
MLK3 Signaling in Cancer Invasion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
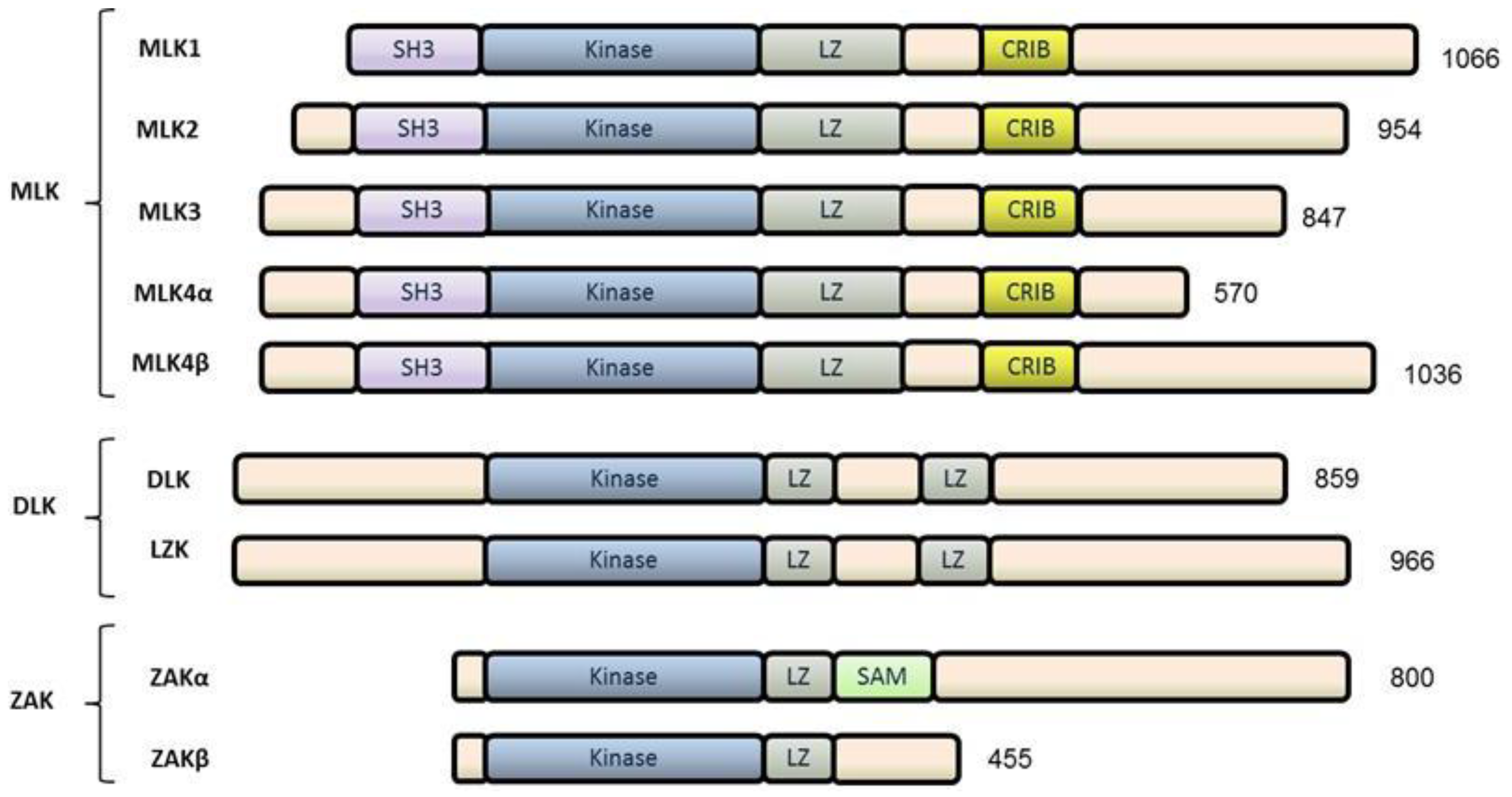
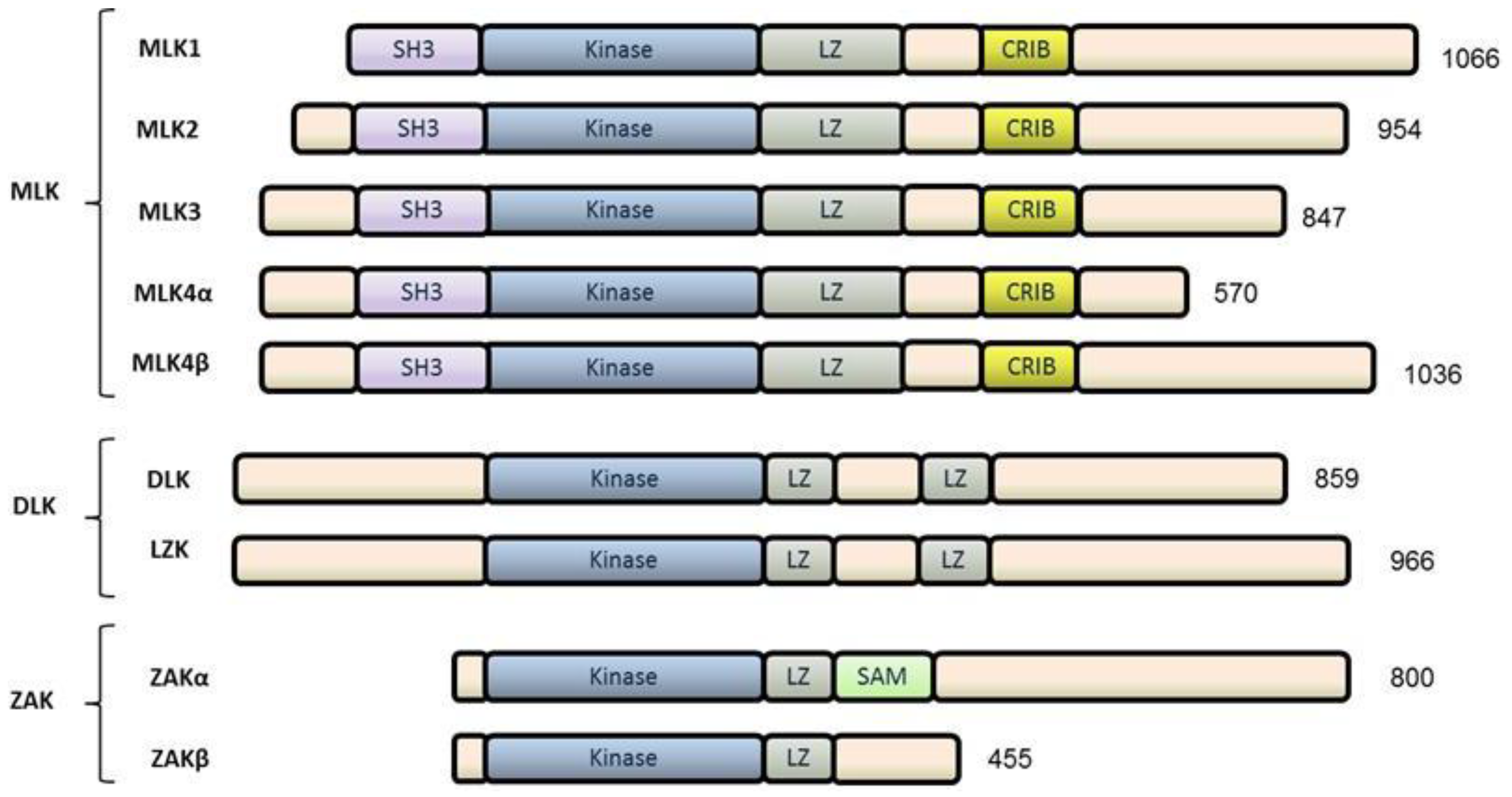
2. The Mixed Lineage Kinase (MLK) Family
2.1. MLKs
2.2. DLKs
2.3. ZAK
3. MLK3
4. MLK3 Regulation
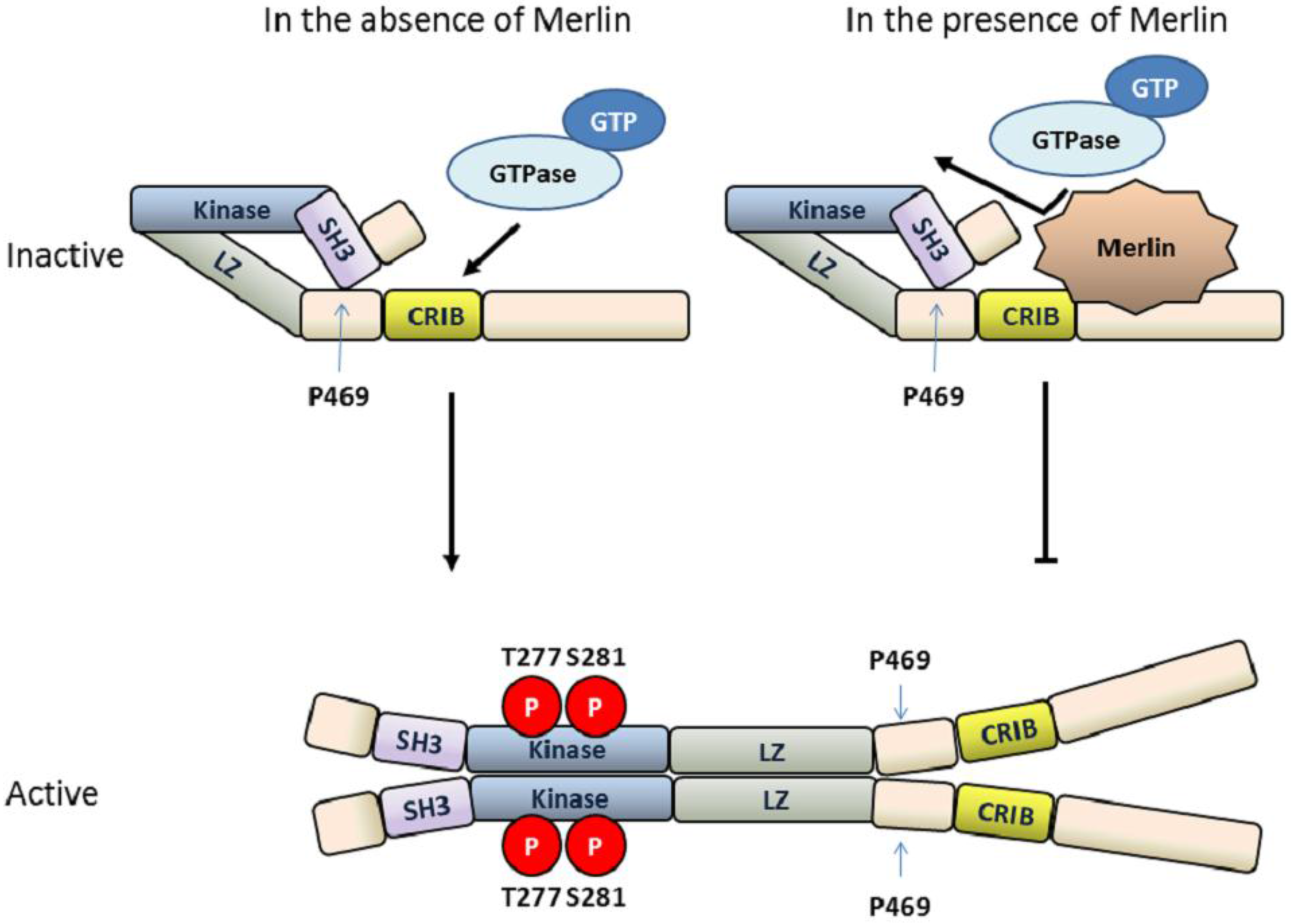
4.1. MLK3 Contains Intrinsic Autoinhibitory Function
4.2. Dimerization and Autophosphorylation of MLK3
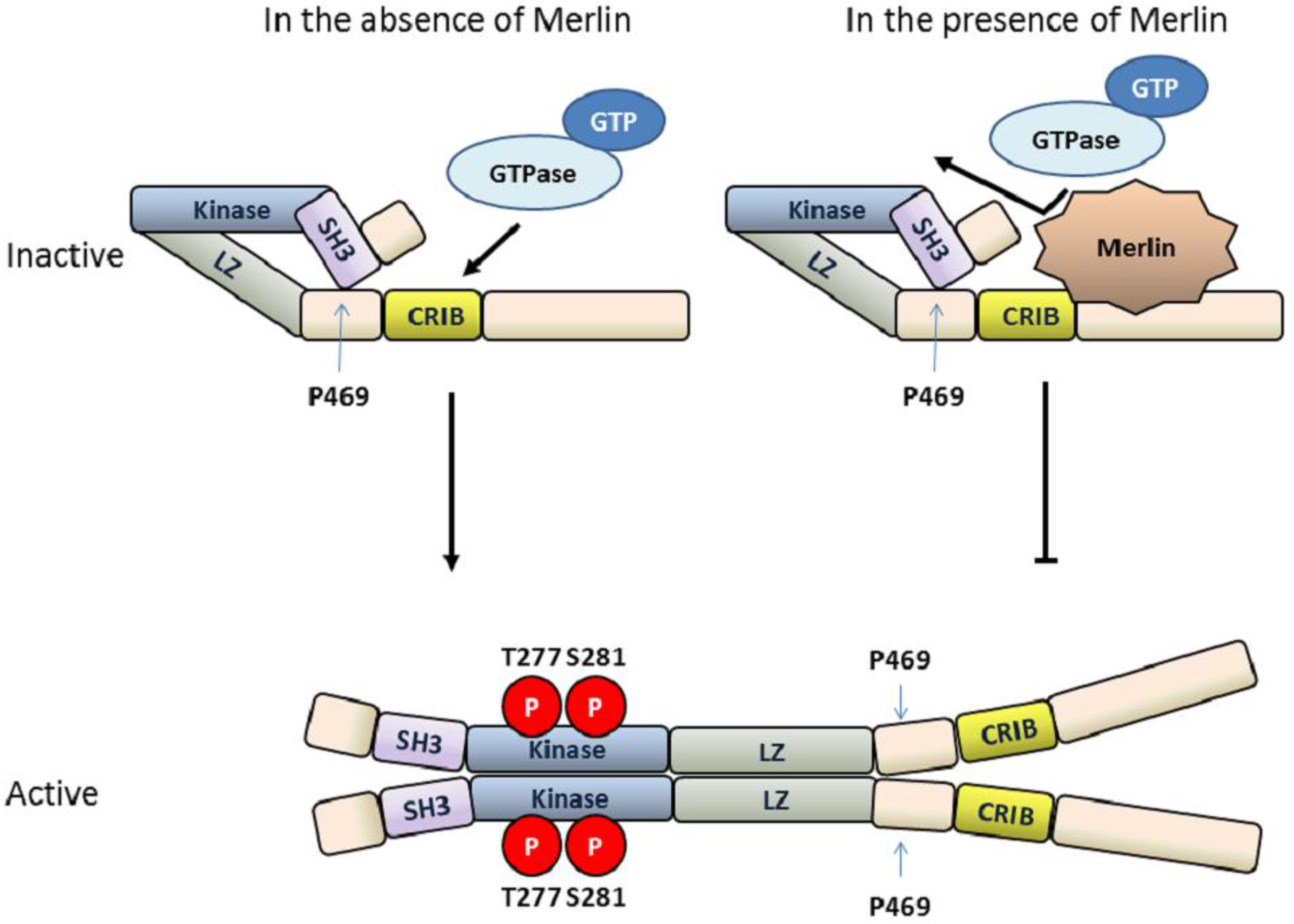
4.3. MLK3 Activity Is Regulated by the Tumor Suppressor Protein Merlin
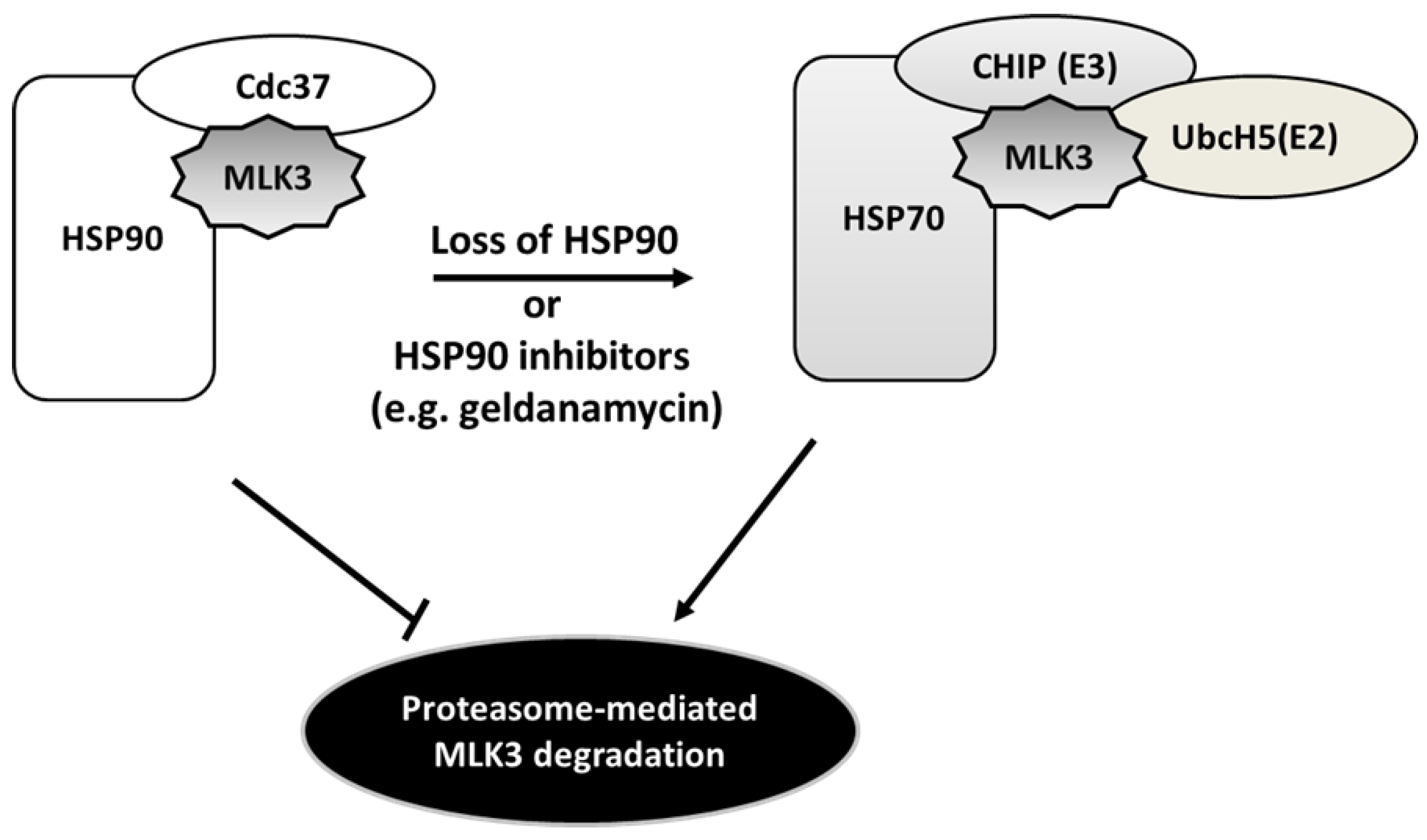
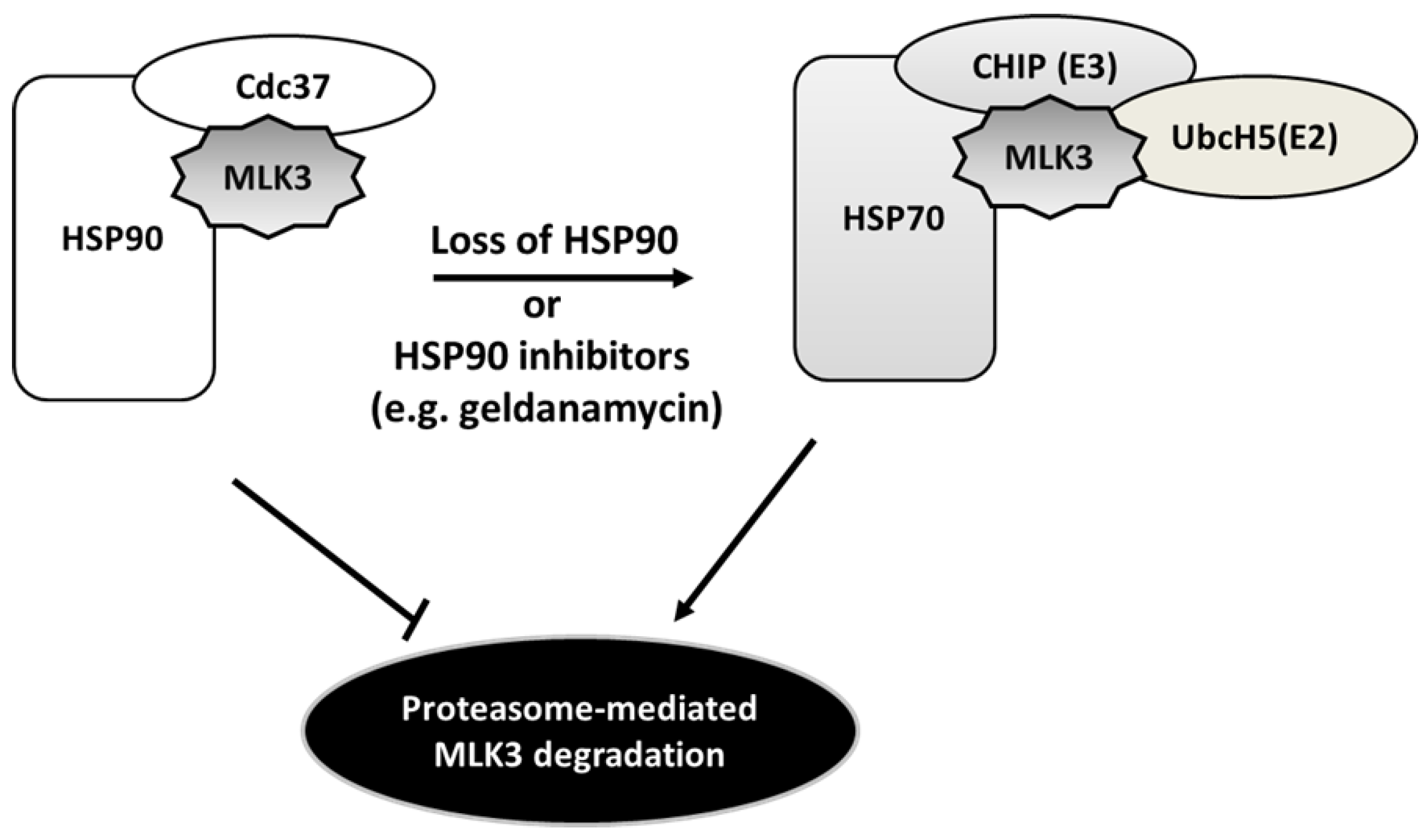
4.4. Heat Shock Protein-Mediated MLK3 Stability
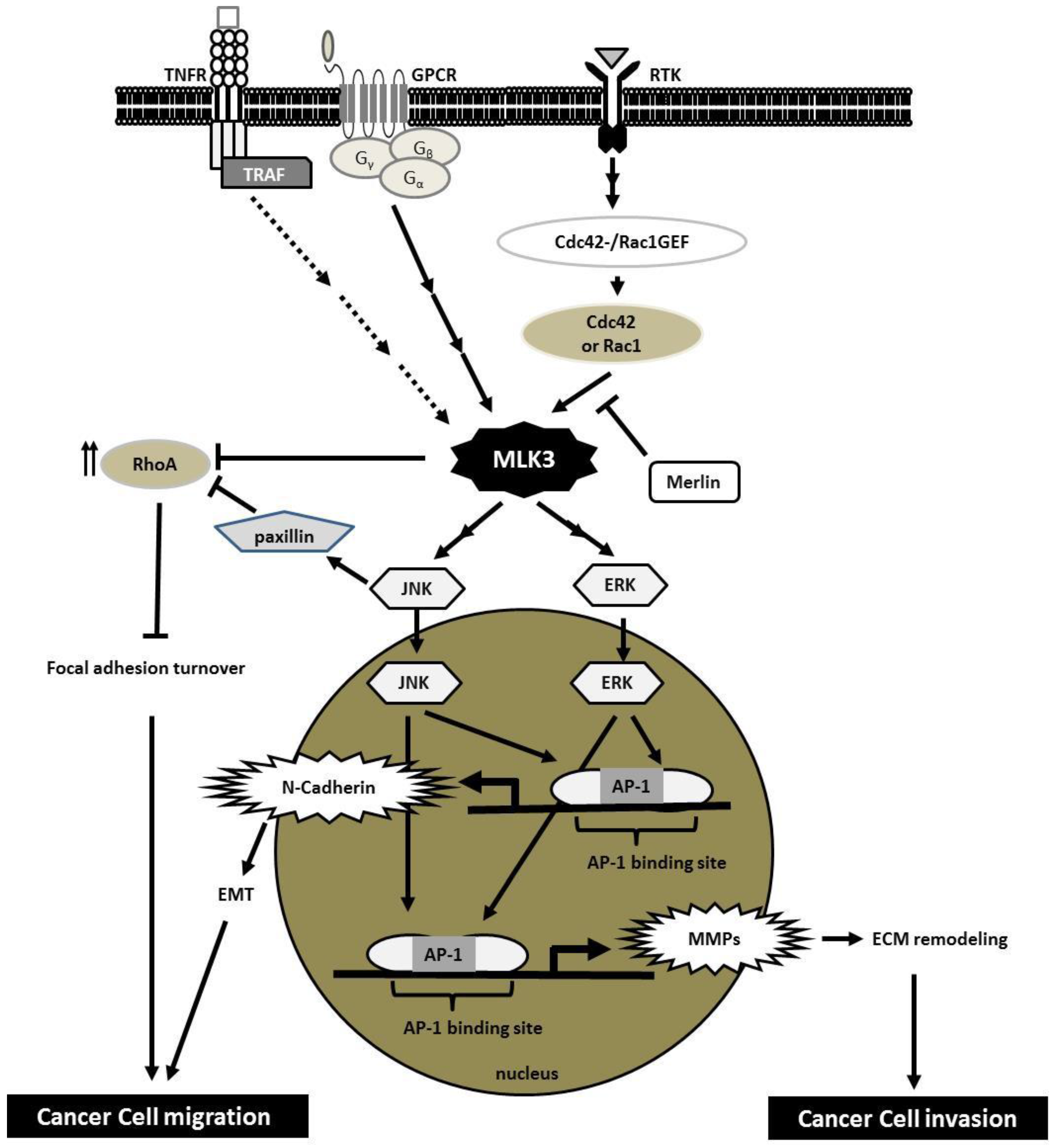
5. MLK3 Transduces Signals from Several Cell-Surface Receptors
5.1. Receptor Tyrosine Kinases (RTKs)
5.2. Chemokine Receptors
5.3. Tumor Necrosis Factor Receptor (TNFR)
6. Implication of MLK3 in Cancer Invasion
6.1. Cancer Cell Migration, Focal Adhesion Dynamics and Epithelial-to-Mesenchymal Transition (EMT)
6.2. Cancer Cell Invasion and Metastasis
7. Other Functions of MLK3
Cancer Cell Proliferation and Viability
8. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Fidler, I.J. Aacr centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef] [PubMed]
- Bacac, M.; Stamenkovic, I. Metastatic cancer cell. Annu. Rev. Pathol. 2008, 3, 221–247. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The pathogenesis of cancer metastasis: The “seed and soil” hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [PubMed]
- Goldsmith, E.J.; Min, X.; He, H.; Zhou, T. Structural studies of MAP Kinase cascade components. Methods Mol. Biol. 2010, 661, 223–237. [Google Scholar] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Gallo, K.A.; Johnson, G.L. Mixed-lineage kinase control of JNK and p38 MAPK pathways. Nat. Rev. Mol. Cell. Biol. 2002, 3, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Gallo, K.A.; Mark, M.R.; Scadden, D.T.; Wang, Z.; Gu, Q.; Godowski, P.J. Identification and characterization of SPRK, a novel src-homology 3 domain-containing proline-rich kinase with serine/threonine kinase activity. J. Biol. Chem. 1994, 269, 15092–15100. [Google Scholar] [PubMed]
- Kashuba, V.I.; Grigorieva, E.V.; Kvasha, S.M.; Pavlova, T.V.; Grigoriev, V.; Protopopov, A.; Kharchenko, O.; Gizatullin, R.; Rynditch, A.V.; Zabarovsky, E.R. Cloning and Initial Functional Characterization of Mlk4α and Mlk4β. Genomics Insights 2011, 4, 1–12. [Google Scholar] [PubMed]
- Marusiak, A.A.; Edwards, Z.C.; Hugo, W.; Trotter, E.W.; Girotti, M.R.; Stephenson, N.L.; Kong, X.; Gartside, M.G.; Fawdar, S.; Hudson, A.; et al. Mixed lineage kinases activate MEK independently of RAF to mediate resistance to RAF inhibitors. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Ezoe, K.; Lee, S.T.; Strunk, K.M.; Spritz, R.A. PTK1, a novel protein kinase required for proliferation of human melanocytes. Oncogene 1994, 9, 935–938. [Google Scholar] [PubMed]
- Ing, Y.L.; Leung, I.W.; Heng, H.H.; Tsui, L.C.; Lassam, N.J. MLK-3: Identification of a widely-expressed protein kinase bearing an SH3 domain and a leucine zipper-basic region domain. Oncogene 1994, 9, 1745–1750. [Google Scholar] [PubMed]
