
Targeted Therapy in Locally Advanced and Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma (LA-R/M HNSCC)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
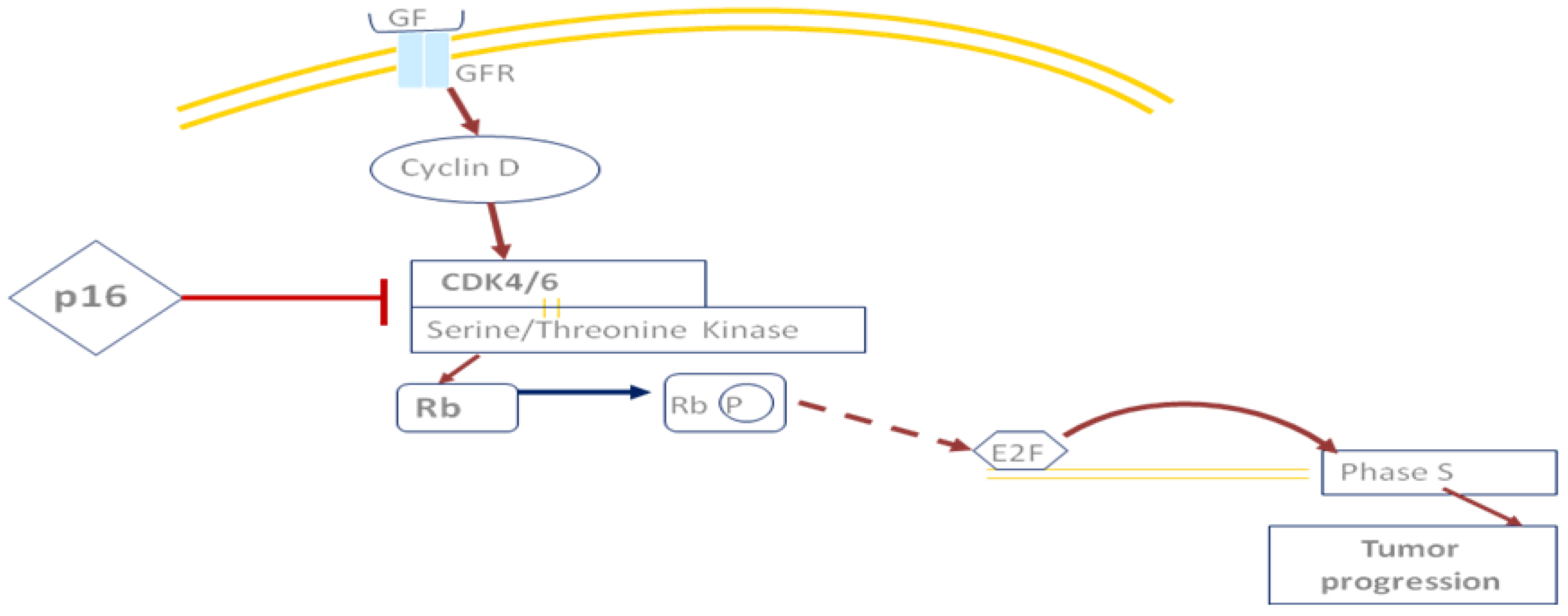
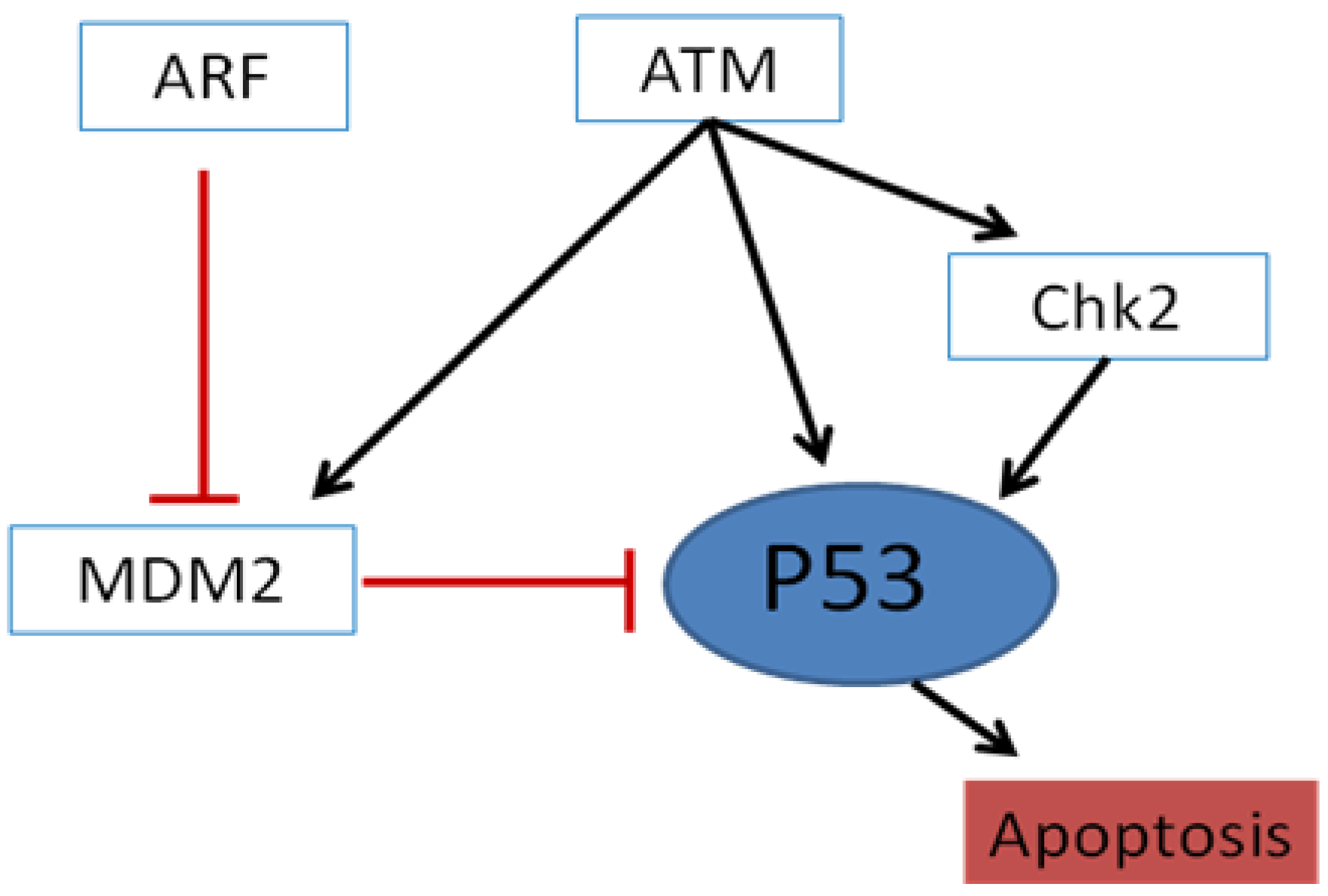
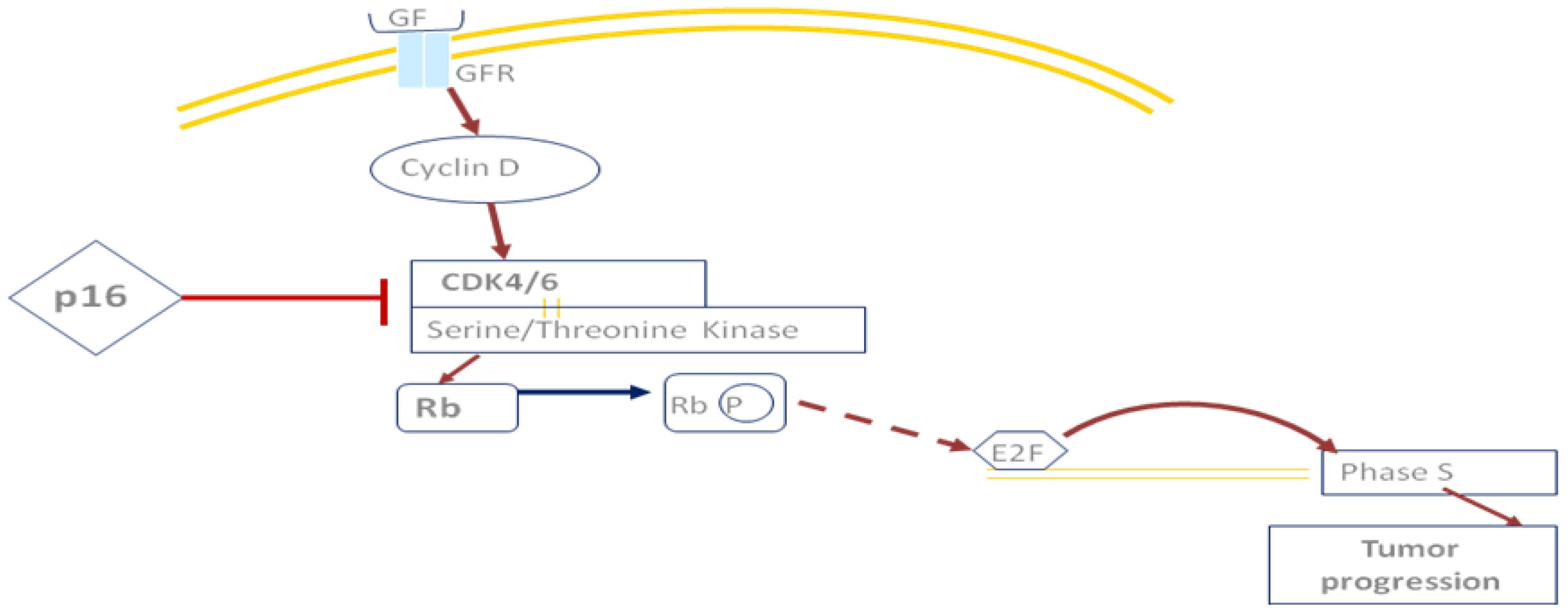
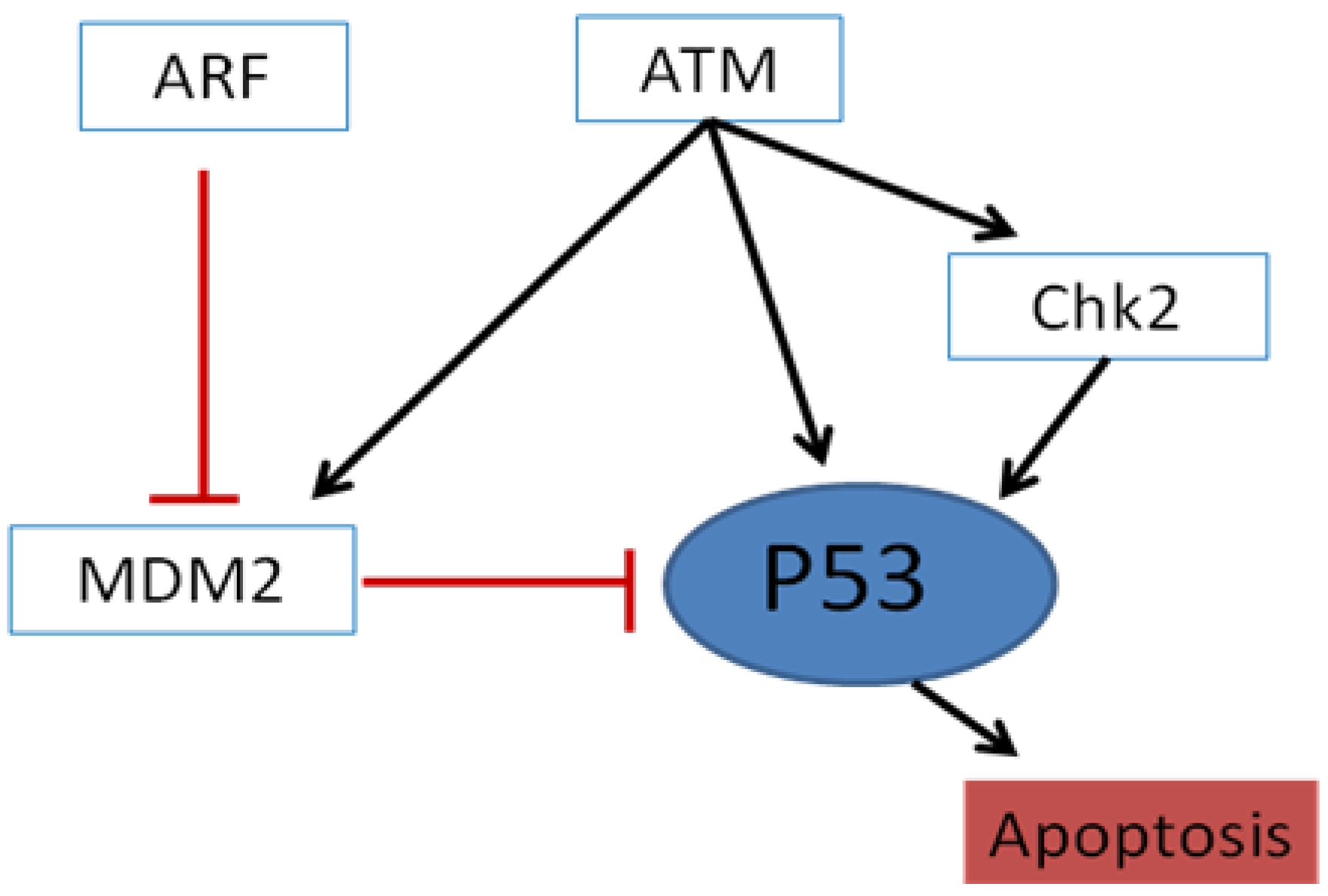
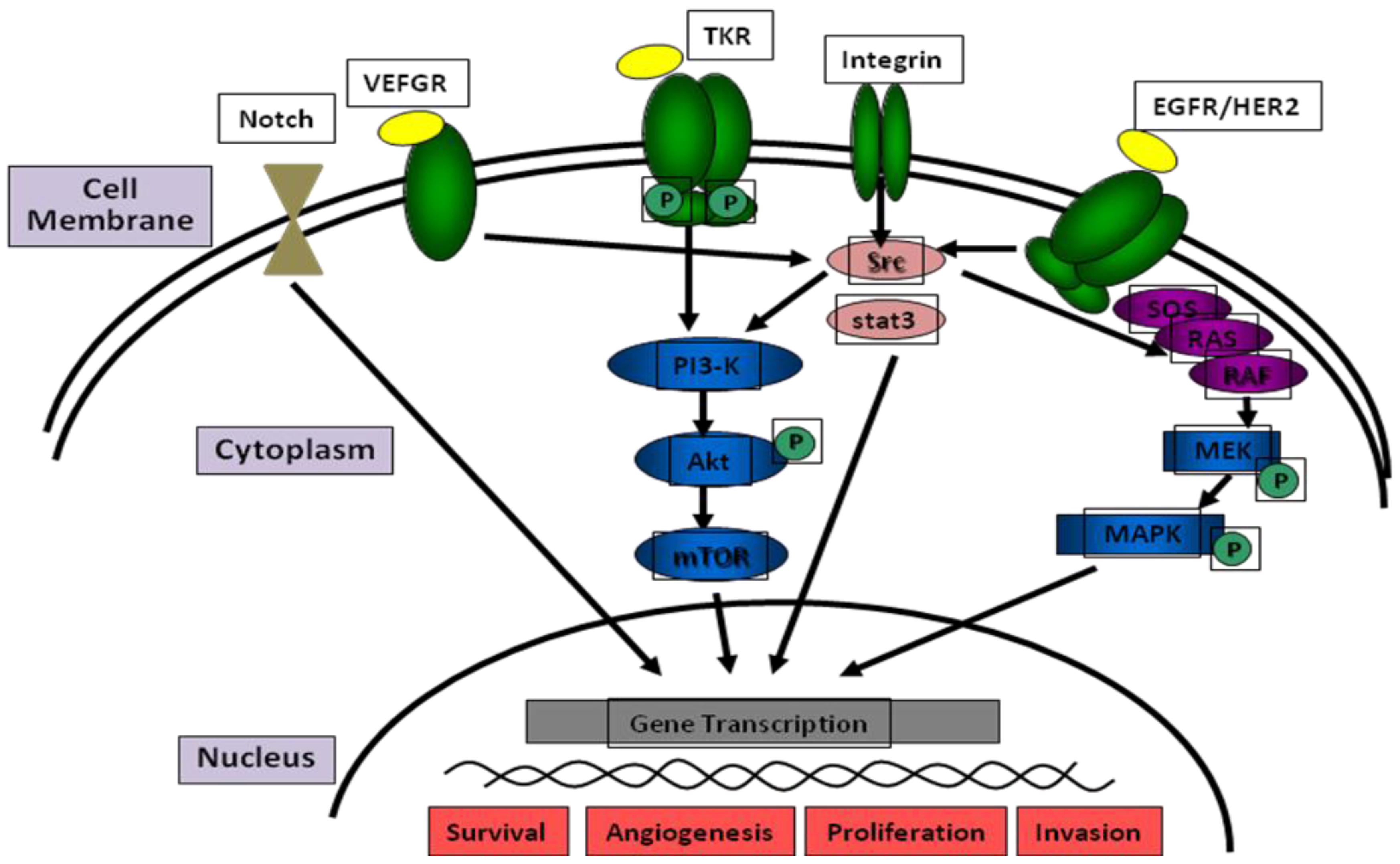
2. The HNSCC Genome and Molecular Pathways
3. Clinical Evidence for Targeted Treatments for HNSCC
3.1. EGFR
3.1.1. Anti-EGFR Monoclonal Antibodies: Cetuximab
3.1.2. Cetuximab and Radiotherapy
3.1.3. Cetuximab as a Single Agent and in Combination with Chemotherapy in Recurrent/Metastatic SCCHN
3.1.4. Actual Regimens
3.1.5. Anti-EGFR non-Cetuximab Monoclonal Antibodies
3.1.6. Small Molecule Tyrosine Kinase Inhibitors (TKIs)
3.2. VEGF Pathway: Targeting VEGF and PDGFR
3.3. Targeting the PI3K Pathway
3.3.1. mTOR Inhibitors
3.3.2. Pan-PI3K Inhibitors
3.4. Targeting Src
3.5. Promising Pathways
4. Inmmunotherapy
4.1. Introduction
4.2. Immunotherapy Approaches
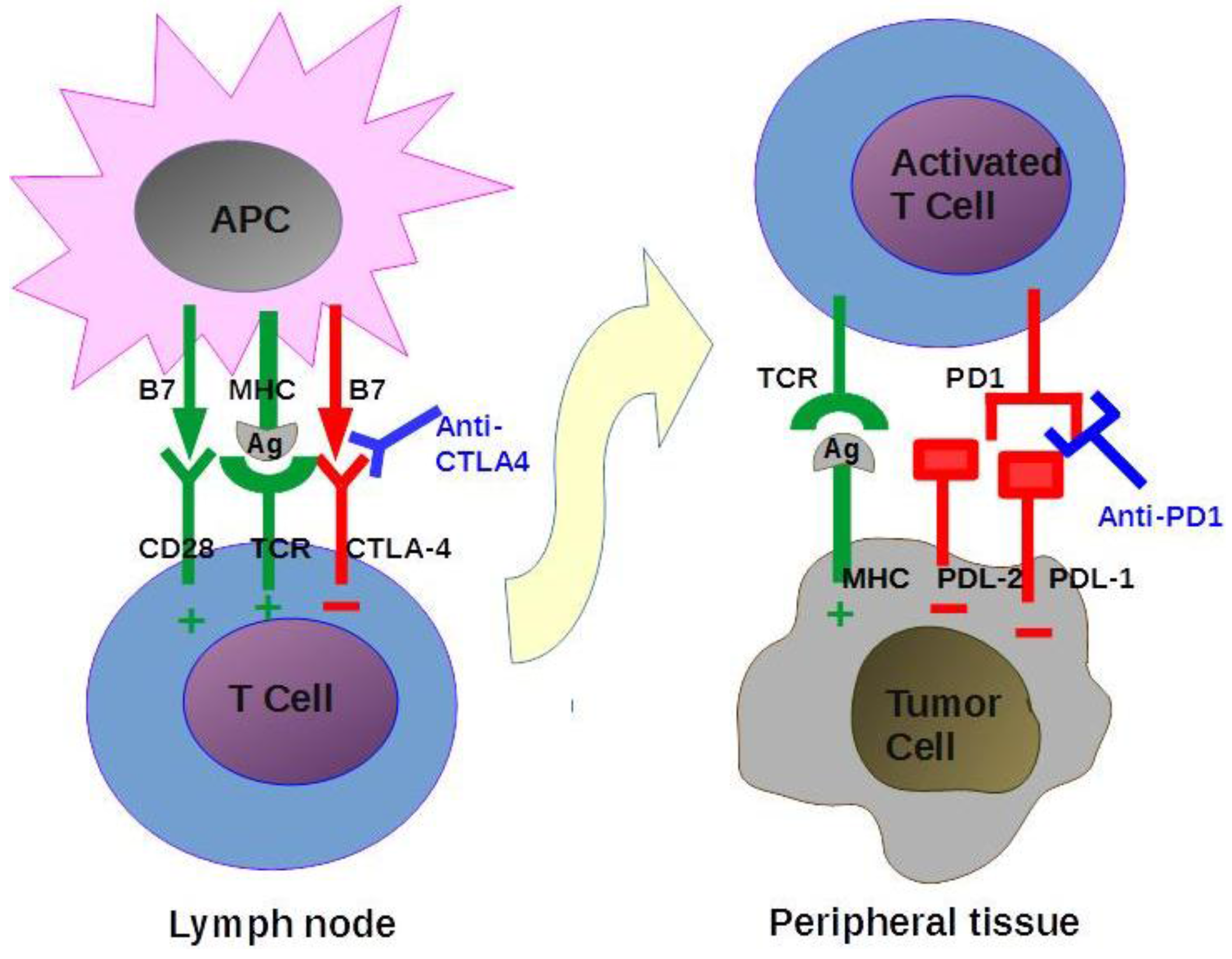
4.2.1. Immune Checkpoint Modulation
4.2.2. Cytokine Therapies
4.2.3. Adoptive T Cell Therapies
4.2.4. Cancer Vaccines
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Vokes, E.E.; Weichselbaum, R.R.; Lippman, S.M.; Hong, W.K. Head and neck cancer. N. Engl. J. Med. 1993, 328, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Adelstein, D.J.; Lavertu, P.; Saxton, J.P.; Secic, M.; Wood, B.G.; Wanamaker, J.R.; Eliachar, I.; Strome, M.; Larto, M.A. Mature results of a phase III randomized trial comparing concurrent chemoradiotherapy with radiation therapy alone in patients with stage III and IV squamous cell carcinoma of the head and neck. Cancer 2000, 88, 876–883. [Google Scholar] [CrossRef]
- Renan, M.J. How many mutations are required for tumorigenesis? Implications from human cancer data. Mol. Carcinog. 1993, 7, 139–146. [Google Scholar] [PubMed]
- Van Dyke, D.L.; Worsham, M.J.; Benninger, M.S.; Krause, C.J.; Baker, S.R.; Wolf, G.T.; Drumheller, T.; Tilley, B.C.; Carey, T.E. Recurrent cytogenetic abnormalities in squamous cell carcinomas of the head and neck region. Genes Chromosomes Cancer 1994, 9, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar]
- Okami, K.; Reed, A.L.; Cairns, P.; Koch, W.M.; Westra, W.H.; Wehage, S.; Jen, J.; Sidransky, D. Cyclin D1 amplification is independent of p16 inactivation in head and neck squamous cell carcinoma. Oncogene 1999, 18, 3541–3545. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Califano, J.A. Molecular biology and immunology of head and neck cancer. Surg. Oncol. Clin. N. Am. 2015, 24, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Isaacsson Velho, P.H.; Castro, G., Jr.; Chung, C.H. Targeting the PI3K Pathway in Head and Neck Squamous Cell Carcinoma. Am. Soc. Clin. Oncol. Educ. Book 2015, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Psyrri, A.; Seiwert, T.Y.; Jimeno, A. Molecular pathways in head and neck cancer: EGFR, PI3K, and more. Am. Soc. Clin. Oncol. Educ. Book 2013, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Pezzuto, F.; Buonaguro, L.; Caponigro, F.; Ionna, F.; Starita, N.; Annunziata, C.; Buonaguro, F.M.; Tornesello, M.L. Update on Head and Neck Cancer: Current Knowledge on Epidemiology, Risk Factors, Molecular Features and Novel Therapies. Oncology 2015, 89, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Nagatsuka, H.; Ishiwari, Y.; Tsujigiwa, H.; Nakano, K.; Nagai, N. Quantitation of epidermal growth factor receptor gene amplification by competitive polymerase chain reaction in pre-malignant and malignant oral epithelial lesions. Oral Oncol. 2001, 37, 599–604. [Google Scholar] [CrossRef]
