
Functional Roles of E6 and E7 Oncoproteins in HPV-Induced Malignancies at Diverse Anatomical Sites

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
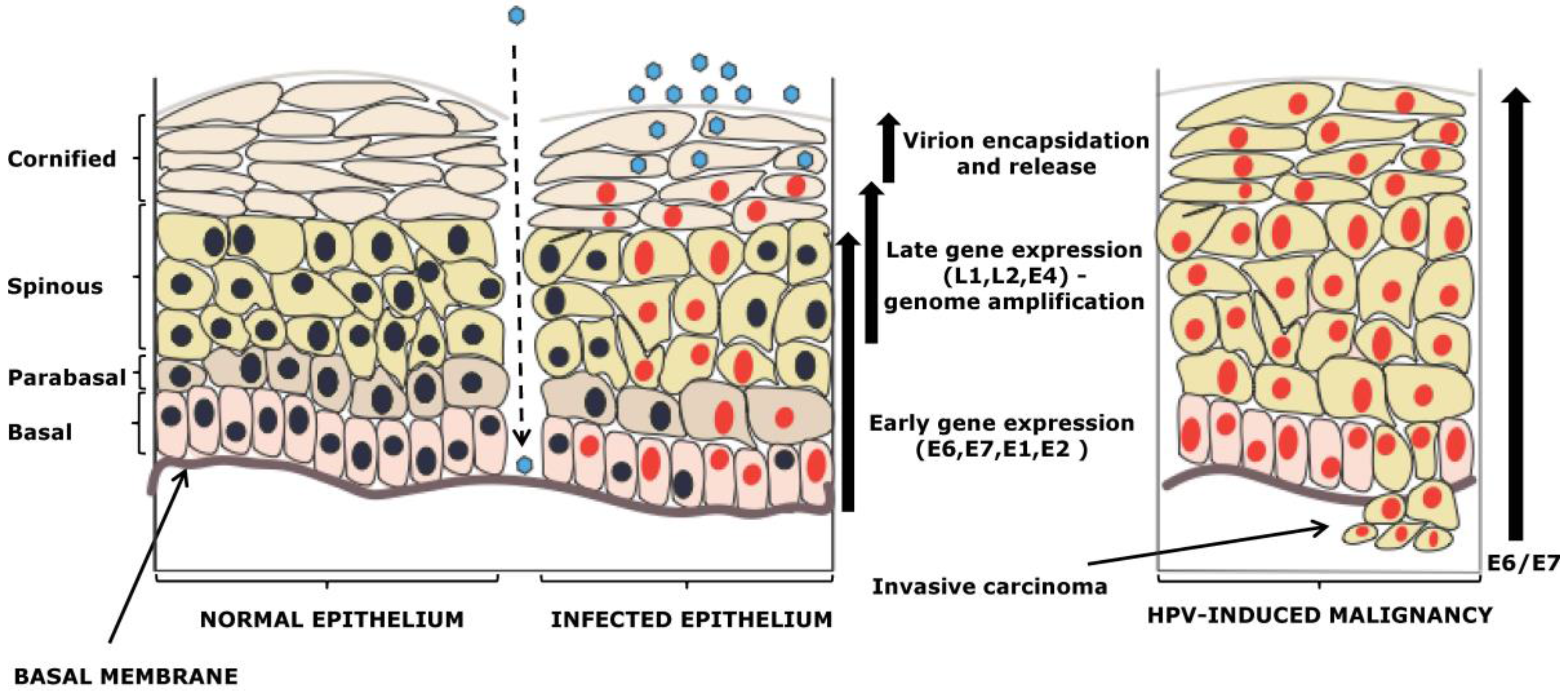
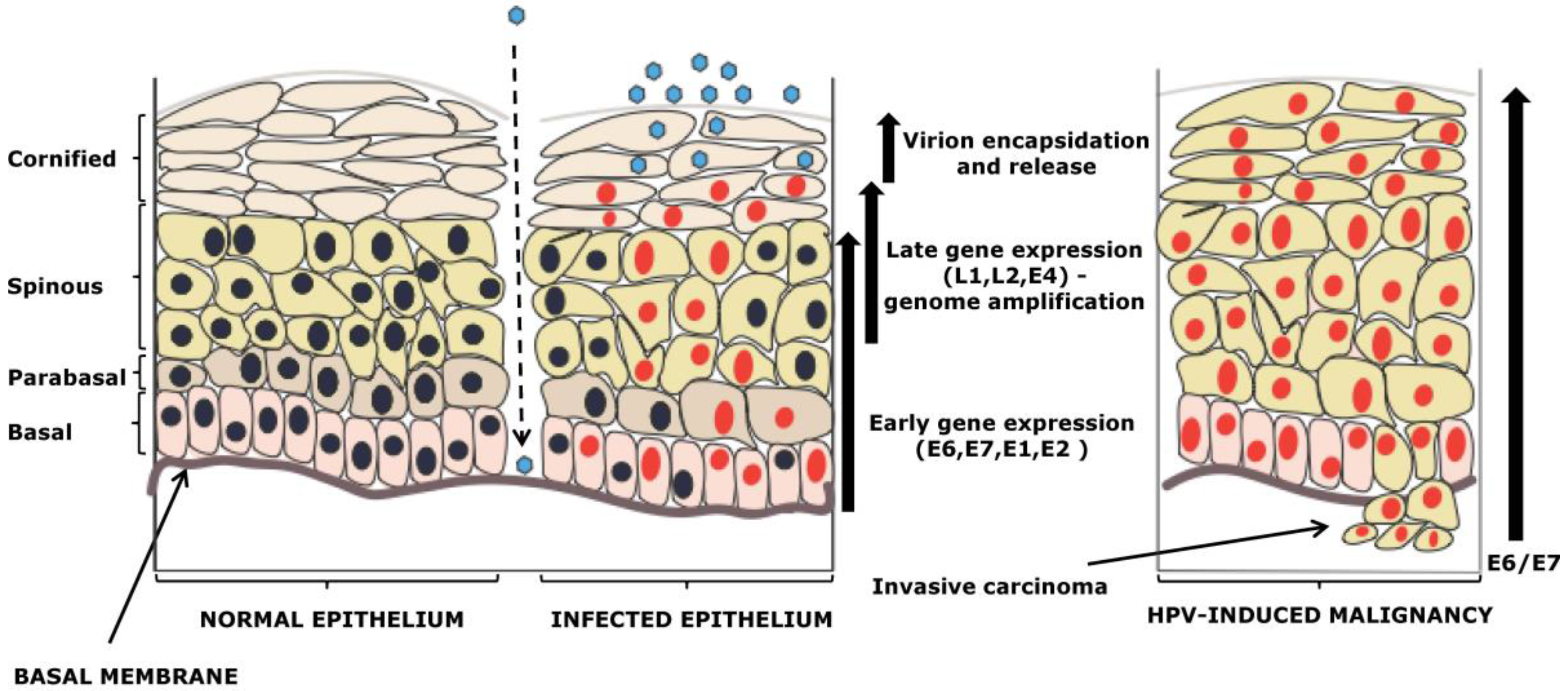
2. Viral Life Cycle Differences between Alpha and Beta-HPV Types
3. The Role of the High Risk Alpha E6 and E7 in Pathogenesis
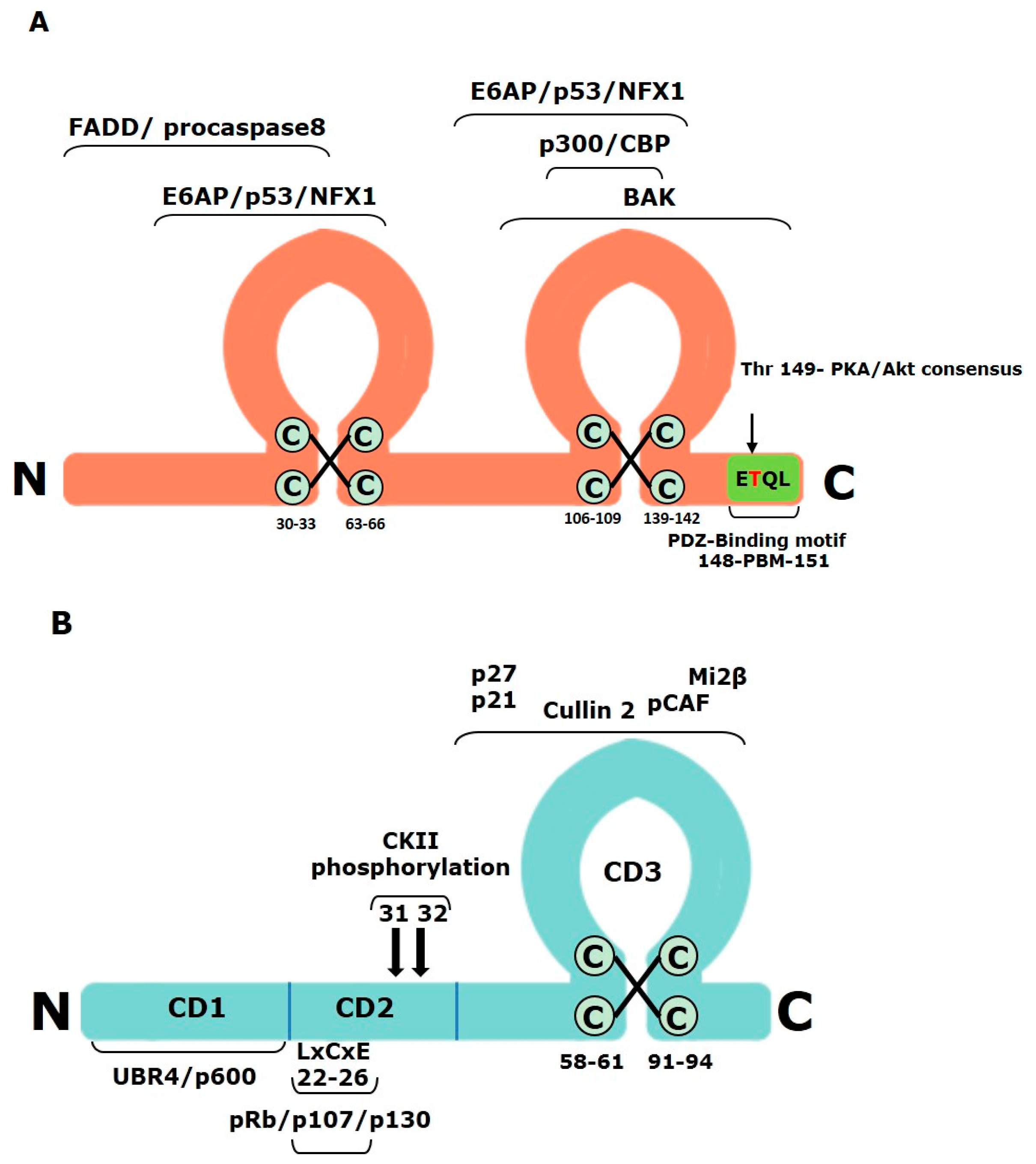
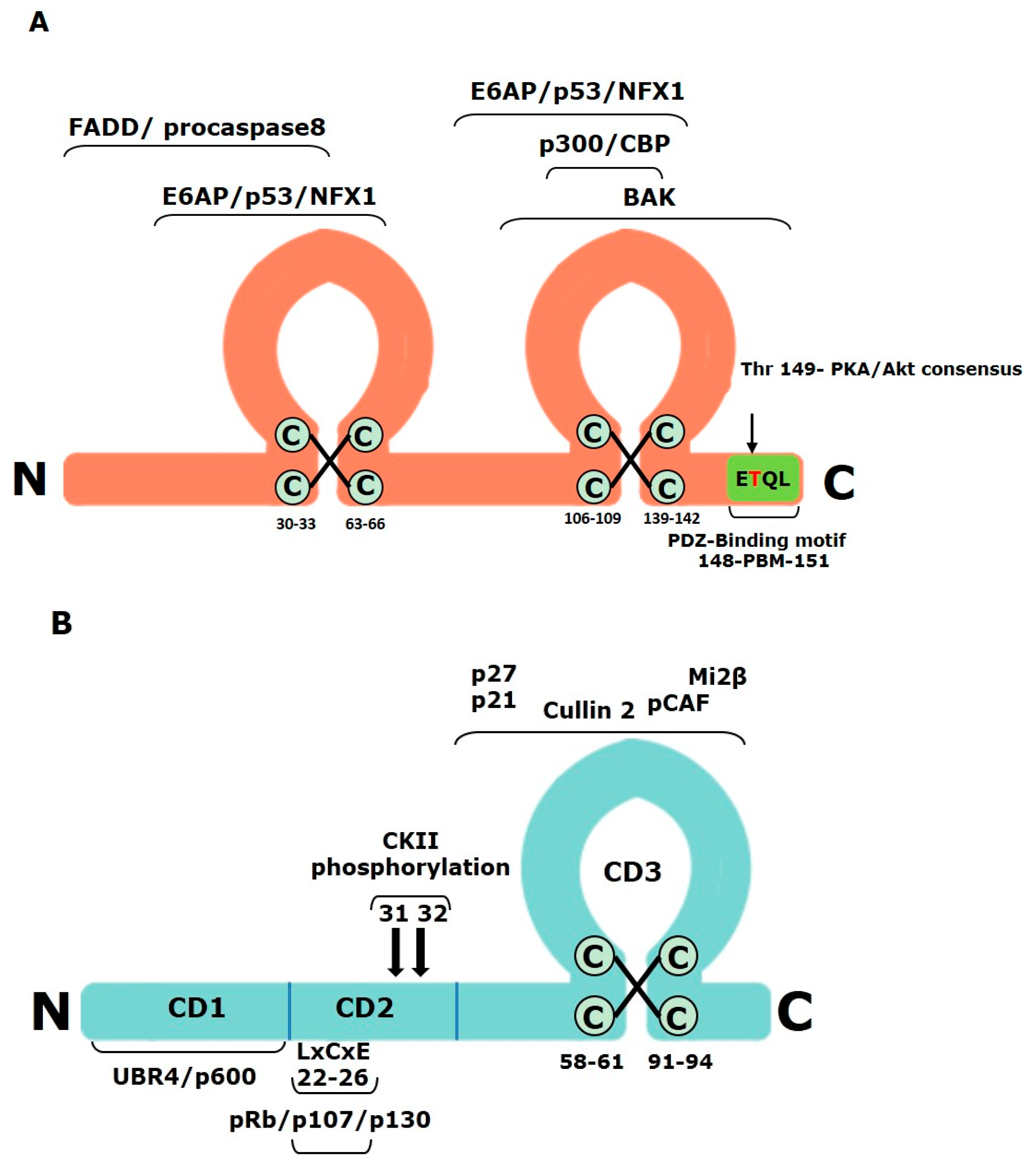
3.1. Alpha-HPV E6
3.1.1. p53 and Components of the Proteasome Ubiquitin Pathway as Interacting Partners of Alpha-HPV E6
3.1.2. Alpha-HPV E6 and Apoptosis
3.1.3. Alpha-HPV E6 and DNA Replication
3.1.4. Alpha-HPV E6 and PDZ Domain-Containing Proteins as Interacting Partners
3.1.5. Phospho-Regulation of Alpha-HPV E6 PDZ-Binding Motif
3.2. Alpha-HPV E7
3.2.1. Conserved Domains of Alpha-HPV E7
3.2.2. Alpha-HPV E7 and Centrosomal Abnormalities
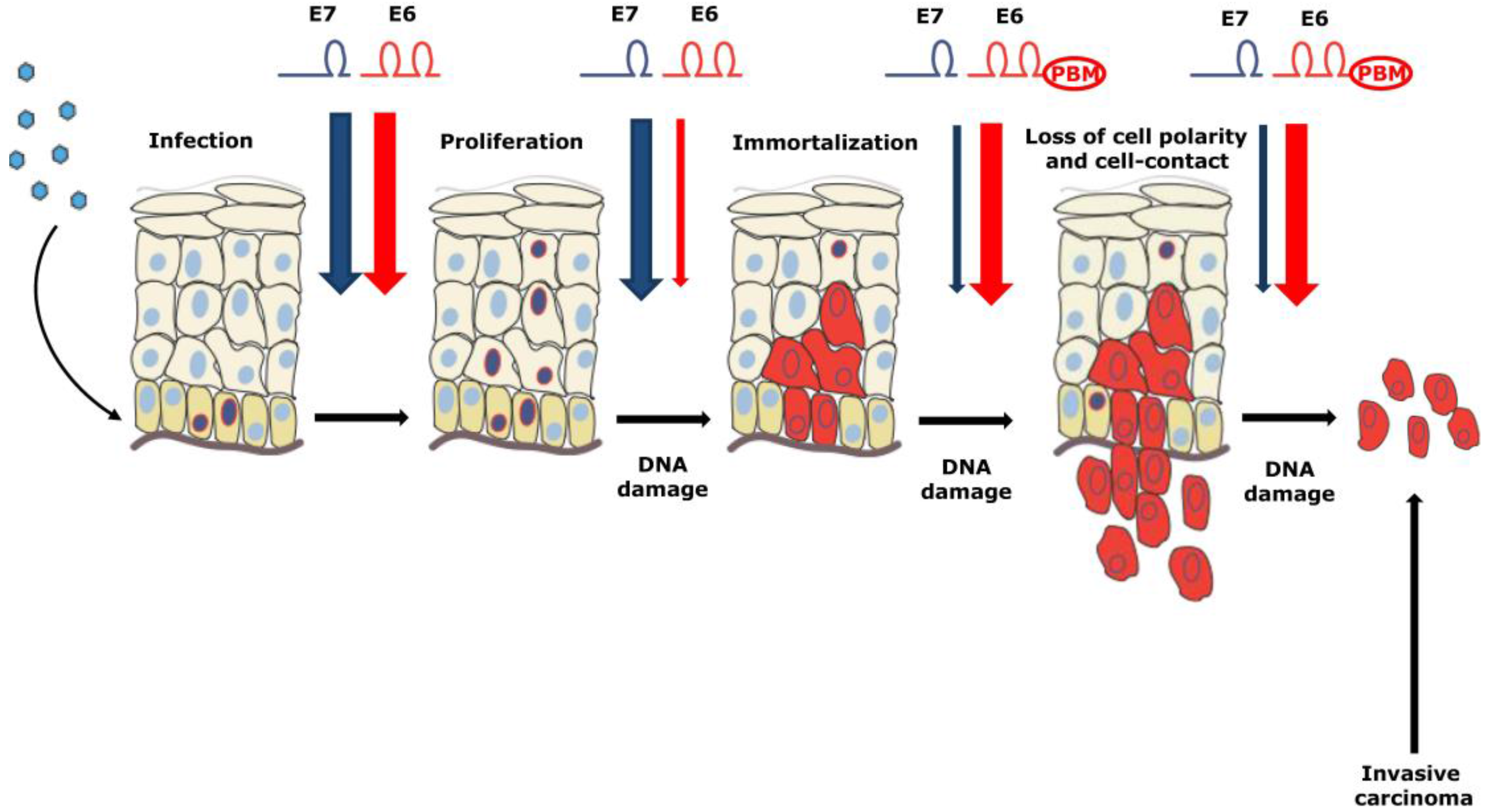
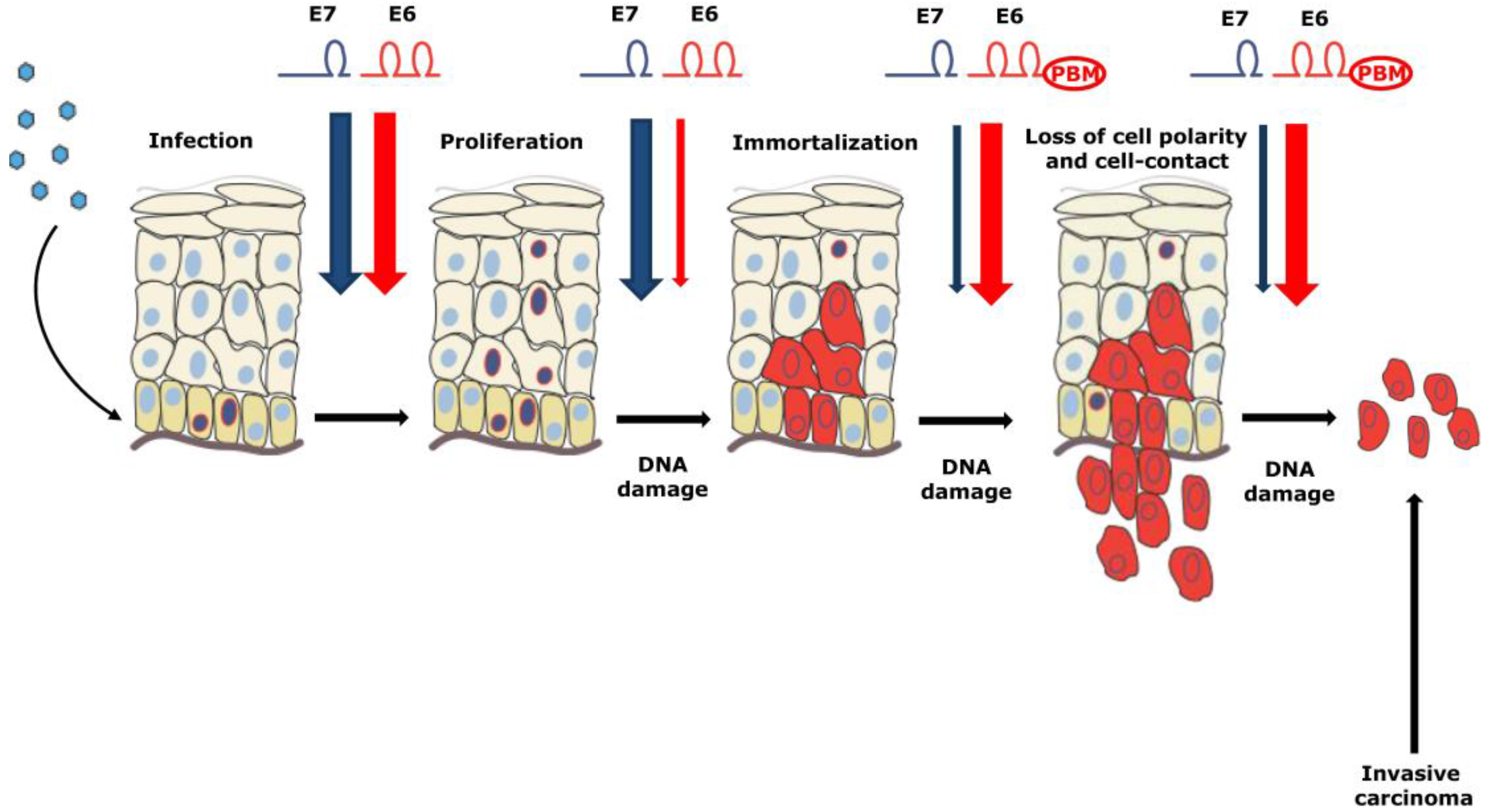
4. High Risk α-HPV Infections at Different Anatomical Sites
4.1. Cervix
4.2. Head-and-Neck Region
5. The Beta E6 and E7 Proteins
5.1. Beta-HPV E6
5.1.1. Ubiquitin Ligases and p53 as Interacting Partners of Beta-HPV E6
5.1.2. Beta-HPV E6 and NOTCH Signaling
5.1.3. Beta-HPV E6 and p53 Gene Regulation
5.1.4. Beta-HPV E6 and Telomerase
5.1.5. Beta-HPV E6 and Apoptosis
5.2. Beta-HPV E7
6. β-HPV Infections of Cutaneous Epithelia
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ganti, K.; Broniarczyk, J.; Manoubi, W.; Massimi, P.; Mittal, S.; Pim, D.; Szalmas, A.; Thatte, J.; Thomas, M.; Tomaić, V.; et al. The human papillomavirus E6 PDZ binding motif: From life cycle to malignancy. Viruses 2015, 7, 3530–3551. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Bray, F. Chapter 2: The burden of HPV-related cancers. Vaccine 2006, 24, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer (IARC). Monographs on the Evaluation of Carcinogenic Risks to Humans Volume 90 Human Papillomaviruses; IARC Press: Lyon, France, 2007; Volume 90. [Google Scholar]
- Bernard, H.U.; Burk, R.D.; Chen, Z.; van Doorslaer, K.; zur Hausen, H.; de Villiers, E.M. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010, 401, 70–79. [Google Scholar] [CrossRef] [PubMed]
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Narayan, N.; Pim, D.; Tomaić, V.; Massimi, P.; Nagasaka, K.; Kranjec, C.; Gammoh, N.; Banks, L. Human papillomaviruses, cervical cancer and cell polarity. Oncogene 2008, 27, 7018–7030. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Anderson, W.F.; Lortet-Tieulent, J.; Curado, M.P.; Ferlay, J.; Franceschi, S.; Rosenberg, P.S.; Bray, F.; Gillison, M.L. Worldwide trends in incidence rates for oral cavity and oropharyngeal cancers. J. Clin. Oncol. 2013, 31, 4550–4559. [Google Scholar] [CrossRef] [PubMed]
- Combes, J.D.; Chen, A.A.; Franceschi, S. Prevalence of human papillomavirus in cancer of the oropharynx by gender. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2954–2958. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Pfister, H.J. Beta genus papillomaviruses and skin cancer. Virology 2015, 479, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Orth, G. Epidermodysplasia verruciformis: A model for understanding the oncogenicity of human papillomaviruses. Ciba Found. Symp. 1986, 120, 157–174. [Google Scholar] [PubMed]
- Quint, K.D.; Genders, R.E.; de Koning, M.N.; Borgogna, C.; Gariglio, M.; Bouwes Bavinck, J.N.; Doorbar, J.; Feltkamp, M.C. Human Beta-papillomavirus infection and keratinocyte carcinomas. J. Pathol. 2015, 235, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. Model systems of human papillomavirus-associated disease. J. Pathol. 2016, 238, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.; Storey, A. E6 proteins from diverse cutaneous HPV types inhibit apoptosis in response to UV damage. Oncogene 2000, 19, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Robinson, K.; Howie, H.L.; Galloway, D.A. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS Pathog. 2012, 8, e1002807. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.M.; Munger, K. The viral etiology of skin cancer. J. Investig. Dermatol. 2014, 34, E29–E32. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.; Laimins, L.A. In vitro systems for the study and propagation of human papillomaviruses. Curr. Top. Microbiol. Immunol. 1994, 186, 199–215. [Google Scholar] [PubMed]