- Liu, T.C.; Huang, C.J.; Chu, Y.C.; Wei, C.C.; Chou, C.C.; Chou, M.Y.; Chou, C.K.; Yang, J.J. Cloning and expression of ZAK, a mixed lineage kinase-like protein containing a leucine-zipper and a sterile-alpha motif. Biochem. Biophys. Res. Commun. 2000, 274, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Leung, I.W.; Lassam, N. Dimerization via tandem leucine zippers is essential for the activation of the mitogen-activated protein kinase kinase kinase, MLK-3. J. Biol. Chem. 1998, 273, 32408–32415. [Google Scholar] [CrossRef] [PubMed]
- Vacratsis, P.O.; Gallo, K.A. Zipper-mediated oligomerization of the mixed lineage kinase SPRK/MLK-3 is not required for its activation by the GTPase cdc 42 but Is necessary for its activation of the JNK pathway. J. Biol. Chem. 2000, 275, 27893–27900. [Google Scholar] [PubMed]
- Zhang, H.; Gallo, K.A. Autoinhibition of mixed lineage kinase 3 through its Src homology 3 domain. J. Biol. Chem. 2001, 276, 45598–45603. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wu, W.; Du, Y.; Santos, S.J.; Conrad, S.E.; Watson, J.T.; Grammatikakis, N.; Gallo, K.A. Hsp90/p50cdc37 is required for mixed-lineage kinase (MLK) 3 signaling. J. Biol. Chem. 2004, 279, 19457–19463. [Google Scholar] [CrossRef] [PubMed]
- Schachter, K.A.; Du, Y.; Lin, A.; Gallo, K.A. Dynamic positive feedback phosphorylation of mixed lineage kinase 3 by JNK reversibly regulates its distribution to Triton-soluble domains. J. Biol. Chem. 2006, 281, 19134–19144. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Zhang, H.; Miller, E.M.; Seibold, S.A.; Chen, W.; Gallo, K.A. Induced, selective proteolysis of MLK3 negatively regulates MLK3/JNK signalling. Biochem. J. 2010, 427, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Zeng, H.; Li, J.; Xiao, L.; He, Y.; Tang, Y.; Li, Y. miR-199a regulates the tumor suppressor mitogen-activated protein kinase kinase kinase 11 in gastric cancer. Biol. Pharm. Bull. 2010, 33, 1822–1827. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Modi, N.; Stewart, A.M.; Hieronimus, R.I.; Liu, J.; Gutmann, D.H.; Chadee, D.N. Regulation of mixed lineage kinase 3 is required for Neurofibromatosis-2-mediated growth suppression in human cancer. Oncogene 2011, 30, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Chadee, D.N.; Kyriakis, J.M. A novel role for mixed lineage kinase 3 (MLK3) in B-Raf activation and cell proliferation. Cell Cycle 2004, 3, 1227–1229. [Google Scholar] [CrossRef] [PubMed]
- Chadee, D.N.; Xu, D.; Hung, G.; Andalibi, A.; Lim, D.J.; Luo, Z.; Gutmann, D.H.; Kyriakis, J.M. Mixed-lineage kinase 3 regulates B-Raf through maintenance of the B-Raf/Raf-1 complex and inhibition by the NF2 tumor suppressor protein. Proc. Natl. Acad. Sci. USA 2006, 103, 4463–4468. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Gallo, K.; Godowski, P.; Hirai, S.I.; Ohno, S.; Zon, L.; Kyriakis, J.M.; Avruch, J. The mixed lineage kinase SPRK phosphorylates and activates the stress-activated protein kinase activator, SEK-1. J. Biol. Chem. 1996, 271, 19025–19028. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.Y.; Kim, B.C.; Xu, Z.; Kim, S.J. Mixed lineage kinase 3 (MLK3)-activated p38 MAP kinase mediates transforming growth factor-beta-induced apoptosis in hepatoma cells. J. Biol. Chem. 2004, 279, 29478–29484. [Google Scholar] [CrossRef] [PubMed]