- Lorimer, I.A. Mutant epidermal growth factor receptors as targets for cancer therapy. Curr. Cancer Drug Targets 2002, 2, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Rubin Grandis, J.; Zeng, Q.; Tweardy, D.J. Retinoic acid normalizes the increased gene transcription rate of TGF-alpha and EGFR in head and neck cancer cell lines. Nat. Med. 1996, 2, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.K.; Ohtsuki, Y.; Furihata, M.; Takeuchi, T.; Iwata, J.; Liang, S.B.; Sonobe, H. Co-overexpression of p53 protein and epidermal growth factor receptor in human papillary thyroid carcinomas correlated with lymph node metastasis, tumor size and clinicopathologic stage. Int. J. Oncol. 1999, 15, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Ang, K.K.; Berkey, B.A.; Tu, X.; Zhang, H.Z.; Katz, R.; Hammond, E.H.; Fu, K.K.; Milas, L. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002, 62, 7350–7356. [Google Scholar] [PubMed]
- Numico, G.; Russi, E.G.; Colantonio, I.; Lantermo, R.A.; Silvestris, N.; Vitiello, R.; Comino, A.; Abrate, M.; Zavattero, C.; Melano, A.; et al. EGFR status and prognosis of patients with locally advanced head and neck cancer treated with chemoradiotherapy. Anticancer Res. 2010, 30, 671–676. [Google Scholar] [PubMed]
- Rodrigo, J.P.; Gonzalez, M.V.; Lazo, P.S.; Ramos, S.; Coto, E.; Alvarez, I.; Garcia, L.A.; Suarez, C. Genetic alterations in squamous cell carcinomas of the hypopharynx with correlations to clinicopathological features. Oral Oncol. 2002, 38, 357–363. [Google Scholar] [CrossRef]
- Crombet, T.; Torres, O.; Rodriguez, V.; Menendez, A.; Stevenson, A.; Ramos, M.; Torres, F.; Figueredo, R.; Veitia, I.; Iznaga, N.; et al. Phase I clinical evaluation of a neutralizing monoclonal antibody against epidermal growth factor receptor in advanced brain tumor patients: Preliminary study. Hybridoma 2001, 20, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Modjtahedi, H.; Hickish, T.; Nicolson, M.; Moore, J.; Styles, J.; Eccles, S.; Jackson, E.; Salter, J.; Sloane, J.; Spencer, L.; et al. Phase I trial and tumour localisation of the anti-EGFR monoclonal antibody ICR62 in head and neck or lung cancer. Br. J. Cancer 1996, 73, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, N.I.; Prewett, M.; Zuklys, K.; Rockwell, P.; Mendelsohn, J. Biological efficacy of a chimeric antibody to the epidermal growth factor receptor in a human tumor xenograft model. Clin. Cancer Res. 1995, 1, 1311–1318. [Google Scholar] [PubMed]
- Wu, X.; Rubin, M.; Fan, Z.; DeBlasio, T.; Soos, T.; Koff, A.; Mendelsohn, J. Involvement of p27KIP1 in G1 arrest mediated by an anti-epidermal growth factor receptor monoclonal antibody. Oncogene 1996, 12, 1397–1403. [Google Scholar]
- Robert, F.; Ezekiel, M.P.; Spencer, S.A.; Meredith, R.F.; Bonner, J.A.; Khazaeli, M.B.; Saleh, M.N.; Carey, D.; LoBulio, A.F.; Wheeler, R.H.; et al. Phase I study of anti—Epidermal growth factor receptor antibody cetuximab in combination with radiation therapy in patients with advanced head and neck cancer. J. Clin. Oncol. 2001, 19, 3234–3243. [Google Scholar] [PubMed]
- Baselga, J.; Pfister, D.; Cooper, M.R.; Cohen, R.; Burtness, B.; Bos, M.; D'Andrea, G.; Seidman, A.; Norton, L.; Gunnett, K.; et al. Phase I studies of anti-epidermal growth factor receptor chimeric antibody C225 alone and in combination with cisplatin. J. Clin. Oncol. 2000, 18, 904–914. [Google Scholar] [PubMed]