- Chow, L.T. Model systems to study the life cycle of human papillomaviruses and HPV-associated cancers. Virol. Sin. 2015, 30, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Allen-Hoffmann, B.L.; Schlosser, S.J.; Ivarie, C.A.; Sattler, C.A.; Meisner, L.F.; O’Connor, S.L. Normal growth and differentiation in a spontaneously immortalized near-diploid human keratinocyte cell line, NIKS. J. Investig. Dermatol. 2000, 114, 444–455. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Meyers, C.; Budgeon, L.R.; Howett, M.K. Induction of productive human papillomavirus type 11 life cycle in epithelial cells grown in organotypic raft cultures. Virology 2006, 347, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Griffin, H.; Wu, Z.; Marnane, R.; Dewar, V.; Molijn, A.; Quint, W.; van Hoof, C.; Struyf, F.; Colau, B.; Jenkins, D.; et al. E4 antibodies facilitate detection and type-assignment of active HPV infection in cervical disease. PLoS ONE 2012, 7, e49974. [Google Scholar] [CrossRef] [PubMed]
- Andrews, E.; Seaman, W.T.; Webster-Cyriaque, J. Oropharyngeal carcinoma in non-smokers and non-drinkers: A role for HPV. Oral Oncol. 2009, 45, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef] [PubMed]
- Fehrmann, F.; Laimins, L.A. Human papillomaviruses: Targeting differentiating epithelial cells for malignant transformation. Oncogene 2003, 22, 5201–5207. [Google Scholar] [CrossRef] [PubMed]
- Genther, S.M.; Sterling, S.; Duensing, S.; Münger, K.; Sattler, C.; Lambert, P.F. Quantitative role of the human papillomavirus type 16 E5 gene during the productive stage of the viral life cycle. J. Virol. 2003, 77, 2832–2842. [Google Scholar] [CrossRef] [PubMed]
- Galloway, D.A.; Laimins, L.A. Human papillomaviruses: Shared and distinct pathways for pathogenesis. Curr. Opin. Virol. 2015, 14, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Lazarczyk, M.; Pons, C.; Mendoza, J.A.; Cassonnet, P.; Jacob, Y.; Favre, M. Regulation of cellular zinc balance as a potential mechanism of EVER-mediated protection against pathogenesis by cutaneous oncogenic human papillomaviruses. J. Exp. Med. 2008, 205, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Lazarczyk, M.; Cassonnet, P.; Pons, C.; Jacob, Y.; Favre, M. The EVER proteins as a natural barrier against papillomaviruses: A new insight into the pathogenesis of human papillomavirus infections. Microbiol. Mol. Biol. Rev. 2009, 73, 348–370. [Google Scholar] [CrossRef] [PubMed]
- Androphy, E.J.; Hubbert, N.L.; Schiller, J.T.; Lowy, D.R. Identification of the HPV-16 E6 protein from transformed mouse cells and human cervical carcinoma cell lines. EMBO J. 1987, 6, 989–992. [Google Scholar] [PubMed]
- Smotkin, D.; Wettstein, F.O. Transcription of human papillomavirus type 16 early genes in a cervical cancer and a cancer-derived cell line and identification of the E7 protein. Proc. Natl. Acad. Sci. USA 1986, 83, 4680–4684. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Johnson, S.H.; Kasperbauer, J.L.; Eckloff, B.W.; Tombers, N.M.; Vasmatzis, G.; Smith, D.I. Mate pair sequencing of oropharyngeal squamous cell carcinomas reveals that HPV integration occurs much less frequently than in cervical cancer. J. Clin. Virol. 2014, 59, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Salas, L.M.; Cullinan, A.E.; Siwkowski, A.; Hampel, A.; DiPaolo, J.A. Inhibition of HPV-16 E6/E7 immortalization of normal keratinocytes by hairpin ribozymes. Proc. Natl. Acad. Sci. USA 1998, 95, 1189–1194. [Google Scholar] [CrossRef] [PubMed]
- Butz, K.; Denk, C.; Ullmann, A.; Scheffner, M.; Hoppe-Seyler, F. Induction of apoptosis in human papillomaviruspositive cancer cells by peptide aptamers targeting the viral E6 oncoprotein. Proc. Natl. Acad. Sci. USA 2000, 97, 6693–6697. [Google Scholar] [CrossRef] [PubMed]
- Butz, K.; Ristriani, T.; Hengstermann, A.; Denk, C.; Scheffner, M.; Hoppe-Seyler, F. siRNA targeting of the viral E6 oncogene efficiently kills human papillomavirus-positive cancer cells. Oncogene 2003, 22, 5938–5945. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.; Sacks, P.G.; Adler-Storthz, K.; Shillitoe, E.J. Effect on cancer cells of plasmids that express antisense RNA of human papillomavirus type 18. Cancer Res. 1992, 52, 4706–4711. [Google Scholar] [PubMed]
- Von Knebel Doeberitz, M.; Rittmüller, C.; zur Hausen, H.; Dürst, M. Inhibition of tumorigenicity of cervical cancer cells in nude mice by HPV E6-E7 anti-sense RNA. Int. J. Cancer. 1992, 51, 831–834. [Google Scholar] [CrossRef] [PubMed]
- Yoshinouchi, M.; Yamada, T.; Kizaki, M.; Fen, J.; Koseki, T.; Ikeda, Y.; Nishihara, T.; Yamato, K. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by E6 siRNA. Mol. Ther. 2003, 8, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.S.; Schlegel, R. The E6 and E7 genes of HPV-18 are sufficient for inducing two-stage in vitro transformation of human keratinocytes. Oncogene 1989, 4, 1529–1532. [Google Scholar] [PubMed]