- Tibbles, L.A.; Ing, Y.L.; Kiefer, F.; Chan, J.; Iscove, N.; Woodgett, J.R.; Lassam, N.J. MLK-3 activates the SAPK/JNK and p38/RK pathways via SEK1 and MKK3/6. EMBO. J. 1996, 15, 7026–7035. [Google Scholar] [PubMed]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Swenson-Fields, K.I.; Sandquist, J.C.; Rossol-Allison, J.; Blat, I.C.; Wennerberg, K.; Burridge, K.; Means, A.R. MLK3 limits activated Gαq signaling to Rho by binding to p63RhoGEF. Mol. Cell 2008, 32, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Gallo, K.A. MLK3 regulates paxillin phosphorylation in chemokine-mediated breast cancer cell migration and invasion to drive metastasis. Cancer Res. 2012, 72, 4130–4140. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, A.; Sakakura, J.; Yagi, R.; Mazaki, Y.; Schaefer, E.; Yano, H.; Sabe, H. Localized suppression of RhoA activity by Tyr31/118-phosphorylated paxillin in cell adhesion and migration. J. Cell Biol. 2002, 159, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Swenson, K.I.; Winkler, K.E.; Means, A.R. A new identity for MLK3 as an NIMA-related, cell cycle–regulated kinase that is localized near centrosomes and influences microtubule organization. Mol. Biol. Cell 2003, 14, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Whitmarsh, A.J. The JIP family of MAPK scaffold proteins. Biochem. Soc. Trans. 2006, 34, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, J.; Whitmarsh, A.J.; Cavanagh, J.; Sharma, M.; Davis, R.J. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol. Cell. Boil. 1999, 19, 7245–7254. [Google Scholar] [CrossRef]
- Verhey, K.J.; Meyer, D.; Deehan, R.; Blenis, J.; Schnapp, B.J.; Rapoport, T.A.; Margolis, B. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol. 2001, 152, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Yoshioka, K.; Akechi, M.; Yamashita, S.; Takamatsu, N.; Sugiyama, K.; Hibi, M.; Nakabeppu, Y.; Shiba, T.; Yamamoto, K.I. JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a Scaffold factor in the JNK signaling pathway. Mol. Cell. Biol. 1999, 19, 7539–7548. [Google Scholar] [CrossRef] [PubMed]
- Kelkar, N.; Standen, C.L.; Davis, R.J. Role of the JIP4 scaffold protein in the regulation of mitogen-activated protein kinase signaling pathways. Mol. Cell. Biol. 2005, 25, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Hadad, M.; Aviram, S.; Darlyuk-Saadon, I.; Cohen-Katsenelson, K.; Whitmarsh, A.J.; Aronheim, A. The association of the JNK scaffold protein, WDR62, with the mixed lineage kinase 3, MLK3. MAP Kinase 2015. [Google Scholar] [CrossRef]
- Hartkamp, J.; Troppmair, J.; Rapp, U.R. The JNK/SAPK activator mixed lineage kinase 3 (MLK3) transforms NIH 3T3 cells in a MEK-dependent fashion. Cancer Res. 1999, 59, 2195–2202. [Google Scholar] [PubMed]
- Sathyanarayana, P.; Barthwal, M.K.; Kundu, C.N.; Lane, M.E.; Bergmann, A.; Tzivion, G.; Rana, A. Activation of the Drosophila MLK by ceramide reveals TNF-α and ceramide as agonists of mammalian MLK3. Mol. Cell 2002, 10, 1527–1533. [Google Scholar] [CrossRef]