- Milas, L.; Mason, K.; Hunter, N.; Petersen, S.; Yamakawa, M.; Ang, K.; Mendelsohn, J.; Fan, Z. In vivo enhancement of tumor radioresponse by C225 antiepidermal growth factor receptor antibody. Clin. Cancer Res. 2000, 6, 701–708. [Google Scholar] [PubMed]
- Huang, S.M.; Bock, J.M.; Harari, P.M. Epidermal growth factor receptor blockade with C225 modulates proliferation, apoptosis, and radiosensitivity in squamous cell carcinomas of the head and neck. Cancer Res. 1999, 59, 1935–1940. [Google Scholar] [PubMed]
- Pfister, D.G.; Su, Y.B.; Kraus, D.H.; Wolden, S.L.; Lis, E.; Aliff, T.B.; Zahalsky, A.J.; Lake, S.; Needle, M.N.; Shaha, A.R.; et al. Concurrent cetuximab, cisplatin, and concomitant boost radiotherapy for locoregionally advanced, squamous cell head and neck cancer: A pilot phase II study of a new combined-modality paradigm. J. Clin. Oncol. 2006, 24, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Forastiere, A.A.; Metch, B.; Schuller, D.E.; Ensley, J.F.; Hutchins, L.F.; Triozzi, P.; Kish, J.A.; McClure, S.; VonFeldt, E.; Williamson, S.K. Randomized comparison of cisplatin plus fluorouracil and carboplatin plus fluorouracil vs. methotrexate in advanced squamous-cell carcinoma of the head and neck: A Southwest Oncology Group study. J. Clin. Oncol. 1992, 10, 1245–1251. [Google Scholar] [PubMed]
- Leon, X.; Hitt, R.; Constenla, M.; Rocca, A.; Stupp, R.; Kovacs, A.F.; Amellal, N.; Bessa, E.H.; Bourhis, J. A retrospective analysis of the outcome of patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck refractory to a platinum-based chemotherapy. Clin. Oncol. (R. Coll. Radiol.) 2005, 17, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Trigo, J.M.; Bourhis, J.; Tortochaux, J.; Cortes-Funes, H.; Hitt, R.; Gascon, P.; Amellal, N.; Harstrick, A.; Echkardt, A. Phase II multicenter study of the antiepidermal growth factor receptor monoclonal antibody cetuximab in combination with platinum-based chemotherapy in patients with platinum-refractory metastatic and/or recurrent squamous cell carcinoma of the head and neck. J. Clin. Oncol. 2005, 23, 5568–5577. [Google Scholar] [PubMed]
- Herbst, R.S.; Arquette, M.; Shin, D.M.; Dicke, K.; Vokes, E.E.; Azarnia, N.; Hong, W.K.; Kies, M.S. Phase II multicenter study of the epidermal growth factor receptor antibody cetuximab and cisplatin for recurrent and refractory squamous cell carcinoma of the head and neck. J. Clin. Oncol. 2005, 23, 5578–5587. [Google Scholar] [CrossRef] [PubMed]
- Burtness, B.; Goldwasser, M.A.; Flood, W.; Mattar, B.; Forastiere, A.A.; Eastern Cooperative Oncology Group. Phase III randomized trial of cisplatin plus placebo compared with cisplatin plus cetuximab in metastatic/recurrent head and neck cancer: An Eastern Cooperative Oncology Group study. J. Clin. Oncol. 2005, 23, 8646–8654. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Hitt, R.; Irigoyen, A.; Cortes-Funes, H.; Grau, J.J.; Garcia-Saenz, J.A.; Cruz-Hernandez, J.J.; Spanish Head and Neck Cancer Cooperative Groupo (TTCC). Phase II study of the combination of cetuximab and weekly paclitaxel in the first-line treatment of patients with recurrent and/or metastatic squamous cell carcinoma of head and neck. Ann. Oncol. 2012, 23, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.E.; Michel, L.S.; Chung, C.H. New promising molecular targets in head and neck squamous cell carcinoma. Curr. Opin. Oncol. 2012, 24, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.M.; Khuri, F.R. Advances in the management of recurrent or metastatic squamous cell carcinoma of the head and neck. Head Neck 2013, 35, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.W.; McDermott, J.D.; Jimeno, A. Novel treatments for head and neck squamous cell carcinoma: Preclinical identification and clinical investigation. Future Oncol. 