- Hawley-Nelson, P.; Vousden, K.H.; Hubbert, N.L.; Lowy, D.R.; Schiller, J.T. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 1989, 8, 3905–3910. [Google Scholar] [PubMed]
- Mantovani, F.; Banks, L. The human papillomavirus E6 protein and its contribution to malignant progression. Oncogene 2001, 20, 7874–7887. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.R.; Duensing, S.; Brake, T.; Münger, K.; Lambert, P.F.; Arbeit, J.M. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003, 63, 4862–4871. [Google Scholar] [PubMed]
- Strati, K.; Lambert, P.F. Role of Rb-dependent and Rb-independent functions of papillomavirus E7 oncogene in head and neck cancer. Cancer Res. 2007, 67, 11585–11593. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Bergant, M.; Boon, S.S.; Ganti, K.; Kranjec, C.; Massimi, P.; Subbaiah, V.K.; Thomas, M.; Tomaić, V.; Banks, L. Human papillomaviruses and the specificity of PDZ domain targeting. FEBS J. 2012, 279, 3530–3537. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.S.; Wettstein, F.O. Transcription of the cottontail rabbit papillomavirus early region and identification of two E6 polypeptides in COS-7 cells. J. Virol. 1987, 61, 2938–2942. [Google Scholar] [PubMed]
- Cole, S.T.; Danos, O. Nucleotide sequence and comparative analysis of the human papillomavirus type 18 genome. Phylogeny of papillomaviruses and repeated structure of the E6 and E7 gene products. J. Mol. Biol. 1987, 193, 599–608. [Google Scholar] [CrossRef]
- Kanda, T.; Watanabe, S.; Zanma, S.; Sato, H.; Furuno, A.; Yoshiike, K. Human papillomavirus type 16 E6 proteins with glycine substitution for cysteine in the metal-binding motif. Virology 1991, 185, 536–543. [Google Scholar] [CrossRef]
- Sherman, L.; Schlegel, R. Serum- and calcium-induced differentiation of human keratinocytes is inhibited by the E6 oncoprotein of human papillomavirus type 16. J. Virol. 1996, 70, 3269–3279. [Google Scholar] [PubMed]
- Nominé, Y.; Masson, M.; Charbonnier, S.; Zanier, K.; Ristriani, T.; Deryckère, F.; Sibler, A.P.; Desplancq, D.; Atkinson, R.A.; Weiss, E.; et al. Structural and functional analysis of E6 oncoprotein: Insights in the molecular pathways of human papillomavirus-mediated pathogenesis. Mol. Cell 2006, 21, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Zanier, K.; Ruhlmann, C.; Melin, F.; Masson, M.; Ould M’hamed Ould Sidi, A.; Bernard, X.; Fischer, B.; Brino, L.; Ristriani, T.; Rybin, V.; et al. E6 proteins from diverse papillomaviruses self-associate both in vitro and in vivo. J. Mol. Biol. 2010, 396, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Zanier, K.; Charbonnier, S.; Sidi, A.O.; McEwen, A.G.; Ferrario, M.G.; Poussin-Courmontagne, P.; Cura, V.; Brimer, N.; Babah, K.O.; Ansari, T.; et al. Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science 2013, 339, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Banks, L. Interaction of viral oncoproteins with cellular target molecules: Infection with high-risk vs low-risk human papillomaviruses. APMIS 2010, 118, 471–493. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef]
- Ashcroft, M.; Vousden, K.H. Regulation of p53 stability. Oncogene 1999, 18, 7637–7643. [Google Scholar] [CrossRef] [PubMed]
- Hengstermann, A.; Linares, L.K.; Ciechanover, A.; Whitaker, N.J.; Scheffner, M. Complete switch from Mdm2 to human papillomavirus E6-mediated degradation of p53 in cervical cancer cells. Proc. Natl. Acad. Sci. USA 2001, 98, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Storey, A.; Thomas, M.; Massimi, P.; Banks, L. Mutational analysis of HPV-18 E6 identifies domains required for p53 degradation in vitro, abolition of p53 transactivation in vivo and immortalisation of primary BMK cells. Oncogene 1994, 9, 1869–1876. [Google Scholar] [PubMed]
- Patel, D.; Huang, S.M.; Baglia, L.A.; McCance, D.J. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999, 18, 5061–5072. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [PubMed]
- Thomas, M.C.; Chiang, C.M. E6 oncoprotein represses p53-dependent gene activation via inhibition of protein acetylation independently of inducing p53 degradation. Mol. Cell 2005, 17, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol. Cell. Biol. 1993, 13, 4918–4927. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol. Cell. Biol. 1993, 13, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Talis, A.L.; Huibregtse, J.M.; Howley, P.M. The role of E6AP in the regulation of p53 protein levels in human papillomavirus (HPV)-positive and HPV-negative cells. J. Biol. Chem. 1998, 273, 6439–6445. [Google Scholar] [CrossRef] [PubMed]