- Chen, J.; Miller, E.M.; Gallo, K.A. MLK3 is critical for breast cancer cell migration and promotes a malignant phenotype in mammary epithelial cells. Oncogene 2010, 29, 4399–4411. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Abi Saab, W.F.; Modi, N.; Stewart, A.M.; Liu, J.; Chadee, D.N. Mixed lineage kinase 3 is required for matrix metalloproteinase expression and invasion in ovarian cancer cells. Exp. Cell Res. 2012, 318, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Chadee, D.N.; Kyriakis, J.M. MLK3 is required for mitogen activation of B-Raf, ERK and cell proliferation. Nat. Cell Biol. 2004, 6, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.; Angel, P.; Schorpp-Kistner, M. AP-1 subunits: Quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [Google Scholar] [CrossRef] [PubMed]
- Talotta, F.; Mega, T.; Bossis, G.; Casalino, L.; Basbous, J.; Jariel-Encontre, I.; Piechaczyk, M.; Verde, P. Heterodimerization with FRA-1 cooperates with the ERK pathway to stabilize c-Jun in response to the RAS oncoprotein. Oncogene 2010, 29, 4732–4740. [Google Scholar] [CrossRef] [PubMed]
- Hehner, S.P.; Hofmann, T.G.; Ushmorov, A.; Dienz, O.; Wing-Lan Leung, I.; Lassam, N.; Scheidereit, C.; Droge, W.; Schmitz, M.L. Mixed-lineage kinase 3 delivers CD3/CD28-derived signals into the IkappaB kinase complex. Mol. Cell. Biol. 2000, 20, 2556–2568. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gallo, K.A.; Conrad, S.E. Targeting mixed lineage kinases in ER-positive breast cancer cells leads to G2/M cell cycle arrest and apoptosis. Oncotarget 2013, 4, 1158–1171. [Google Scholar] [CrossRef] [PubMed]
- Pufall, M.A.; Graves, B.J. Autoinhibitory domains: Modular effectors of cellular regulation. Annu. Rev. Cell Dev. Biol. 2002, 18, 421–462. [Google Scholar] [CrossRef] [PubMed]
- Aitio, O.; Hellman, M.; Kazlauskas, A.; Vingadassalom, D.F.; Leong, J.M.; Saksela, K.; Permi, P. Recognition of tandem PxxP motifs as a unique Src homology 3-binding mode triggers pathogen-driven actin assembly. Proc. Natl. Acad. Sci. USA 2010, 107, 21743–21748. [Google Scholar] [CrossRef] [PubMed]
- Burbelo, P.D.; Drechsel, D.; Hall, A. A conserved binding motif defines numerous candidate target proteins for both Cdc42 and Rac GTPases. J. Biol. Chem. 1995, 270, 29071–29074. [Google Scholar] [PubMed]
- Teramoto, H.; Coso, O.A.; Miyata, H.; Igishi, T.; Miki, T.; Gutkind, J.S. Signaling from the small GTP-binding proteins Rac1 and Cdc42 to the c-Jun N-terminal Kinase/Stress-activated protein kinase pathway. J. Biol. Chem. 1996, 271, 27225–27228. [Google Scholar] [CrossRef] [PubMed]
- Böck, B.C.; Vacratsis, P.O.; Qamirani, E.; Gallo, K.A. Cdc42-induced Activation of the Mixed-Lineage Kinase SPRK in Vivo. J. Biol. Chem. 2000, 275, 14231–14241. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Bock, B.C.; Schachter, K.A.; Chao, M.; Gallo, K.A. Cdc42 induces activation loop phosphorylation and membrane targeting of mixed lineage kinase 3. J. Biol. Chem. 2005, 280, 42984–42993. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Yang, J. Functional mechanisms for human tumor suppressors. J. Cancer 2010, 1, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Petrilli, A.M.; Fernandez-Valle, C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, R.D.; Angelo, L.S.; Kurzrock, R. NF2/Merlin in hereditary neurofibromatosis 2 versus cancer: Biologic mechanisms and clinical associations. Oncotarget 2014, 5, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Chadee, D.N. Inhibition of Cdc42-mediated activation of mixed lineage kinase 3 by the tumor suppressor protein merlin. Small GTPases 2010, 1, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L. Chaperoning oncogenes: Hsp90 as a target of geldanamycin. Handb. Exp. Pharmacol. 2006, 172, 259–277. [Google Scholar] [PubMed]
- Jackson, S.E. Hsp90: Structure and function. Top. Curr. Chem. 2013, 328, 155–240. [Google Scholar] [PubMed]
- Blessing, N.A.; Brockman, A.L.; Chadee, D.N. The E3 ligase CHIP mediates ubiquitination and degradation of mixed-lineage kinase 3. Mol. Cell. Biol. 2014, 34, 3132–3143. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chandana, S.R. Inhibition of MLK3 decreases proliferation and increases antiproliferative activity of epidermal growth factor receptor (EGFR) inhibitor in pancreatic cancer cell lines. Cancer Growth Metastasis 2010, 3, 1–9. [Google Scholar]
- Velho, S.; Pinto, A.; Licastro, D.; Oliveira, M.J.; Sousa, F.; Stupka, E.; Seruca, R. Dissecting the signaling pathways associated with the oncogenic activity of MLK3 P252H mutation. BMC Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Velho, S.; Oliveira, C.; Paredes, J.; Sousa, S.; Leite, M.; Matos, P.; Milanezi, F.; Ribeiro, A.S.; Mendes, N.; Licastro, D.; et al. Mixed lineage kinase 3 gene mutations in mismatch repair deficient gastrointestinal tumours. Hum. Mol. Genet. 2010, 19, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.W.; Kim, I.A.; et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod. Pathol. 2014, 27, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Masuda, H.; Zhang, D.; Bartholomeusz, C.; Doihara, H.; Hortobagyi, G.N.; Ueno, N.T. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res. Treat. 2012, 136, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.E.; Furnari, F.B.; Cavenee, W.K. Targeting EGFR for Treatment of Glioblastoma: Molecular Basis to Overcome Resistance. Curr. Cancer Drug Targets 2012, 12, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Gazdar, A.F. Epidermal growth factor receptor inhibition in lung cancer: The evolving role of individualized therapy. Cancer Metastasis Rev. 2010, 29, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Stiegler, A.L.; Boggon, T.J.; Kobayashi, S.; Halmos, B. EGFR-mutated lung cancer: A paradigm of molecular oncology. Oncotarget 2010, 1, 497–514. [Google Scholar] [CrossRef] [PubMed]