2014, 10, 1065–1080. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.B. Current challenges and clinical investigations of epidermal growth factor receptor (EGFR)- and ErbB family-targeted agents in the treatment of head and neck squamous cell carcinoma (HNSCC). Cancer Treat. Rev. 2014, 40, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Stohlmacher-Williams, J.; Davidenko, I.; Licitra, L.; Winquist, E.; Villanueva, C.; Foa, P.; Rottey, S.; Skladowski, K.; Tahara, M.; et al. Cisplatin and fluorouracil with or without panitumumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck (SPECTRUM): An open-label phase 3 randomised trial. Lancet Oncol. 2013, 14, 697–710. [Google Scholar] [CrossRef]
- Machiels, J.P.; Subramanian, S.; Ruzsa, A.; Repassy, G.; Lifirenko, I.; Flygare, A.; Sorensen, P.; Nielsen, T.; Lisby, S.; Clement, P.M. Zalutumumab plus best supportive care vs. best supportive care alone in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck after failure of platinum-based chemotherapy: An open-label, randomised phase 3 trial. Lancet Oncol. 2011, 12, 333–343. [Google Scholar] [CrossRef]
- Stewart, J.S.; Cohen, E.E.; Licitra, L.; Van Herpen, C.M.; Khorprasert, C.; Soulieres, D.; Vodvarka, P.; Rischin, D.; Garin, A.M.; Hirsch, F.R.; et al. Phase III study of gefitinib compared with intravenous methotrexate for recurrent squamous cell carcinoma of the head and neck. J. Clin. Oncol. 2009, 27, 1864–1871. [Google Scholar] [CrossRef] [PubMed]
- Argiris, A.; Ghebremichael, M.; Gilbert, J.; Lee, J.W.; Sachidanandam, K.; Kolesar, J.M.; Burtness, B.; Forastiere, A.A. Phase III randomized, placebo-controlled trial of docetaxel with or without gefitinib in recurrent or metastatic head and neck cancer: An eastern cooperative oncology group trial. J. Clin. Oncol. 2013, 31, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Soulieres, D.; Senzer, N.N.; Vokes, E.E.; Hidalgo, M.; Agarwala, S.S.; Siu, L.L. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J. Clin. Oncol. 2004, 22, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Siu, L.L.; Soulieres, D.; Chen, E.X.; Pond, G.R.; Chin, S.F.; Francis, P.; Harvey, L.; Klein, M.; Zhang, W.; Dancey, J.; et al. Phase I/II trial of Erlotinib and cisplatin in patients with recurrent or metastatic squamous cell carcinoma of the head and neck. J. Clin. Oncol. 2007, 25, 2178–2183. [Google Scholar] [CrossRef] [PubMed]
- De Souza, J.A.; Davis, D.W.; Zhang, Y.; Khattri, A.; Seiwert, T.Y.; Aktolga, S.; Wong, S.J.; Kozloff, M.F.; Nattam, S.; Lingen, M.W.; et al. A phase II study of lapatinib in recurrent of metastatic squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2012, 18, 2336–2343. [Google Scholar] [CrossRef] [PubMed]
- Seiwert, T.Y.; Fayette, J.; Cupissol, D.; Del Campo, J.M.; Clement, P.M.; Hitt, R.; Degardin, M.; Zhang, W.; Blackman, A.; Ehrnrooth, E.; et al. A randomized, phase II study of afatinib vs. cetuximab in metastatic or recurrent squamous cell carcinoma of the head and neck. Ann. Oncol. 2014, 25, 1813–1820. [Google Scholar] [CrossRef] [PubMed]
- Machiels, J.P.; Licitra, L.F.; Haddad, R.I.; Tahara, M.; Cohen, E.E. Rationale and design of LUX-Head & Neck 1: A randomised, Phase III trial of afatinib vs. methotrexate in patients with recurrent and/or metastatic head and neck squamous cell carcinoma who progressed after platinum-based therapy. BMC Cancer 2014, 14, 473. [Google Scholar] [CrossRef]