- Tomaić, V.; Pim, D.; Banks, L. The stability of the human papillomavirus E6 oncoprotein is E6AP dependent. Virology 2009, 393, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Tomaić, V.; Pim, D.; Thomas, M.; Massimi, P.; Myers, M.P.; Banks, L. Regulation of the human papillomavirus type 18 E6/E6AP ubiquitin ligase complex by the HECT domain-containing protein EDD. J. Virol. 2011, 85, 3120–3127. [Google Scholar] [CrossRef] [PubMed]
- Vos, R.M.; Altreuter, J.; White, E.A.; Howley, P.M. The ubiquitin-specific peptidase USP15 regulates human papillomavirus type 16 E6 protein stability. J. Virol. 2009, 83, 8885–8892. [Google Scholar] [CrossRef] [PubMed]
- Tomaić, V.; Ganti, K.; Pim, D.; Bauer, C.; Blattner, C.; Banks, L. Interaction of HPV E6 oncoproteins with specific proteasomal subunits. Virology 2013, 446, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Krajewski, S.; Krajewska, M.; Reed, J.C. Immunohistochemical analysis of in vivo patterns of Bak expression, a proapoptotic member of the Bcl-2 protein family. Cancer Res. 1996, 56, 2849–2855. [Google Scholar] [PubMed]
- Jackson, S.; Harwood, C.; Thomas, M.; Banks, L.; Storey, A. Role of Bak in UV-induced apoptosis in skin cancer and abrogation by HPV E6 proteins. Genes Dev. 2000, 14, 3065–3073. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Banks, L. Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene. 1998, 17, 2943–2954. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Banks, L. Human papillomavirus (HPV) E6 interactions with Bak are conserved amongst E6 proteins from high and low risk HPV types. J. Gen. Virol. 1999, 80, 1513–1517. [Google Scholar] [CrossRef] [PubMed]
- Borbély, A.A.; Murvai, M.; Kónya, J.; Beck, Z.; Gergely, L.; Li, F.; Veress, G. Effects of human papillomavirus type 16 oncoproteins on survivin gene expression. J. Gen. Virol. 2006, 87, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Filippova, M.; Song, H.; Connolly, J.L.; Dermody, T.S.; Duerksen-Hughes, P.J. The human papillomavirus 16 E6 protein binds to tumor necrosis factor (TNF) R1 and protects cells from TNF-induced apoptosis. J. Biol. Chem. 2002, 277, 21730–21739. [Google Scholar] [CrossRef] [PubMed]
- Filippova, M.; Parkhurst, L.; Duerksen-Hughes, P.J. The human papillomavirus 16 E6 protein binds to Fas-associated death domain and protects cells from Fas-triggered apoptosis. J. Biol. Chem. 2004, 279, 25729–25744. [Google Scholar] [CrossRef] [PubMed]
- Filippova, M.; Johnson, M.M.; Bautista, M.; Filippov, V.; Fodor, N.; Tungteakkhun, S.S.; Williams, K.; Duerksen-Hughes, P.J. The large and small isoforms of human papillomavirus type 16 E6 bind to and differentially affect procaspase 8 stability and activity. J. Virol. 2007, 81, 4116–4129. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Senescence and immortalization: Role of telomeres and telomerase. Carcinogenesis 2005, 26, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Snijders, P.J.; van Duin, M.; Walboomers, J.M.; Steenbergen, R.D.; Risse, E.K.; Helmerhorst, T.J.; Verheijen, R.H.; Meijer, C.J. Telomerase activity exclusively in cervical carcinomas and a subset of cervical intraepithelial neoplasia grade III lesions: Strong association with elevated messenger RNA levels of its catalytic subunit and high-risk human papillomavirus DNA. Cancer Res. 1998, 58, 3812–3818. [Google Scholar] [PubMed]
- Klingelhutz, A.J.; Foster, S.A.; McDougall, J.K. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 1996, 380, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Veldman, T.; Horikawa, I.; Barrett, J.C.; Schlegel, R. Transcriptional activation of the telomerase hTERT gene by human papillomavirus type 16 E6 oncoprotein. J. Virol. 2001, 75, 4467–4472. [Google Scholar] [CrossRef] [PubMed]
- Gewin, L.; Galloway, D.A. E box-dependent activation of telomerase by human papillomavirus type 16 E6 does not require induction of c-myc. J. Virol. 2001, 75, 7198–7201. [Google Scholar] [CrossRef] [PubMed]
- Veldman, T.; Liu, X.; Yuan, H.; Schlegel, R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 8211–8216. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yuan, H.; Fu, B.; Disbrow, G.L.; Apolinario, T.; Tomaić, V.; Kelley, M.L.; Baker, C.C.; Huibregtse, J.; Schlegel, R. The E6AP ubiquitin ligase is required for transactivation of the hTERT promoter by the human papillomavirus E6 oncoprotein. J. Biol. Chem. 2005, 280, 10807–10816. [Google Scholar] [CrossRef] [PubMed]
