|---|---|---|---|
| NCT00053963 | 2002 | FR901228 (HDACi) | Refractory disease (0–21 years) | 1 | completed |
| NCT00217412 | 2005 | Vorinostat (HDACi) | Relapsed or refractory disease (1–21 years) | 1 | completed |
| NCT00882206 | 2009 | Vorinostat (HDACi) | Relapsed or refractory disease (2–60 years) | 2 | completed |
| NCT01251965 | 2010 | Ruxolitinib (JAK1/JAK2 inhibitor) | Relapsed or refractory disease (14 years or older) | 1/2 | completed |
| NCT01321346 | 2011 | Panobinostat (HDACi) | Refractory disease (8–21 years) | 1 | completed |
| NCT02141828 | 2014 | EPZ-5676 (DOT1L blocker) | Relapsed or refractory disease (0–18 years) MLL-rearranged | 1 | completed |
| NCT02419755 | 2015 | Vorinostat (HDACi) | Relapsed or refractory disease (0–21 years) MLL-rearranged | 2 | recruiting |
| NCT02420717 | 2015 | Ruxolitinib (JAK1/JAK2 inhibitor) | Ph-like (10 years or older) | 2 | recruiting |
| NCT02723994 | 2016 | Ruxolitinib (JAK1/JAK2 inhibitor) | CRLF2-rearranged and/or JAK Pathway-mutant (1–21 years) | 2 | recruiting |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janczar, S.; Janczar, K.; Pastorczak, A.; Harb, H.; Paige, A.J.W.; Zalewska-Szewczyk, B.; Danilewicz, M.; Mlynarski, W. The Role of Histone Protein Modifications and Mutations in Histone Modifiers in Pediatric B-Cell Progenitor Acute Lymphoblastic Leukemia. Cancers 2017, 9, 2. https://doi.org/10.3390/cancers9010002
Janczar S, Janczar K, Pastorczak A, Harb H, Paige AJW, Zalewska-Szewczyk B, Danilewicz M, Mlynarski W. The Role of Histone Protein Modifications and Mutations in Histone Modifiers in Pediatric B-Cell Progenitor Acute Lymphoblastic Leukemia. Cancers. 2017; 9(1):2. https://doi.org/10.3390/cancers9010002
Chicago/Turabian StyleJanczar, Szymon, Karolina Janczar, Agata Pastorczak, Hani Harb, Adam J. W. Paige, Beata Zalewska-Szewczyk, Marian Danilewicz, and Wojciech Mlynarski. 2017. "The Role of Histone Protein Modifications and Mutations in Histone Modifiers in Pediatric B-Cell Progenitor Acute Lymphoblastic Leukemia" Cancers 9, no. 1: 2. https://doi.org/10.3390/cancers9010002
APA StyleJanczar, S., Janczar, K., Pastorczak, A., Harb, H., Paige, A. J. W., Zalewska-Szewczyk, B., Danilewicz, M., & Mlynarski, W. (2017). The Role of Histone Protein Modifications and Mutations in Histone Modifiers in Pediatric B-Cell Progenitor Acute Lymphoblastic Leukemia. Cancers, 9(1), 2. https://doi.org/10.3390/cancers9010002