
Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy

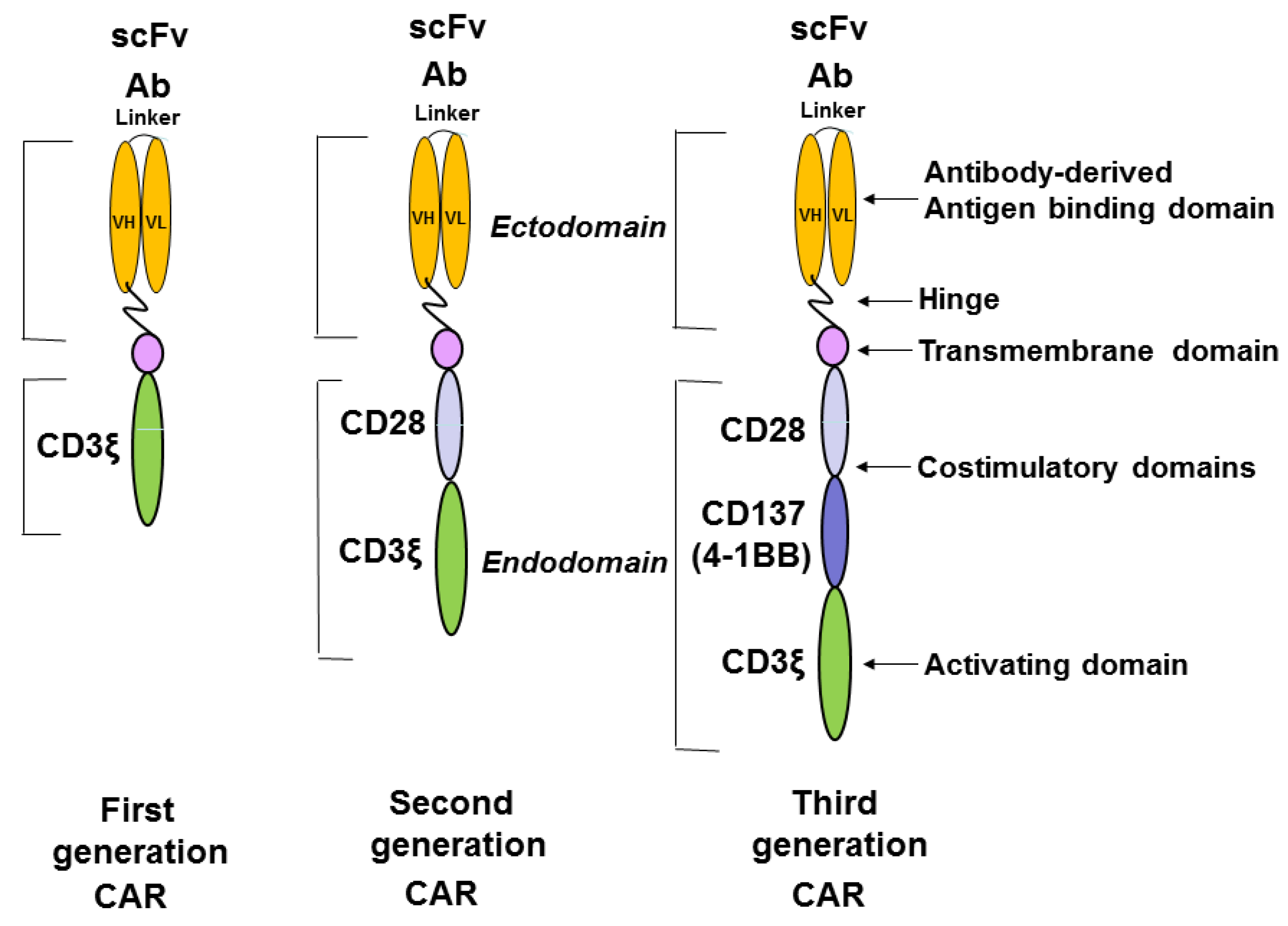
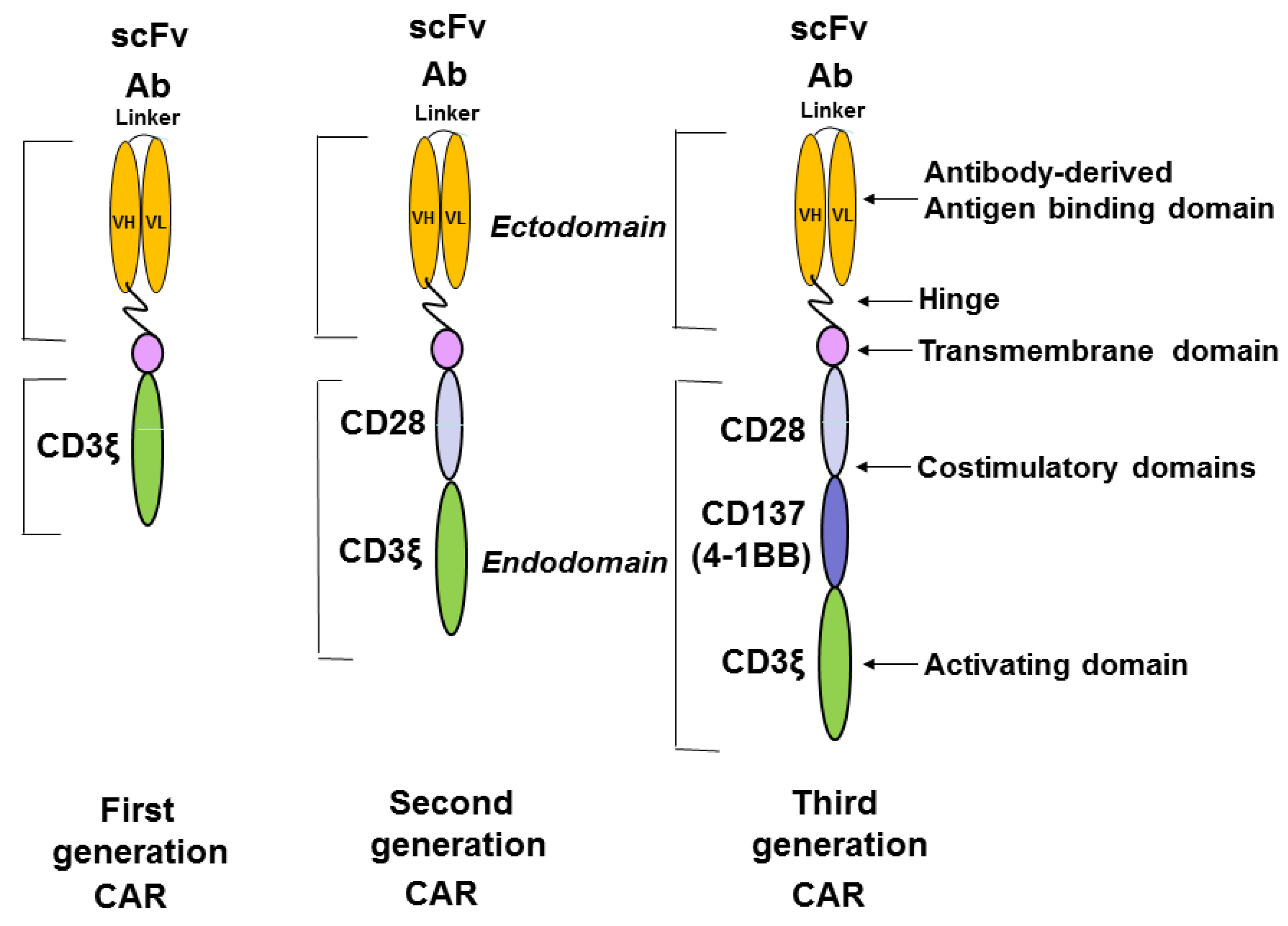{kind=link}
{kind=link}
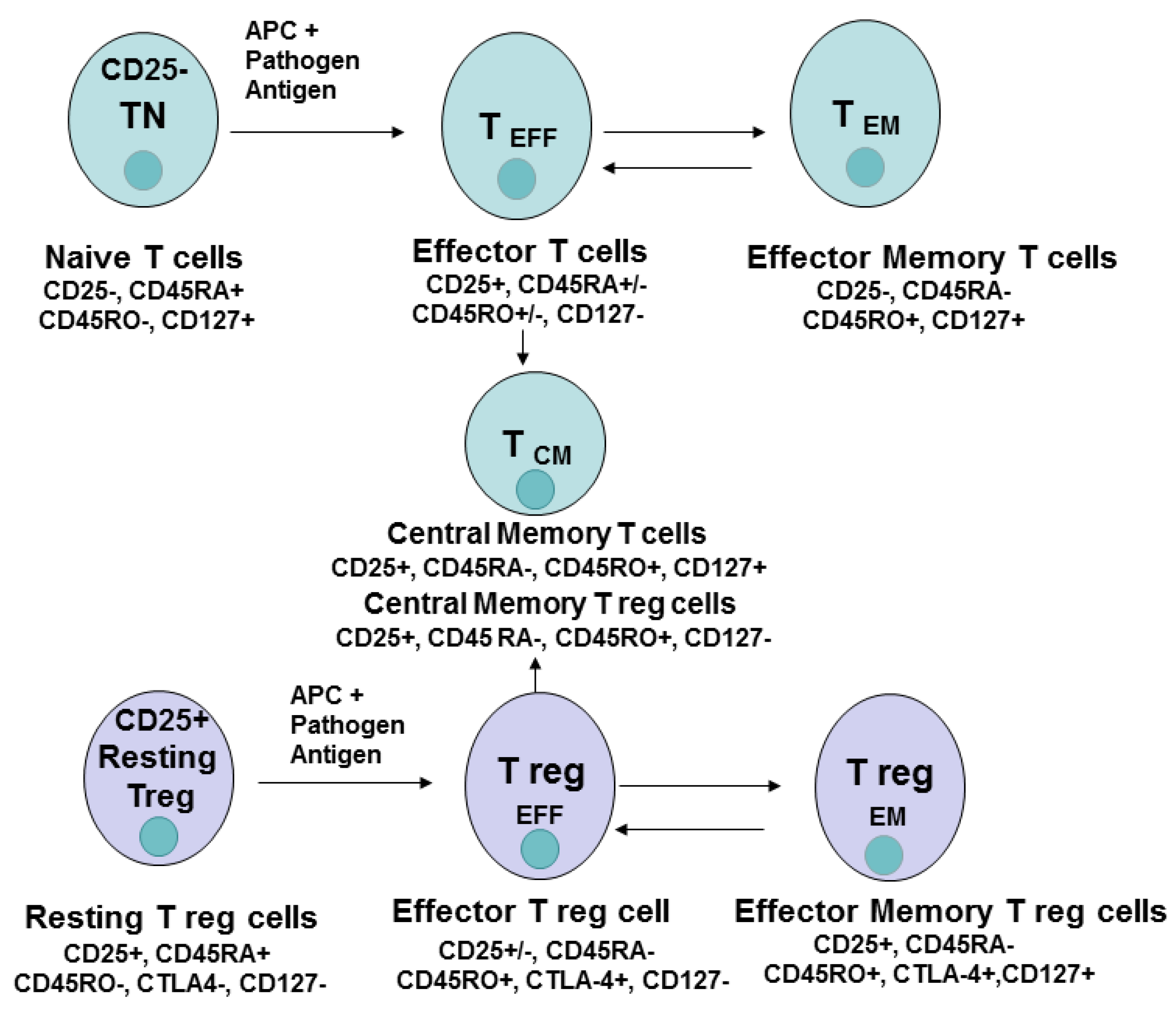{kind=link}
{kind=link}
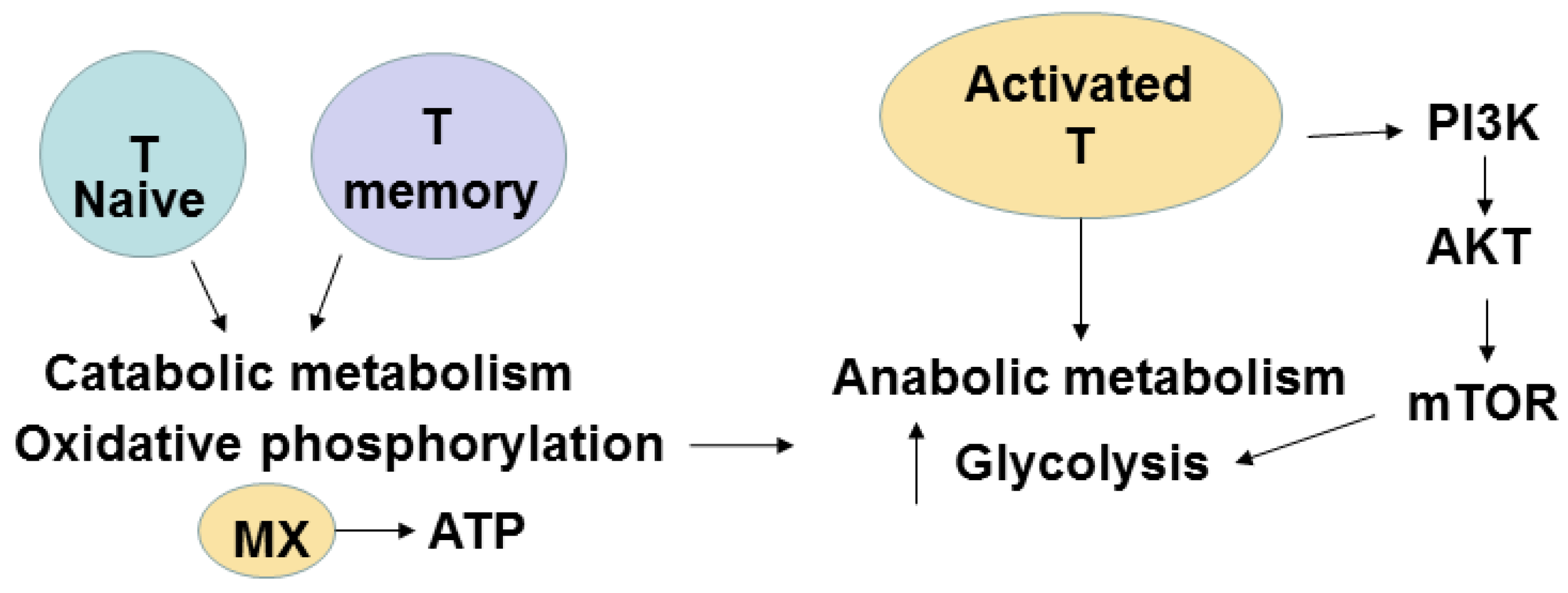{kind=link}
{kind=link}
Abstract
:1. Introduction
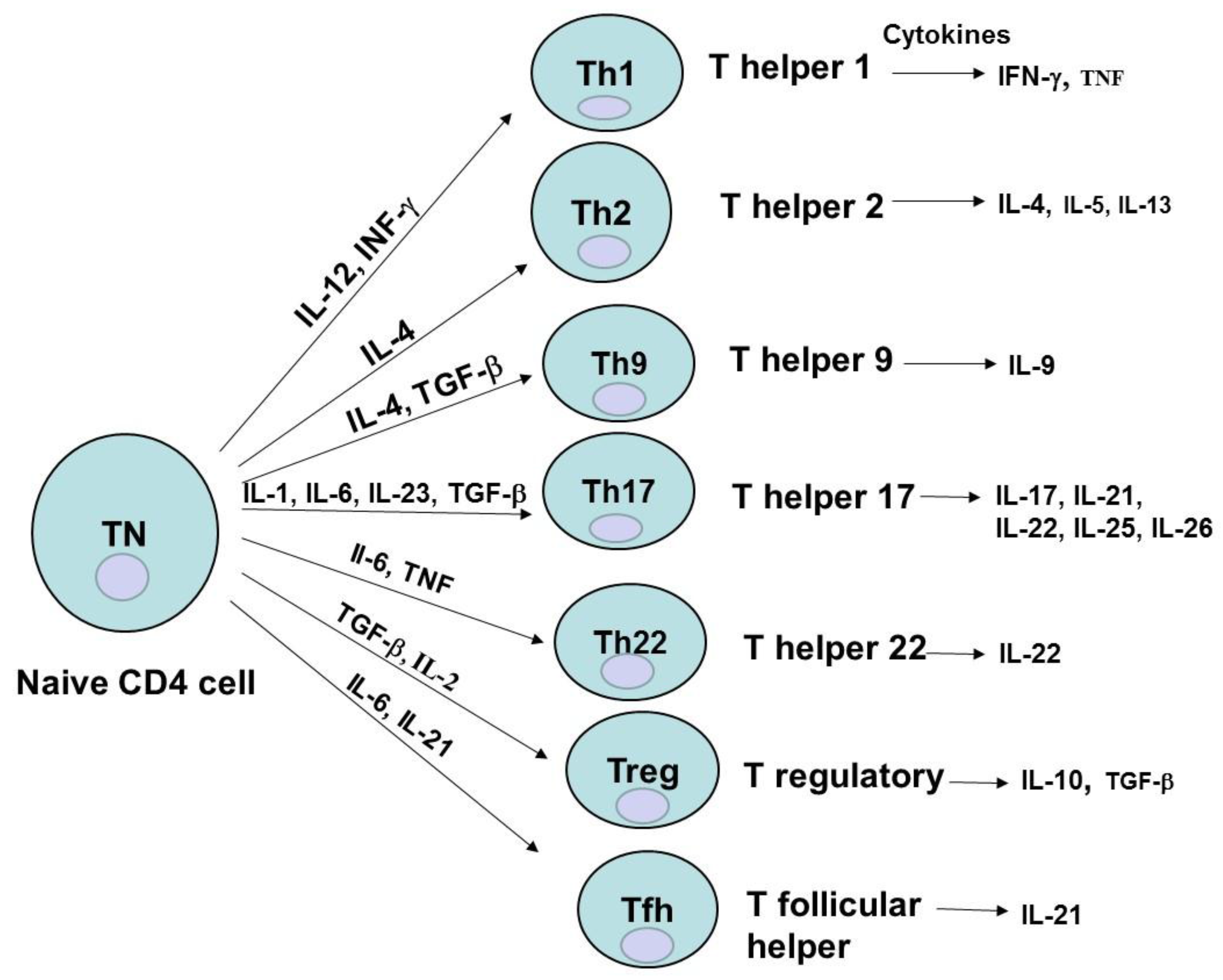
2. CD4 Cell Subsets
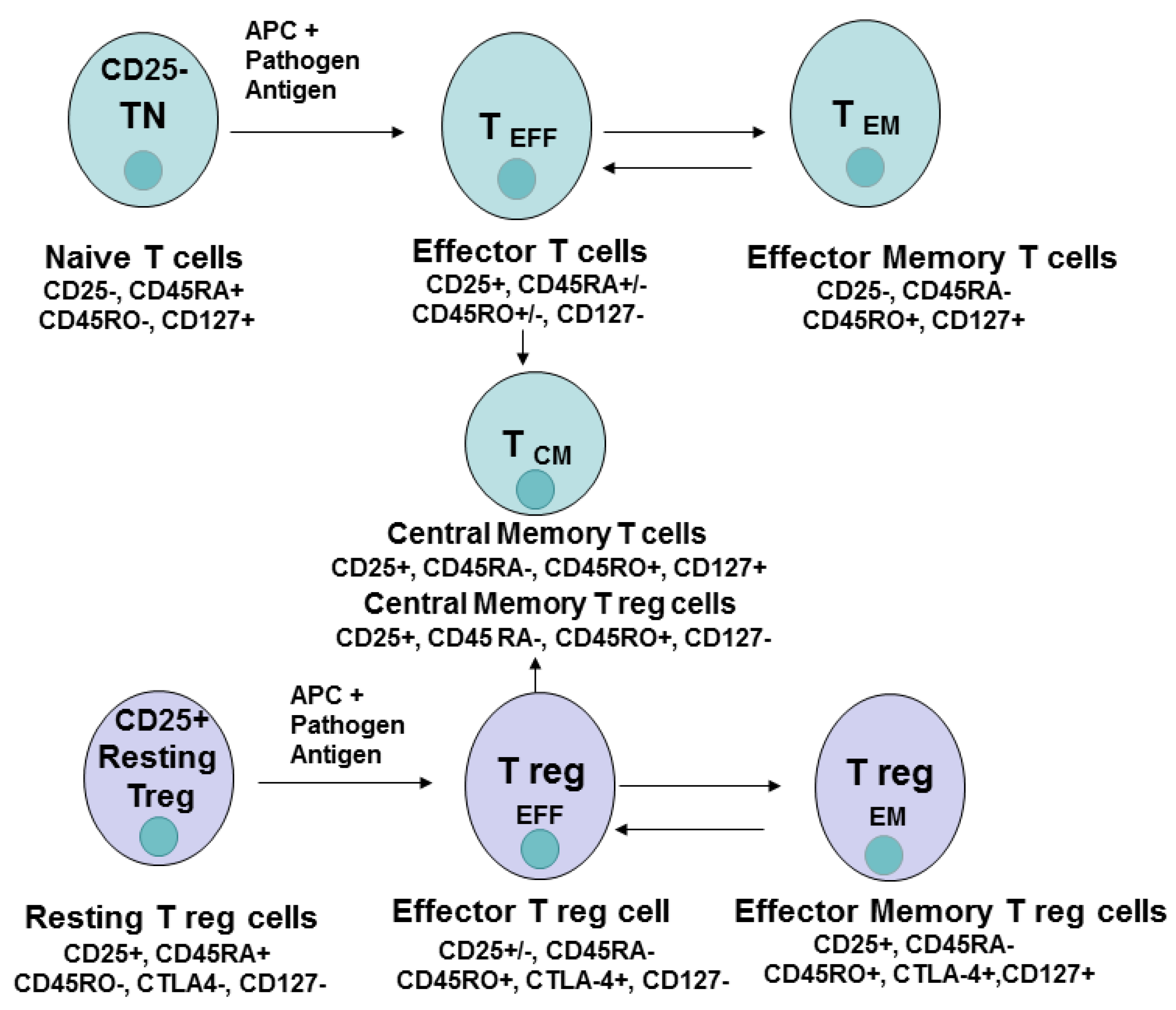
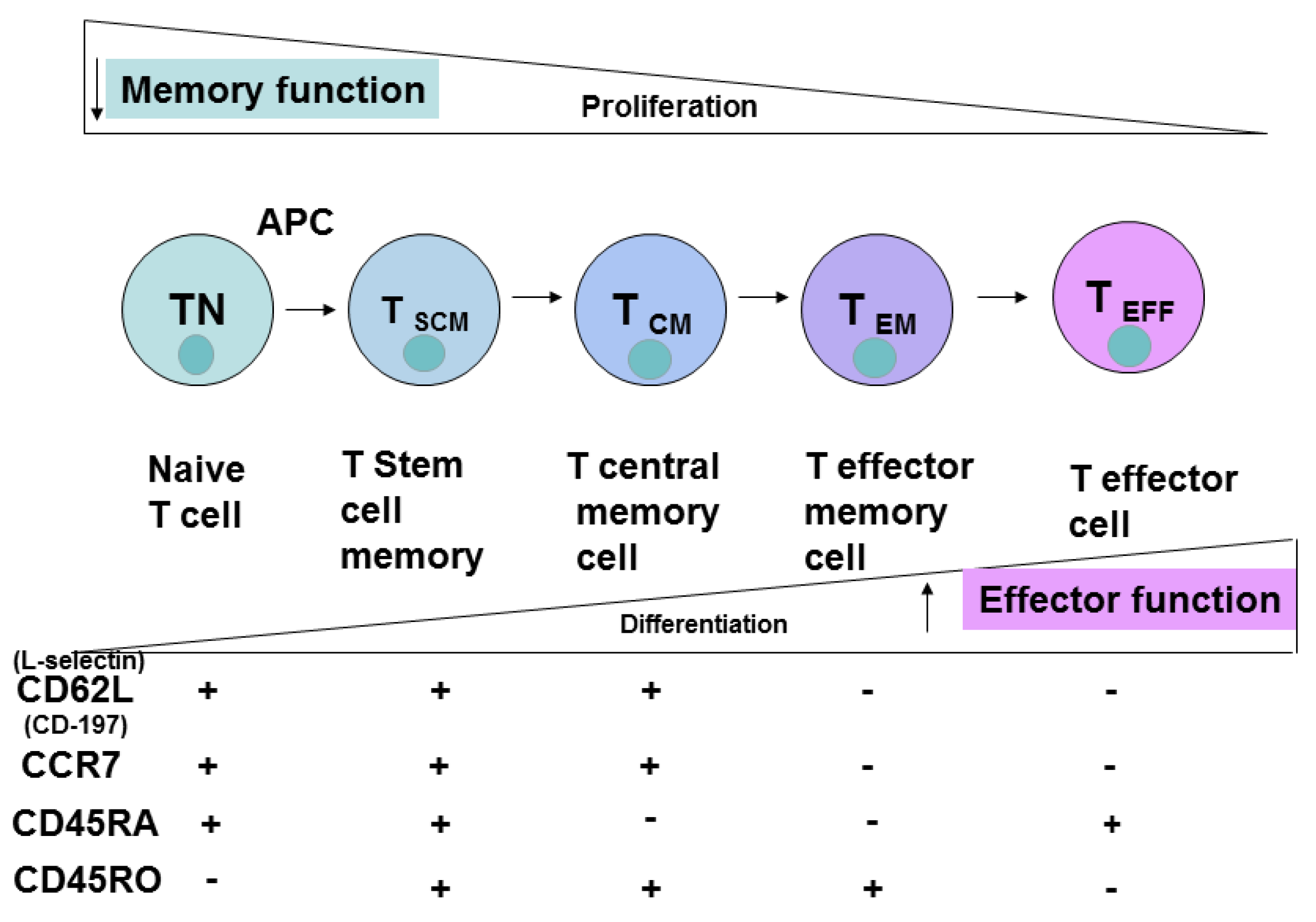
3. CD4 Cell Differentiation, Memory, Effector Cells
4. CD8 Cell Subsets and Cell Differentiation
5. Extracellular T Cell Markers
6. Epigenetic and Genetic Profiles
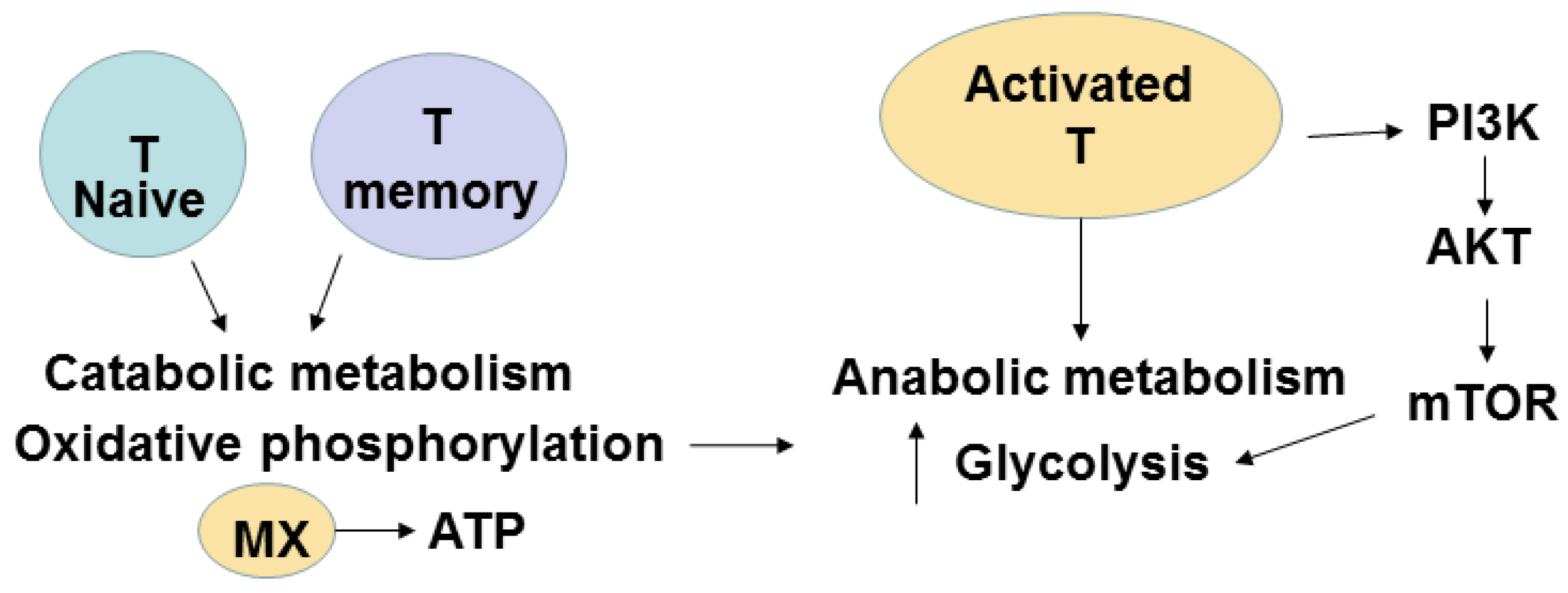
7. Metabolic Pathways of T Cells
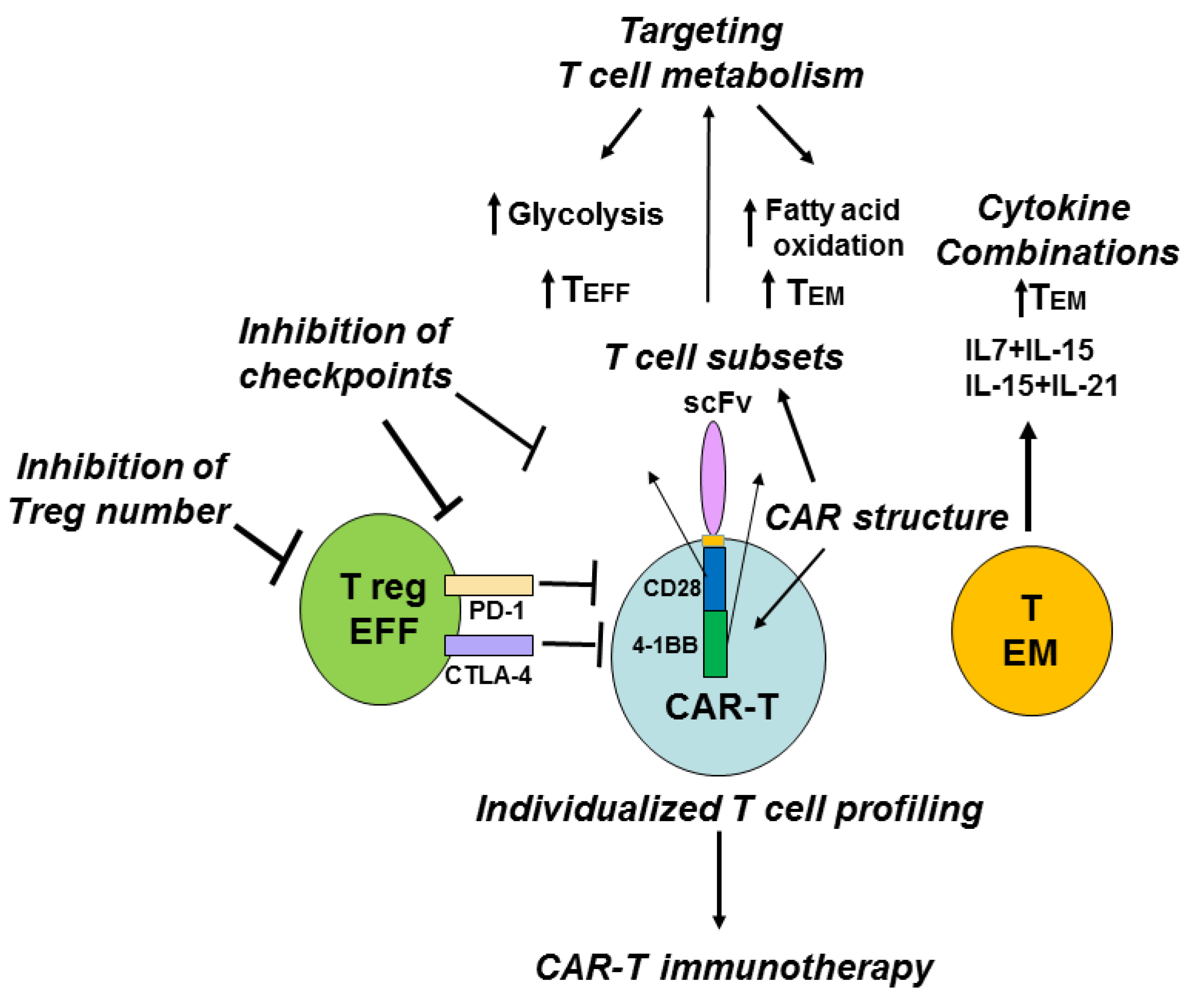
8. Role of Different T Cell Subsets, Treg Cells, Immune Checkpoints, Metabolic Pathways, Cytokines and T Cell Profiling in Potential Improvement of CAR-T Immunotherapy
9. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maus, M.V.; Grupp, S.A.; Porter, D.L.; June, C.H. Antibody-modified T cells: Cars take the front seat for hematologic malignancies. Blood 2014, 123, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Kakarla, S.; Gottschalk, S. Car T cells for solid tumors: Armed and ready to go? Cancer J. 2014, 20, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G. The emergence of T-bodies/car T cells. Cancer J. 2014, 20, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, M.; Ramos, C.A.; Durett, A.; Liu, E.; Dakhova, O.; Liu, H.; Creighton, C.J.; Gee, A.P.; Heslop, H.E.; et al. Closely related T-memory stem cells correlate with in vivo expansion of car.CD19-T cells and are preserved by IL-7 and IL-15. Blood 2014, 123, 3750–3759. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Klebanoff, C.A.; Restifo, N.P. Paths to stemness: Building the ultimate antitumour T cell. Nat. Rev. Cancer 2012, 12, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Liadi, I.; Singh, H.; Romain, G.; Rey-Villamizar, N.; Merouane, A.; Adolacion, J.R.; Kebriaei, P.; Huls, H.; Qiu, P.; Roysam, B.; et al. Individual motile CD4(+) T cells can participate in efficient multikilling through conjugation to multiple tumor cells. Cancer Immunol. Res. 2015, 3, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified Tcells derived from defined CD8(+) and CD4(+) subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Turovskaya, O.; Kim, G.; Madan, R.; Karp, C.L.; Cheroutre, H.; Kronenberg, M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 2009, 10, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Cheadle, E.J.; Sheard, V.; Rothwell, D.G.; Bridgeman, J.S.; Ashton, G.; Hanson, V.; Mansoor, A.W.; Hawkins, R.E.; Gilham, D.E. Differential role of Th1 and tTh2 cytokines in autotoxicity driven by CD19-specific second-generation chimeric antigen receptor T cells in a mouse model. J. Immunol. 2014, 192, 3654–3665. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. Car T cells transform to trucks: Chimeric antigen receptor-redirected T cells engineered to deliver inducible IL-12 modulate the tumour stroma to combat cancer. Cancer Immunol. Immunother. 2012, 61, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Kopecky, C.; Hombach, A.A.; Abken, H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011, 71, 5697–5706. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, M.D.; Way, S.S.; Abbas, A.K. Regulatory T cell memory. Nat. Rev. Immunol. 2016, 16, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Jensen, M.C.; Lansdorp, P.M.; Gough, M.; Elliott, C.; Riddell, S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Investig. 2008, 118, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Moeller, M.; Haynes, N.M.; Kershaw, M.H.; Jackson, J.T.; Teng, M.W.; Street, S.E.; Cerutti, L.; Jane, S.M.; Trapani, J.A.; Smyth, M.J.; et al. Adoptive transfer of gene-engineered CD4+ helper T cells induces potent primary and secondary tumor rejection. Blood 2005, 106, 2995–3003. [Google Scholar] [CrossRef] [PubMed]
- Shedlock, D.J.; Shen, H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science 2003, 300, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Arvey, A.; Chinen, T.; van der Veeken, J.; Gasteiger, G.; Rudensky, A.Y. Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 2014, 158, 749–763. [Google Scholar] [CrossRef] [PubMed]
- Best, J.A.; Blair, D.A.; Knell, J.; Yang, E.; Mayya, V.; Doedens, A.; Dustin, M.L.; Goldrath, A.W.; The Immunological Genome Project Consortium. Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat. Immunol. 2013, 14, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Zediak, V.P.; Johnnidis, J.B.; Wherry, E.J.; Berger, S.L. Cutting edge: Persistently open chromatin at effector gene loci in resting memory CD8+ T cells independent of transcriptional status. J. Immunol. 2011, 186, 2705–2709. [Google Scholar] [CrossRef] [PubMed]