- Sekaric, P.; Cherry, J.J.; Androphy, E.J. Binding of human papillomavirus type 16 E6 to E6AP is not required for activation of hTERT. J. Virol. 2008, 82, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Luo, W.; Elzi, D.J.; Grandori, C.; Galloway, D.A. NFX1 interacts with mSin3A/histone deacetylase to repress hTERT transcription in keratinocytes. Mol. Cell. Biol. 2008, 28, 4819–4828. [Google Scholar] [CrossRef] [PubMed]
- Songyang, Z.; Fanning, A.S.; Fu, C.; Xu, J.; Marfatia, S.M.; Chishti, A.H.; Crompton, A.; Chan, A.C.; Anderson, J.M.; Cantley, L.C. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science 1997, 275, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Javier, R.T. Cell polarity proteins: Common targets for tumorigenic human viruses. Oncogene 2008, 27, 7031–7046. [Google Scholar] [CrossRef] [PubMed]
- Van Ham, M.; Hendriks, W. PDZ domains-glue and guide. Mol. Biol. Rep. 2003, 30, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Kiyono, T.; Hiraiwa, A.; Fujita, M.; Hayashi, Y.; Akiyama, T.; Ishibashi, M. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the Drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 11612–11616. [Google Scholar] [CrossRef] [PubMed]
- Watson, R.A.; Thomas, M.; Banks, L.; Roberts, S. Activity of the human papillomavirus E6 PDZ-binding motif correlates with an enhanced morphological transformation of immortalized human keratinocytes. J. Cell Sci. 2003, 116, 4925–4934. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.L.; Nguyen, M.M.; Lee, D.; Griep, A.E.; Lambert, P.F. The PDZ ligand domain of the human papillomavirus type 16 E6 protein is required for E6’s induction of epithelial hyperplasia in vivo. J. Virol. 2003, 77, 6957–6964. [Google Scholar] [CrossRef] [PubMed]
- Delury, C.P.; Marsh, E.K.; James, C.D.; Boon, S.S.; Banks, L.; Knight, G.L.; Roberts, S. The role of protein kinase A regulation of the E6 PDZ-binding domain during the differentiation-dependent life cycle of human papillomavirus type 18. J. Virol. 2013, 87, 9463–9472. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Laimins, L.A. Role of the PDZ domain-binding motif of the oncoprotein E6 in the pathogenesis of human papillomavirus type 31. J. Virol. 2004, 78, 12366–12377. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Myers, M.P.; Massimi, P.; Guarnaccia, C.; Banks, L. Analysis of Multiple HPV E6 PDZ Interactions Defines Type-Specific PDZ Fingerprints That Predict Oncogenic Potential. PLoS Pathog. 2016, 12, e1005789. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Glaunsinger, B.; Pim, D.; Javier, R.; Banks, L. HPV E6 and MAGUK protein interactions: Determination of the molecular basis for specific protein recognition and degradation. Oncogene 2001, 20, 5431–5439. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Massimi, P.; Navarro, C.; Borg, J.P.; Banks, L. The hScrib/Dlg apico-basal control complex is differentially targeted by HPV-16 and HPV-18 E6 proteins. Oncogene 2005, 24, 6222–6230. [Google Scholar] [CrossRef] [PubMed]
- Kranjec, C.; Tomaić, V.; Massimi, P.; Nicolaides, L.; Doorbar, J.; Banks, L. The high-risk HPV E6 target scribble (hScrib) is required for HPV E6 expression in cervical tumour-derived cell lines. Papillomavirus Res. 2016, 2, 70–77. [Google Scholar] [CrossRef]
- Zhan, L.; Rosenberg, A.; Bergami, K.C.; Yu, M.; Xuan, Z.; Jaffe, A.B.; Allred, C.; Muthuswamy, S.K. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 2008, 135, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Henry, G.D.; Hegde, R.S.; Baleja, J.D. Solution structure of the hDlg/SAP97 PDZ2 domain and its mechanism of interaction with HPV-18 papillomavirus E6 protein. Biochemistry 2007, 46, 10864–10874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dasgupta, J.; Ma, R.Z.; Banks, L.; Thomas, M.; Chen, X.S. Structures of a human papillomavirus (HPV) E6 polypeptide bound to MAGUK proteins: Mechanisms of targeting tumor suppressors by a high-risk HPV oncoprotein. J. Virol. 2007, 81, 3618–3626. [Google Scholar] [CrossRef] [PubMed]
- Tomaić, V.; Gardiol, D.; Massimi, P.; Ozbun, M.; Myers, M.; Banks, L. Human and primate tumour viruses use PDZ binding as an evolutionarily conserved mechanism of targeting cell polarity regulators. Oncogene 2009, 28, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Boon, S.S.; Banks, L. High-risk human papillomavirus E6 oncoproteins interact with 14–3-3ζ in a PDZ binding motif-dependent manner. J. Virol. 2013, 87, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Muslin, A.J.; Tanner, J.W.; Allen, P.M.; Shaw, A.S. Interaction of 14–3-3 with signaling proteins is mediated by the recognition of phosphoserine. Cell 1996, 84, 889–897. [Google Scholar] [CrossRef]