- Machiels, J.P.; Haddad, R.I.; Fayette, J.; Licitra, L.F.; Tahara, M.; Vermorken, J.B.; Clement, P.M.; Gauler, T.; Cupissol, D.; Grau, J.J.; et al. Afatinib vs. methotrexate as second-line treatment in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck progressing on or after platinum-based therapy (LUX-Head & Neck 1): An open-label, randomised phase 3 trial. Lancet Oncol. 2015, 16, 583–594. [Google Scholar] [PubMed]
- Abdul Razak, A.R.; Soulieres, D.; Laurie, S.A.; Hotte, S.J.; Singh, S.; Winquist, E.; Chia, S.; LeTourneau, C.; Nguyen-Tan, P.F.; Chen, E.X.; et al. A phase II trial of dacomitinib, an oral pan-human EGF receptor (HER) inhibitor, as first-line treatment in recurrent and/or metastatic squamous-cell carcinoma of the head and neck. Ann. Oncol. 2013, 24, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kwon, H.J.; Jung, I.; Yun, M.R.; Ahn, M.J.; Kang, B.W.; Sun, J.M.; Kim, S.B.; Yoon, D.H.; Park, K.U.; et al. Phase II clinical and exploratory biomarker study of dacomitinib in patients with recurrent and/or metastatic squamous cell carcinoma of head and neck. Clin. Cancer Res. 2015, 21, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Vassilakopoulou, M.; Psyrri, A.; Argiris, A. Targeting angiogenesis in head and neck cancer. Oral Oncol. 2015, 51, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Argiris, A.; Kotsakis, A.P.; Hoang, T.; Worden, F.P.; Savvides, P.; Gibson, M.K.; Gyanchandani, R.; Blumenschein, G.R., Jr.; Chen, H.X.; Grandis, J.R.; et al. Cetuximab and bevacizumab: Preclinical data and phase II trial in recurrent or metastatic squamous cell carcinoma of the head and neck. Ann. Oncol. 2013, 24, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Argiris, A.; Karamouzis, M.V.; Gooding, W.E.; Branstetter, B.F.; Zhong, S.; Raez, L.E.; Savvides, P.; Romkes, M. Phase II trial of pemetrexed and bevacizumab in patients with recurrent or metastatic head and neck cancer. J. Clin. Oncol. 2011, 29, 1140–1145. [Google Scholar] [CrossRef] [PubMed]
- Machiels, J.P.; Henry, S.; Zanetta, S.; Kaminsky, M.C.; Michoux, N.; Rommel, D.; Schmitz, S.; Bompas, E.; Dillies, A.F.; Faivre, S.; et al. Phase II study of sunitinib in recurrent or metastatic squamous cell carcinoma of the head and neck: GORTEC 2006-01. J. Clin. Oncol. 2010, 28, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.K.; Moon, J.; Huang, C.H.; Guaglianone, P.P.; LeBlanc, M.; Wolf, G.T.; Urba, S.G. Phase II evaluation of sorafenib in advanced and metastatic squamous cell carcinoma of the head and neck: Southwest Oncology Group Study S0420. J. Clin. Oncol. 2010, 28, 3330–3335. [Google Scholar] [CrossRef] [PubMed]
- Limaye, S.; Riley, S.; Zhao, S.; O'Neill, A.; Posner, M.; Adkins, D.; Jaffa, Z.; Clark, J.; Haddad, R. A randomized phase II study of docetaxel with or without vandetanib in recurrent or metastatic squamous cell carcinoma of head and neck (SCCHN). Oral Oncol. 2013, 49, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Califano, J.A. Sequencing the head and neck cancer genome: Implications for therapy. Ann. N. Y. Acad. Sci. 2014, 1333, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, V.; Keilholz, U.; Boehm, A.; Guntinas-Lichius, O.; Hennemann, B.; Schmoll, H.J.; Ivanyi, P.; Abbas, M.; Lehmann, U.; Koch, A.; et al. TEMHEAD: A single-arm multicentre phase II study of temsirolimus in platin- and cetuximab refractory recurrent and/or metastatic squamous cell carcinoma of the head and neck (SCCHN) of the German SCCHN Group (AIO). Ann. Oncol. 2015, 26, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.E.; Arias-Pulido, H.; Lee, S.J.; Fekrazad, M.H.; Ozawa, H.; Fertig, E.; Howard, J.; Bishop, J.; Wang, H.; Olson, G.T.; et al. A phase II study of temsirolimus and erlotinib in patients with recurrent and/or metastatic, platinum-refractory head and neck squamous cell carcinoma. Oral Oncol. 