- Van der Windt, G.J.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, M.; Liu, J.; Mehta, G.U.; Patel, S.J.; Roychoudhuri, R.; Crompton, J.G.; Klebanoff, C.A.; Ji, Y.; Li, P.; Yu, Z.; et al. Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. 2016, 23, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013. [Google Scholar] [CrossRef] [PubMed]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Abken, H. Adoptive therapy with car redirected T cells: The challenges in targeting solid tumors. Immunotherapy 2015, 7, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Weber, M. Cancer-killing car therapies gain speed. Cancer Discov. 2015. [Google Scholar] [CrossRef]
- Abate-Daga, D.; Rosenberg, S.A.; Morgan, R.A. Pancreatic cancer: Hurdles in the engineering of CAR-based immunotherapies. Oncoimmunology 2014. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Eshhar, Z. Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: Counteracting off-tumor toxicities for safe car T cell therapy. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 59–83. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Qiao, J.; Fu, Y.X. Immunotherapy and tumor microenvironment. Cancer Lett. 2016, 370, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Restifo, N.P. Sorting through subsets: Which T-cell populations mediate highly effective adoptive immunotherapy? J. Immunother. 2012, 35, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Berger, C.; Sommermeyer, D.; Hanafi, L.A.; Pender, B.; Robinson, E.M.; Melville, K.; Budiarto, T.M.; Steevens, N.N.; Chaney, C.; et al. Anti-CD19 chimeric antigen receptor-modified T cell therapy for B Cell non-hodgkin lymphoma and chronic lymphocytic leukemia: Fludarabine and cyclophosphamide lymphodepletion improves in vivo expansion and persistence of CAR-T cells and clinical outcomes. In Proceedings of the 57th American Society of Hematology (ASH) Annual Meeting, Orlando, FL, USA, 5–8 December 2015.