- Boon, S.S.; Tomaić, V.; Thomas, M.; Roberts, S.; Banks, L. Cancer-causing human papillomavirus E6 proteins display major differences in the phospho-regulation of their PDZ interactions. J. Virol. 2015, 89, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, M.C.; Frattini, M.G.; Grossman, S.R.; Laimins, L.A. Human papillomavirus type 18 E7 protein requires intact Cys-X-X-Cys motifs for zinc binding, dimerization, and transformation but not for Rb binding. J. Virol. 1993, 67, 3142–3150. [Google Scholar] [PubMed]
- Rawls, J.A.; Pusztai, R.; Green, M. Chemical synthesis of human papillomavirus type 16 E7 oncoprotein: Autonomous protein domains for induction of cellular DNA synthesis and for trans activation. J. Virol. 1990, 64, 6121–6129. [Google Scholar] [PubMed]
- Patrick, D.R.; Oliff, A.; Heimbrook, D.C. Identification of a novel retinoblastoma gene product binding site on human papillomavirus type 16 E7 protein. J. Biol. Chem. 1994, 269, 6842–6850. [Google Scholar] [PubMed]
- Oh, K.J.; Kalinina, A.; Wang, J.; Nakayama, K.; Nakayama, K.I.; Bagchi, S. The papillomavirus E7 oncoprotein is ubiquitinated by UbcH7 and Cullin 1- and Skp2-containing E3 ligase. J. Virol. 2004, 78, 5338–5346. [Google Scholar] [CrossRef] [PubMed]
- Firzlaff, J.M.; Galloway, D.A.; Eisenman, R.N.; Lüscher, B. The E7 protein of human papillomavirus type 16 is phosphorylated by casein kinase II. New Biol. 1989, 1, 44–53. [Google Scholar] [PubMed]
- Barbosa, M.S.; Edmond, C.; Fisher, C.; Schiller, J.T.; Lowy, D.R.; Vousden, K.H. The region of the HPV E7 oncoprotein homologous to adenovirus E1a and Sv40 large T antigen contains separate domains for Rb binding and casein kinase II phosphorylation. EMBO J. 1990, 9, 153–160. [Google Scholar] [PubMed]
- Massimi, P.; Banks, L. Differential phosphorylation of the HPV-16 E7 oncoprotein during the cell cycle. Virology 2000, 276, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Banks, L.; Edmonds, C.; Vousden, K.H. Ability of the HPV16 E7 protein to bind RB and induce DNA synthesis is not sufficient for efficient transforming activity in NIH3T3 cells. Oncogene 1990, 5, 1383–1389. [Google Scholar] [PubMed]
- Brokaw, J.L.; Yee, C.L.; Münger, K. A mutational analysis of the amino terminal domain of the human papillomavirus type 16 E7 oncoprotein. Virology 1994, 205, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Demers, G.W.; Espling, E.; Harry, J.B.; Etscheid, B.G.; Galloway, D.A. Abrogation of growth arrest signals by human papillomavirus type 16 E7 is mediated by sequences required for transformation. J. Virol. 1996, 70, 6862–6869. [Google Scholar] [PubMed]
- Huh, K.W.; DeMasi, J.; Ogawa, H.; Nakatani, Y.; Howley, P.M.; Münger, K. Association of the human papillomavirus type 16 E7 oncoprotein with the 600-kDa retinoblastoma protein-associated factor, p600. Proc. Natl. Acad. Sci. USA 2005, 102, 11492–11497. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.M.; McCance, D.J. Down regulation of the interleukin-8 promoter by human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 and P/CAF. J. Virol. 2002, 76, 8710–8721. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, Y.; Konishi, H.; Vassilev, A.; Kurooka, H.; Ishiguro, K.; Sawada, J.; Ikura, T.; Korsmeyer, S.J.; Qin, J.; Herlitz, A.M. p600, a unique protein required for membrane morphogenesis and cell survival. Proc. Natl. Acad. Sci. USA 2005, 102, 15093–15098. [Google Scholar] [CrossRef] [PubMed]
- DeMasi, J.; Huh, K.W.; Nakatani, Y.; Münger, K.; Howley, P.M. Bovine papillomavirus E7 transformation function correlates with cellular p600 protein binding. Proc. Natl. Acad. Sci. USA 2005, 102, 11486–11491. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Kramer, R.E.; Tan, M.J.; Hayes, S.D.; Harper, J.W.; Howley, P.M. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol. 2012, 86, 13174–13186. [Google Scholar] [CrossRef] [PubMed]
- Firzlaff, J.M.; Lüscher, B.; Eisenman, R.N. Negative charge at the casein kinase II phosphorylation site is important for transformation but not for Rb protein binding by the E7 protein of human papillomavirus type 16. Proc. Natl. Acad. Sci. USA 1991, 88, 5187–5191. [Google Scholar] [CrossRef] [PubMed]
- Chien, W.M.; Parker, J.N.; Schmidt-Grimminger, D.C.; Broker, T.R.; Chow, L.T. Casein kinase II phosphorylation of the human papillomavirus-18 E7 protein is critical for promoting S-phase entry. Cell Growth Differ. 2000, 11, 425–435. [Google Scholar] [PubMed]
- Dyson, N.; Howley, P.M.; Münger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Slansky, J.E.; Farnham, P.J. Introduction to the E2F family: Protein structure and gene regulation. Curr. Top. Microbiol. Immunol. 1996, 208, 1–30. [Google Scholar] [PubMed]