2013, 49, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Massarelli, E.; Lin, H.; Ginsberg, L.E.; Tran, H.T.; Lee, J.J.; Canales, J.R.; Williams, M.D.; Blumenschein, G.R., Jr.; Lu, C.; Heymach, J.V.; et al. Phase II trial of everolimus and erlotinib in patients with platinum-resistant recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2015, 26, 1476–1480. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Bauman, J.E.; Weissman, C.; Adkins, D.; Schnadig, I.; Beauregard, P.; Bowles, D.W.; Spira, A.; Levy, B.; Seetharamu, N.; et al. A randomized, phase 2 trial of docetaxel with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Oral Oncol. 2015, 51, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Shirai, K.; Choi, M.; Laskin, J.; Kochenderfer, M.; Spira, A.; Cline-Burkhardt, V.; Winquist, E.; Hausman, D.; Walker, L.; et al. A randomized, phase II trial of cetuximab with or without PX-866, an irreversible oral phosphatidylinositol 3-kinase inhibitor, in patients with relapsed or metastatic head and neck squamous cell cancer. Ann. Oncol. 2015, 26, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Fury, M.G.; Baxi, S.; Shen, R.; Kelly, K.W.; Lipson, B.L.; Carlson, D.; Stambuk, H.; Haque, S.; Pfister, D.G. Phase II study of saracatinib (AZD0530) for patients with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC). Anticancer Res. 2011, 31, 249–253. [Google Scholar] [PubMed]
- Brooks, H.D.; Glisson, B.S.; Bekele, B.N.; Johnson, F.M.; Ginsberg, L.E.; El-Naggar, A.; Culotta, K.S.; Takebe, N.; Wright, J.; Tran, H.T.; et al. Phase 2 study of dasatinib in the treatment of head and neck squamous cell carcinoma. Cancer 2011, 117, 2112–2119. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J.; Darnell, J.E., Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–2473. [Google Scholar] [CrossRef] [PubMed]
- Song, J.I.; Grandis, J.R. STAT signaling in head and neck cancer. Oncogene 2000, 19, 2489–2495. [Google Scholar] [CrossRef] [PubMed]
- Roithmaier, S.; Haydon, A.M.; Loi, S.; Esmore, D.; Griffiths, A.; Bergin, P.; Williams, T.J.; Schwarz, M.A. Incidence of malignancies in heart and/or lung transplant recipients: A single-institution experience. J. Heart Lung Transplant. 2007, 26, 845–849. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Gooden, M.J.; de Bock, G.H.; Leffers, N.; Daemen, T.; Nijman, H.W. The prognostic influence of tumour-infiltrating lymphocytes in cancer: A systematic review with meta-analysis. Br. J. Cancer 2011, 105, 93–103. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar] [PubMed]
- Schoenfeld, J.D. Immunity in head and neck cancer. Cancer Immunol. Res. 2015, 3, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Malm, I.J.; Bruno, T.C.; Fu, J.; Zeng, Q.; Taube, J.M.; Westra, W.; Pardoll, D.; Drake, C.G.; Kim, Y.J. Expression profile and in vitro blockade of programmed death-1 in human papillomavirus-negative head and neck squamous cell carcinoma. Head Neck 2015, 37, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Duray, A.; Demoulin, S.; Hubert, P.; Delvenne, P.; Saussez, S. Immune suppression in head and neck cancers: A review. Clin. Dev. Immunol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Chakraborty, T.; Chakraborty, K.; Pal, S.; Baral, R. Dysregulation in immune functions is reflected in tumor cell cytotoxicity by peripheral blood mononuclear cells from head and neck squamous cell carcinoma patients. Cancer Immun. 2008, 8, 10. [Google Scholar] [PubMed]