- Yewale, C.; Baradia, D.; Vhora, I.; Patil, S.; Misra, A. Epidermal growth factor receptor targeting in cancer: A review of trends and strategies. Biomaterials 2013, 34, 8690–8707. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Yarden, Y. EGF–ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Tebbutt, N.; Pedersen, M.W.; Johns, T.G. Targeting the ERBB family in cancer: Couples therapy. Nat. Rev. Cancer 2013, 13, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Marmor, M.D.; Skaria, K.B.; Yarden, Y. Signal transduction and oncogenesis by ErbB/HER receptors. Int. J. Radiat. Oncol. Biol. Physics. 2004, 58, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Gajria, D.; Chandarlapaty, S. HER2-amplified breast cancer: Mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev. Anticancer Ther. 2011, 11, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zhang, W.; Jiang, T. Oncogene addiction in gliomas: Implications for molecular targeted therapy. J. Exp. Clin. Cancer Res. 2011, 30, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Lam, D. Oncogenic driver mutations in lung cancer. Transl. Respir. Med. 2013, 1, 1–8. [Google Scholar] [CrossRef]
- Chong, C.R.; Janne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.B.; Kumler, I.; Nielsen, D.L. A systematic review of trastuzumab and lapatinib in the treatment of women with brain metastases from HER2-positive breast cancer. Cancer Treat. Rev. 2013, 39, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Fukumoto, Y.; Chaika, N.; Svoboda, R.; Wheelock, M.J.; Johnson, K.R. Collagen I–mediated up-regulation of N-cadherin requires cooperative signals from integrins and discoidin domain receptor 1. J. Cell Boil. 2008, 180, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, M.; Tenca, P.; Frittoli, E.; Faretta, M.; Tocchetti, A.; di Fiore, P.P.; Scita, G. Mechanisms through which Sos-1 coordinates the activation of Ras and Rac. J. Cell Biol. 2002, 156, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Tamás, P.; Solti, Z.; Bauer, P.; Illés, A.; Sipeki, S.; Bauer, A.; Faragó, A.; Downward, J.; Buday, L. Mechanism of epidermal growth factor regulation of Vav2, a guanine nucleotide exchange factor for Rac. J. Biol. Chem. 2003, 278, 5163–5171. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Hu, B.; Vuori, K.; Sarkaria, J.N.; Furnari, F.B.; Cavenee, W.K.; Cheng, S.Y. EGFRvIII stimulates glioma growth and invasion through PKA-dependent serine phosphorylation of Dock180. Oncogene 2014, 33, 2504–2512. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Li, Y.; Yin, Y.; Zhang, W.; Hou, Y.; Zhang, L.; Li, Z.; Xie, B.; Gao, W.Q.; Sarkaria, J.N.; et al. Protein kinase A-dependent phosphorylation of Dock180 at serine residue 1250 is important for glioma growth and invasion stimulated by platelet derived-growth factor receptor alpha. Neurooncology 2015, 17, 832–842. [Google Scholar]
- Zhu, G.; Fan, Z.; Ding, M.; Zhang, H.; Mu, L.; Ding, Y.; Zhang, Y.; Jia, B.; Chen, L.; Chang, Z.; et al. An EGFR/PI3K/AKT axis promotes accumulation of the Rac1-GEF Tiam1 that is critical in EGFR-driven tumorigenesis. Oncogene 2015, 34, 5971–5982. [Google Scholar] [CrossRef] [PubMed]
- Kakinuma, T.; Hwang, S.T. Chemokines, chemokine receptors, and cancer metastasis. J. Leukoc. Biol. 2006, 79, 639–651. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Lang, R.; Wei, J.; Fan, Y.; Cui, L.; Gu, F.; Guo, X.; Pringle, G.A.; Zhang, X.; Fu, L. Increased expression of SDF-1/CXCR4 is associated with lymph node metastasis of invasive micropapillary carcinoma of the breast. Histopathology 2009, 54, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, S.; Yanagawa, T.; Fan, J.; Katoh, R. Expression of CXCR4 and its ligand SDF-1 in intestinal-type gastric cancer is associated with lymph node and liver metastasis. Anticancer Res. 2009, 29, 4751–4758. [Google Scholar] [PubMed]