- Imai, Y.; Matsushima, Y.; Sugimura, T.; Terada, M. Purification and characterization of human papillomavirus type 16 E7 protein with preferential binding capacity to the underphosphorylated form of retinoblastoma gene product. J. Virol. 1991, 65, 4966–4972. [Google Scholar] [PubMed]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar] [PubMed]
- Huh, K.; Zhou, X.; Hayakawa, H.; Cho, J.Y.; Libermann, T.A.; Jin, J.; Harper, J.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 2007, 81, 9737–9747. [Google Scholar] [CrossRef] [PubMed]
- Morris, E.J.; Dyson, N.J. Retinoblastoma protein partners. Adv. Cancer Res. 2001, 82, 1–54. [Google Scholar] [PubMed]
- Davies, R.; Hicks, R.; Crook, T.; Morris, J.; Vousden, K. Human papillomavirus type 16 E7 associates with a histone H1 kinase and with p107 through sequences necessary for transformation. J. Virol. 1993, 67, 2521–2528. [Google Scholar] [PubMed]
- Baus, F.; Gire, V.; Fisher, D.; Piette, J.; Dulić, V. Permanent cell cycle exit in G2 phase after DNA damage in normal human fibroblasts. EMBO J. 2003, 22, 3992–4002. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, D.; Vairo, G.; Chittenden, T.; Xiao, Z.X.; Xu, G.; Wydner, K.L.; DeCaprio, J.A.; Lawrence, J.B.; Livingston, D.M. E2F-4, a new member of the E2F transcription factor family, interacts with p107. Genes Dev. 1994, 8, 2665–2679. [Google Scholar] [CrossRef] [PubMed]
- Hijmans, E.M.; Voorhoeve, P.M.; Beijersbergen, R.L.; van’t Veer, L.J.; Bernards, R. E2F-5, a new E2F family member that interacts with p130 in vivo. Mol. Cell. Biol. 1995, 15, 3082–3089. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, J.M.; Fairchild, B.; Verona, R.; Moberg, K.; Andon, N.; Lees, J.A. E2F-6, a member of the E2F family that can behave as a transcriptional repressor. Proc. Natl. Acad. Sci. USA 1998, 95, 2850–2855. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin-Drubin, M.E.; Huh, K.W.; Münger, K. Human papillomavirus type 16 E7 oncoprotein associates with E2F6. J. Virol. 2008, 82, 8695–8705. [Google Scholar] [CrossRef] [PubMed]
- Helt, A.M.; Galloway, D.A. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J. Virol. 2001, 75, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Thompson, D.A.; Münger, K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology 1997, 239, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, T.F.; Nguyen, D.X.; Thrash, B.R.; McCance, D.J. E7 abolishes raf-induced arrest via mislocalization of p21 (Cip1). Mol. Cell. Biol. 2002, 22, 7041–7052. [Google Scholar] [CrossRef] [PubMed]
- Brehm, A.; Nielsen, S.J.; Miska, E.A.; McCance, D.J.; Reid, J.L.; Bannister, A.J.; Kouzarides, T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999, 18, 2449–2458. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Laribee, R.N.; Klemsz, M.J.; Roman, A. Human papillomavirus type 16 E7 protein increases acetylation of histone H3 in human foreskin keratinocytes. Virology 2004, 329, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Massimi, P.; Pim, D.; Banks, L. Human papillomavirus type 16 E7 binds to the conserved carboxy-terminal region of the TATA box binding protein and this contributes to E7 transforming activity. J. Gen. Virol. 1997, 78, 2607–2613. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; De-Medina, T.; Syken, J.; Vidal, M.; Münger, K. A novel human DnaJ protein, hTid-1, a homolog of the Drosophila tumor suppressor protein Tid56, can interact with the human papillomavirus type 16 E7 oncoprotein. Virology 1998, 247, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Bornens, M. Centrosome composition and microtubule anchoring mechanisms. Curr. Opin. Cell Biol. 2002, 14, 25–34. [Google Scholar] [CrossRef]
- Sluder, G.; Nordberg, J.J. The good, the bad and the ugly: The practical consequences of centrosome amplification. Curr. Opin. Cell Biol. 2004, 16, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Centrosome aberrations: Cause or consequence of cancer progression? Nat. Rev. Cancer 2002, 2, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, J.L.; Whitehead, C.M.; Lingle, W.L.; Barrett, S.L. Centrosomes and cancer. Biol. Cell 1999, 91, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Tsou, M.F.; Stearns, T. Mechanism limiting centrosome duplication to once per cell cycle. Nature 2006, 442, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Lingle, W.L.; Lutz, W.H.; Ingle, J.N.; Maihle, N.J.; Salisbury, J.L. Centrosome hypertrophy in human breast tumors: Implications for genomic stability and cell polarity. Proc. Natl. Acad. Sci. USA 1998, 95, 2950–2955. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S. A tentative classification of centrosome abnormalities in cancer. Cell Biol. Int. 2005, 29, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Münger, K. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 2002, 62, 7075–7082. [Google Scholar] [PubMed]