- Brocks, C.P.; Pries, R.; Frenzel, H.; Ernst, M.; Schlenke, P.; Wollenberg, B. Functional alteration of myeloid dendritic cells through head and neck cancer. Anticancer Res. 2007, 27, 817–824. [Google Scholar] [PubMed]
- Molling, J.W.; Langius, J.A.; Langendijk, J.A.; Leemans, C.R.; Bontkes, H.J.; van der Vliet, H.J.; von Blomberg, B.M.; Scheper, R.J.; van den Eertwegh, A.J. Low levels of circulating invariant natural killer T cells predict poor clinical outcome in patients with head and neck squamous cell carcinoma. J. Clin. Oncol. 2007, 25, 862–868. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.A.; Yoon, H.J.; Lee, J.I.; Hong, S.P.; Hong, S.D. Relationship between the expressions of PD-L1 and tumor-infiltrating lymphocytes in oral squamous cell carcinoma. Oral Oncol. 2011, 47, 1148–1153. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, T.S.; Gasparoto, T.H.; Costa, M.R.; de Melo, E.F., Jr.; Ikoma, M.R.; Damante, J.H.; Cavassani, K.A.; garlet, G.P.; da Silva, J.S.; Campanelli, A.P.; et al. Enhanced programmed death 1 (PD-1) and PD-1 ligand (PD-L1) expression in patients with actinic cheilitis and oral squamous cell carcinoma. Cancer Immunol. Immunother. 2011, 60, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Nordfors, C.; Grun, N.; Tertipis, N.; Ahrlund-Richter, A.; Haeggblom, L.; Sivars, L.; Du, J.; Nyberg, T.; Marklund, L.; Munck.Wikland, E.; et al. CD8+ and CD4+ tumour infiltrating lymphocytes in relation to human papillomavirus status and clinical outcome in tonsillar and base of tongue squamous cell carcinoma. Eur. J. Cancer 2013, 49, 2522–2530. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.J.; Thirdborough, S.M.; Mellows, T.; Riley, C.; Harris, S.; Suchak, K.; Webb, A.; Hampton, C.; Patel, N.N.; Randall, C.J.; et al. Tumour-infiltrating lymphocytes predict for outcome in HPV-positive oropharyngeal cancer. Br. J. Cancer 2014, 110, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Pretscher, D.; Distel, L.V.; Grabenbauer, G.G.; Wittlinger, M.; Buettner, M.; Niedobitek, G. Distribution of immune cells in head and neck cancer: CD8+ T-cells and CD20+ B-cells in metastatic lymph nodes are associated with favourable outcome in patients with oro- and hypopharyngeal carcinoma. BMC Cancer 2009, 9, 292. [Google Scholar] [CrossRef] [PubMed]
- Badoual, C.; Hans, S.; Merillon, N.; van Ryswick, C.; Ravel, P.; Benhamouda, N.; Levionnois, E.; Nizard, M.; Si-Mohamed, A.; Besnier, N.; et al. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res. 2013, 73, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Ukpo, O.C.; Thorstad, W.L.; Lewis, J.S., Jr. B7-H1 expression model for immune evasion in human papillomavirus-related oropharyngeal squamous cell carcinoma. Head Neck Pathol. 2013, 7, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R.; Weber, J.S.; Joshua, A.M.; Hwu, W.J.; Gangadhar, T.C.; et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Haddad, R.I.; Gupta, S.; Mehra, R.; Tahara, M.; Berger, R.; Lee, S.-H.; Burtness, B.; Le, D.T.; Heath, K.; et al. Antitumor activity and safety of pembrolizumab in patients with advanced squamous cell carcinoma of the head and neck (SCCHN). In Proceedings of the 2015 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015.
- Seiwert, T.; Burtness, B.; Weiss, J.; Eder, J.P.; Yearley, J.; Murphy, E.; Nebozhyn, M.; McClanahan, T.; Ayers, M.; Lunceford, J.K.; et al. Inflamed-phenotype gene expression signatures to predict benefit from the anti-PD-1 antibody pembrolizumab in PD-L1+ head and neck cancer patients. In Proceedings of the 2015 ASCO Annual Meeting, Chicago, IL, USA, 29 May–2 June 2015.