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Sommermeyer, D.; Pender, B.; Robinson, E.M.; Melville, K.; Budiarto, T.M.; Steevens, N.N.; Chaney, C.; et al. Addition of fludarabine to cyclophosphamide lymphodepletion improves in vivo expansion of CD19 chimeric antigen receptor-modified T cells and clinical outcome in adults with B Cell acute lymphoblastic leukemia. In Proceedings of the 57th American Society of Hematology (ASH) Annual Meeting, Orlando, FL, USA, 5–8 December 2015.
- Popplewell, L.; Wang, X.; Naranjo, A.; Blanchard, S.; Wagner, J.; Wong, C.L.; Urak, R.; Chang, W.C.; Khaled, S.K.; Siddiqi, T.; et al. Phase I studies of cellular immunotherapy using central memory derived-CD19-specific T cells following autologous stem cell transplantation for patients with high-risk intermediate grade B-lineage non-hodgkin lymphoma. In Proceedings of the 57th American Society of Hematology (ASH) Annual Meeting, Orlando, FL, USA, 5–8 December 2015.
- Kofler, D.M.; Chmielewski, M.; Rappl, G.; Hombach, A.; Riet, T.; Schmidt, A.; Hombach, A.A.; Wendtner, C.M.; Abken, H. CD28 costimulation impairs the efficacy of a redirected T-cell antitumor attack in the presence of regulatory T cells which can be overcome by preventing Lck activation. Mol. Ther. 2011, 19, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Rosenberg, S.A. IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood 2006, 107, 2409–2414. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Ahmadzadeh, M.; Lu, Y.C.; Liewehr, D.J.; Dudley, M.E.; Liu, F.; Schrump, D.S.; Steinberg, S.M.; Rosenberg, S.A.; Robbins, P.F. Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood 2012, 119, 5688–5696. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J., Jr. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; McGettigan, S.E.; Frigault, M.J.; Lee, J.; Posey, A.D., Jr.; Scholler, J.; Scholler, N.; et al. ICOS-based chimeric antigen receptors program bipolar Th17/Th1 cells. Blood 2014, 124, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Lanitis, E.; Poussin, M.; Hagemann, I.S.; Coukos, G.; Sandaltzopoulos, R.; Scholler, N.; Powell, D.J., Jr. Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol. Ther. 2012, 20, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Condomines, M.; Arnason, J.; Benjamin, R.; Gunset, G.; Plotkin, J.; Sadelain, M. Tumor-targeted human t cells expressing CD28-based chimeric antigen receptors circumvent CTLA-4 inhibition. PLoS ONE 2015, 10, e0130518. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Kershaw, M.H.; Darcy, P.K. Blockade of PD-1 immunosuppression boosts CAR T-cell therapy. Oncoimmunology 2013. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Finkelstein, S.E.; Klebanoff, C.A.; Antony, P.A.; Palmer, D.C.; Spiess, P.J.; Hwang, L.N.; Yu, Z.; Wrzesinski, C.; Heimann, D.M.; et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 2005, 202, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in car T cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Ji, Y.; Gattinoni, L.; Zhang, L.; Yu, Z.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer Immunol. Immunother. 2013, 62, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Lamers, C.H.; van Steenbergen-Langeveld, S.; van Brakel, M.; Groot-van Ruijven, C.M.; van Elzakker, P.M.; van Krimpen, B.; Sleijfer, S.; Debets, R. T cell receptor-engineered T cells to treat solid tumors: T cell processing toward optimal T cell fitness. Hum. Gene Ther. Methods 2014, 25, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Finkelstein, S.E.; Surman, D.R.; Lichtman, M.K.; Gattinoni, L.; Theoret, M.R.; Grewal, N.; Spiess, P.J.; Antony, P.A.; Palmer, D.C.; et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1969–1974. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T.; Brown, M.P. Different cytokine and stimulation conditions influence the expansion and immune phenotype of third-generation chimeric antigen receptor T cells specific for tumor antigen GD2. Cytotherapy 2015, 17, 487–495. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. https://doi.org/10.3390/cancers8030036
Golubovskaya V, Wu L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers. 2016; 8(3):36. https://doi.org/10.3390/cancers8030036
Chicago/Turabian StyleGolubovskaya, Vita, and Lijun Wu. 2016. "Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy" Cancers 8, no. 3: 36. https://doi.org/10.3390/cancers8030036
APA StyleGolubovskaya, V., & Wu, L. (2016). Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers, 8(3), 36. https://doi.org/10.3390/cancers8030036