- Guo, L.; Cui, Z.M.; Zhang, J.; Huang, Y. Chemokine axes CXCL12/CXCR4 and CXCL16/CXCR6 correlate with lymph node metastasis in epithelial ovarian carcinoma. Chin. J. Cancer 2011, 30, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, J.; Cui, Z.M.; Zhao, J.; Zheng, Y. Expression of the CXCL12/CXCR4 and CXCL16/CXCR6 axes in cervical intraepithelial neoplasia and cervical cancer. Chin. J. Cancer 2013, 32, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.Y.; Gao, Z.H.; Qu, X.J. A review of CXCR4/CXCL12 axis in colorectal cancer. Biomed. Aging Pathol. 2014, 4, 285–290. [Google Scholar] [CrossRef]
- Furusato, B.; Mohamed, A.; Uhlen, M.; Rhim, J.S. CXCR4 and cancer. Pathol. Int. 2010, 60, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 Pathway in Cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef] [PubMed]
- Bremer, E. Targeting of the tumor necrosis factor receptor superfamily for cancer immunotherapy. ISRN Oncol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, R.; Ten Hagen, T.L.; Eggermont, A.M. TNF-alpha in cancer treatment: Molecular insights, antitumor effects, and clinical utility. Oncologist 2006, 11, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Sondarva, G.; Kundu, C.N.; Mehrotra, S.; Mishra, R.; Rangasamy, V.; Sathyanarayana, P.; Ray, R.S.; Rana, B.; Rana, A. TRAF2-MLK3 interaction is essential for TNF-alpha-induced MLK3 activation. Cell Res. 2010, 20, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Korchnak, A.C.; Zhan, Y.; Aguilar, M.T.; Chadee, D.N. Cytokine-induced activation of mixed lineage kinase 3 requires TRAF2 and TRAF6. Cell Signal. 2009, 21, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Rana, B.; Mishra, R.; Sondarva, G.; Rangasamy, V.; Das, S.; Viswakarma, N.; Kanthasamy, A. Mixed lineage kinase-c-Jun N-terminal kinase axis: A potential therapeutic target in cancer. Genes Cancer 2013, 4, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Stronach, B.; Perrimon, N. Activation of the JNK pathway during dorsal closure in Drosophila requires the mixed lineage kinase, slipper. Genes Dev. 2002, 16, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal adhesion kinase: In command and control of cell motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Webb, D.J.; Horwitz, A.R. Cell migration at a glance. J. Cell Sci. 2005, 118, 4917–4919. [Google Scholar] [CrossRef] [PubMed]
- Nobes, C.D.; Hall, A. Rho, rac and cdc42 GTPases: Regulators of actin structures, cell adhesion and motility. Biochem. Soc. Trans. 1995, 23, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Jacobson, K.; Schaller, M.D. A role for JNK-paxillin signaling in cell migration. Cell Cycle 2004, 3, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Wildenberg, G.A.; Dohn, M.R.; Carnahan, R.H.; Davis, M.A.; Lobdell, N.A.; Settleman, J.; Reynolds, A.B. p120-Catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell 2006, 127, 1027–1039. [Google Scholar] [CrossRef] [PubMed]