- Seiwert, T.Y.; Burtness, B.; Weiss, J.; Gluck, I.; Eder, J.P.; Pai, S.I.; Dolled-Filhart, M.; Emancipator, K.; Pathiraja, K.; Gause, C.; et al. A phase Ib study of MK-3475 in patients with human papillomavirus (HPV)-associated and non-HPV-associated head and neck (H/N) cancer. J. Clin. Oncol. 2014, 32, 6011. [Google Scholar]
- Segal, N.H.; Antonia, S.J.; Brahmer, J.R.; Maio, M.; Blake-Haskins, A.; Li, X.; Vasselli, J.; Ibrahim, R.A.; Lutzky, J.; Khleif, S.; et al. Preliminary data from a multi-arm expansion study of MEDI4736, an anti-PD-L1 antibody. J. Clin. Oncol. 2014, 32, 5s. [Google Scholar]
- Brown, D.R.; Kjaer, S.K.; Sigurdsson, K.; Iversen, O.-E.; Hernandez-Avila, M.; Wheeler, C.M.; Perez, G.; Koutsky, L.A.; Tay, E.H.; Garcia, P.; et al. The impact of quadrivalent human papillomavirus (HPV; types 6, 11, 16, and 18) L1 virus-like particle vaccine on infection and disease due to oncogenic nonvaccine HPV types in generally HPV-naive women aged 16–26 years. J. Infect. Dis 2009, 199, 926–935. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Pries, R.; Wulff, S.; Wollenberg, B. Toll-like receptor modulation in head and neck cancer. Crit. Rev. Immunol. 2008, 28, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Rich, A.M.; Hussaini, H.M.; Parachuru, V.P.; Seymour, G.J. Toll-like receptors and cancer, particularly oral squamous cell carcinoma. Front. Immunol. 2014, 5, 464. [Google Scholar] [CrossRef] [PubMed]
- Kanzler, H.; Barrat, F.J.; Hessel, E.M.; Coffman, R.L. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat. Med. 2007, 13, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Idera Pharmaceuticals Announces Top-Line Results from a Phase 2 Clinical Trial of IMO-2055 in Treatment of Patients with Advanced Head and Neck Cancer. Available online: http://ir.iderapharma.com/phoenix.zhtml?c=208904&p=irol-newsArticle&ID=1690928&highlight= (accessed on 3 May 2012).
- Northfelt, D.W.; Ramanathan, R.K.; Cohen, P.A.; Von Hoff, D.D.; Weiss, G.J.; Dietsch, G.N.; Manjarrez, K.L.; Randall, T.D.; Hershberg, R.M. A phase I dose-finding study of the novel Toll-like receptor 8 agonist VTX-2337 in adult subjects with advanced solid tumors or lymphoma. Clin. Cancer Res. 2014, 20, 3683–3691. [Google Scholar] [CrossRef] [PubMed]
- June, C.H. Adoptive T cell therapy for cancer in the clinic. J. Clin. Invest. 2007, 117, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Tsang, J.; Beagley, L.; Chua, D.; Lee, V.; Li, V.; Moss, D.J.; Coman, W.; Chan, K.H.; Nicholls, J.; et al. Effective treatment of metastatic forms of Epstein-Barr virus-associated nasopharyngeal carcinoma with a novel adenovirus-based adoptive immunotherapy. Cancer Res. 2012, 72, 1116–1125. [Google Scholar] [CrossRef] [PubMed]
- Comoli, P.; Pedrazzoli, P.; Maccario, R.; Basso, S.; Carminati, O.; Labirio, M.; Schiavo, R.; Secondino, S.; Frasson, C.; Perotti, C.; et al. Cell therapy of stage IV nasopharyngeal carcinoma with autologous Epstein-Barr virus-targeted cytotoxic T lymphocytes. J. Clin. Oncol. 2005, 23, 8942–8949. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C.A.; Narala, N.; Vyas, G.M.; Leen, A.M.; Gerdemann, U.; Sturgis, E.M.; Anderson, M.K.; Savoldo, B.; Heslop, H.E.; Brenner, M.K.; et al. Human papillomavirus type 16 E6/E7-specific cytotoxic T lymphocytes for adoptive immunotherapy of HPV-associated malignancies. J. Immunother. 2013, 36, 66–76. [Google Scholar] [CrossRef]
- Kreimer, A.R.; Struyf, F.; del Rosario-Raymundo, M.R.; Hildesheim, A.; Skinner, S.R.; Wacholder, S.; Garland, S.M.; Herrero, R.; David, M.P.; Wheeler, C.M.; et al. Efficacy of fewer than three doses of an HPV-16/18 AS04-adjuvanted vaccine: Combined analysis of data from the Costa Rica Vaccine and PATRICIA trials. Lancet Oncol. 2015, 16, 775–786. [Google Scholar] [CrossRef]
- Ndiaye, C.; Mena, M.; Alemany, L.; Arbyn, M.; Castellsague, X.; Laporte, L.; Bosch, F.X.; de Sanjose, S.; Trottier, H. HPV DNA, E6/E7 mRNA, and p16INK4a detection in head and neck cancers: A systematic review and meta-analysis. Lancet Oncol. 2014, 15, 1319–1331. [Google Scholar] [CrossRef]
- Devaraj, K.; Gillison, M.L.; Wu, T.C. Development of HPV vaccines for HPV-associated head and neck squamous cell carcinoma. Crit. Rev. Oral Biol. Med. 2003, 14, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Cory, L.; Chu, C. ADXS-HPV: A therapeutic Listeria vaccination targeting cervical cancers expressing the HPV E7 antigen. Hum. Vaccin. Immunother. 2014, 10, 3190–3195. [Google Scholar] [CrossRef] [PubMed]
- Reuschenbach, M.; Karbach, J.; Faulstich, F.; Sauer, M.; Kloor, M.; Rafiyan, M.R.; Pauligk, C.; AlBatran, S.; Jaeger, E.; von Knebel Doeberitz, M. A phase I/IIa clinical trial using a p16INK4a derived peptide as vaccine in patients with advanced human papillomavirus-associated cancer. In Proceedings of the Twelfth Annual AACR International Conference on Frontiers in Cancer Prevention Research, National Harbor, MD, USA, 27–30 October 2013.
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Echarri, M.J.; Lopez-Martin, A.; Hitt, R. Targeted Therapy in Locally Advanced and Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma (LA-R/M HNSCC). Cancers 2016, 8, 27. https://doi.org/10.3390/cancers8030027
Echarri MJ, Lopez-Martin A, Hitt R. Targeted Therapy in Locally Advanced and Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma (LA-R/M HNSCC). Cancers. 2016; 8(3):27. https://doi.org/10.3390/cancers8030027
Chicago/Turabian StyleEcharri, María José, Ana Lopez-Martin, and Ricardo Hitt. 2016. "Targeted Therapy in Locally Advanced and Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma (LA-R/M HNSCC)" Cancers 8, no. 3: 27. https://doi.org/10.3390/cancers8030027
APA StyleEcharri, M. J., Lopez-Martin, A., & Hitt, R. (2016). Targeted Therapy in Locally Advanced and Recurrent/Metastatic Head and Neck Squamous Cell Carcinoma (LA-R/M HNSCC). Cancers, 8(3), 27. https://doi.org/10.3390/cancers8030027