- Royer, C.; Lu, X. Epithelial cell polarity: A major gatekeeper against cancer? Cell Death Differ. 2011, 18, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The Collagen Family. Cold Spring Harbor Perspect. Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Exposito, J.Y.; Valcourt, U.; Cluzel, C.; Lethias, C. The Fibrillar Collagen Family. Int. J. Mol. Sci. 2010, 11, 407–426. [Google Scholar] [CrossRef] [PubMed]
- Tanjore, H.; Kalluri, R. The Role of Type IV Collagen and Basement Membranes in Cancer Progression and Metastasis. Am. J. Pathol. 2006, 168, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Abi Saab, W.F.; Brown, M.S.; Chadee, D.N. MLK4β functions as a negative regulator of MAPK signaling and cell invasion. Oncogenesis 2012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lu, L.; Xiong, Y.; Qin, W.; Zhang, Y.; Qian, Y.; Jiang, H.; Liu, W. MLK3 promotes melanoma proliferation and invasion and is a target of microRNA-125b. Clin. Exp. Dermatol. 2014, 39, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Yu, M.C.; Tung, W.H.; Chen, T.T.; Yu, C.C.; Weng, C.M.; Tsai, Y.J.; Bai, K.J.; Hong, C.Y.; Chien, M.H.; et al. Connective tissue growth factor induces collagen I expression in human lung fibroblasts through the RAC1/MLK3/JNK/AP-1 pathway. Biochim. Biophys. Acta 2013, 1833, 2823–2833. [Google Scholar] [CrossRef] [PubMed]
- Cronan, M.R.; Nakamura, K.; Johnson, N.L.; Granger, D.A.; Cuevas, B.D.; Wang, J.G.; Mackman, N.; Scott, J.E.; Dohlman, H.G.; Johnson, G.L. Defining MAP3 kinases required for MDA-MB-231 cell tumor growth and metastasis. Oncogene 2012, 31, 3889–3900. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Woessner, J.F. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991, 5, 2145–2154. [Google Scholar] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.; Der, C. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed]
- Kebebew, E.; Weng, J.; Bauer, J.; Ranvier, G.; Clark, O.H.; Duh, Q.-Y.; Shibru, D.; Bastian, B.; Griffin, A. The prevalence and prognostic value of BRAF mutation in thyroid cancer. Ann. Surg. 2007, 246, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Larkin, J. Vemurafenib: A new treatment for BRAF-V600 mutated advanced melanoma. Cancer Manag. Res. 2012, 4, 243–252. [Google Scholar] [PubMed]
- Ozanne, B.W.; Spence, H.J.; McGarry, L.C.; Hennigan, R.F. Transcription factors control invasion: AP-1 the first among equals. Oncogene 2007, 26, 1–10. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rattanasinchai, C.; Gallo, K.A. MLK3 Signaling in Cancer Invasion. Cancers 2016, 8, 51. https://doi.org/10.3390/cancers8050051
Rattanasinchai C, Gallo KA. MLK3 Signaling in Cancer Invasion. Cancers. 2016; 8(5):51. https://doi.org/10.3390/cancers8050051
Chicago/Turabian StyleRattanasinchai, Chotirat, and Kathleen A. Gallo. 2016. "MLK3 Signaling in Cancer Invasion" Cancers 8, no. 5: 51. https://doi.org/10.3390/cancers8050051
APA StyleRattanasinchai, C., & Gallo, K. A. (2016). MLK3 Signaling in Cancer Invasion. Cancers, 8(5), 51. https://doi.org/10.3390/cancers8050051