- Duensing, S.; Duensing, A.; Crum, C.P.; Münger, K. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001, 61, 2356–2360. [Google Scholar] [PubMed]
- Shinmura, K.; Bennett, R.A.; Tarapore, P.; Fukasawa, K. Direct evidence for the role of centrosomally localized p53 in the regulation of centrosome duplication. Oncogene 2007, 26, 2939–2944. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Münger, K. Human papillomavirus type 16 E7 oncoprotein can induce abnormal centrosome duplication through a mechanism independent of inactivation of retinoblastoma protein family members. J. Virol. 2003, 77, 12331–12335. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Duensing, A.; Lee, D.C.; Edwards, K.M.; Piboonniyom, S.O.; Manuel, E.; Skaltsounis, L.; Meijer, L.; Münger, K. Cyclin-dependent kinase inhibitor indirubin-3′-oxime selectively inhibits human papillomavirus type 16 E7-induced numerical centrosome anomalies. Oncogene 2004, 23, 8206–8215. [Google Scholar] [CrossRef] [PubMed]
- Duensing, A.; Liu, Y.; Spardy, N.; Bartoli, K.; Tseng, M.; Kwon, J.A.; Teng, X.; Duensing, S. RNA polymerase II transcription is required for human papillomavirus type 16 E7- and hydroxyurea-induced centriole overduplication. Oncogene 2007, 26, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Egawa, K.; Griffin, H.; Doorbar, J. Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses 2015, 7, 3863–3890. [Google Scholar] [CrossRef] [PubMed]
- Darragh, T.M.; Winkler, B. Anal cancer and cervical cancer screening: Key differences. Cancer Cytopathol. 2011, 119, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Isaacson Wechsler, E.; Wang, Q.; Roberts, I.; Pagliarulo, E.; Jackson, D.; Untersperger, C.; Coleman, N.; Griffin, H.; Doorbar, J. Reconstruction of human papillomavirus type 16-mediated early-stage neoplasia implicates E6/E7 deregulation and the loss of contact inhibition in neoplastic progression. J. Virol. 2012, 86, 6358–6364. [Google Scholar] [CrossRef] [PubMed]
- Mirkovic, J.; Howitt, B.E.; Roncarati, P.; Demoulin, S.; Suarez-Carmona, M.; Hubert, P.; McKeon, F.D.; Xian, W.; Li, A.; Delvenne, P.; et al. Carcinogenic HPV infection in the cervical squamo-columnar junction. J. Pathol. 2015, 236, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.E.; Smedts, F.M.; Ploeger, D.; Helmerhorst, T.J.; Ramaekers, F.C.; Arends, J.W.; Hopman, A.H. Distribution pattern and marker profile show two subpopulations of reserve cells in the endocervical canal. Int. J. Gynecol. Pathol. 2009, 28, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Herfs, M.; Vargas, S.O.; Yamamoto, Y.; Howitt, B.E.; Nucci, M.R.; Hornick, J.L.; McKeon, F.D.; Xian, W.; Crum, C.P. A novel blueprint for ‘top down’ differentiation defines the cervical squamocolumnar junction during development, reproductive life, and neoplasia. J. Pathol. 2013, 229, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Banks, L. Molecular mechanisms underlying human papillomavirus E6 and E7 oncoprotein-induced cell transformation. Mutat. Res. Rev. Mutat. Res. 2016. [Google Scholar] [CrossRef]
- Ottinger, M.; Smith, J.A.; Schweiger, M.R.; Robbins, D.; Powell, M.L.; You, J.; Howley, P.M. Cell-type specific transcriptional activities among different papillomavirus long control regions and their regulation by E2. Virology 2009, 395, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Dok, R.; Nuyts, S. HPV Positive Head and Neck Cancers: Molecular Pathogenesis and Evolving Treatment Strategies. Cancers 2016. [Google Scholar] [CrossRef] [PubMed]
- Hübbers, C.U.; Akgül, B. HPV and cancer of the oral cavity. Virulence 2015, 6, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Mirghani, H.; Amen, F.; Moreau, F.; Lacau St Guily, J. Do high-risk human papillomaviruses cause oral cavity squamous cell carcinoma? Oral Oncol. 2015, 51, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Westra, W.H. The morphologic profile of HPV-related head and neck squamous carcinoma: Implications for diagnosis, prognosis, and clinical management. Head Neck Pathol. 2012, 6, S48–S54. [Google Scholar] [CrossRef] [PubMed]
- St Guily, J.L.; Clavel, C.; Okaïs, C.; Prétet, J.L.; Beby-Defaux, A.; Agius, G.; Birembaut, P.; Jacquard, A.C.; Léocmach, Y.; Soubeyrand, B.; et al. Human papillomavirus genotype distribution in tonsil cancers. Head Neck Oncol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.K.; Wise-Draper, T.M.; Wells, S.I. Human papillomavirus induced transformation in cervical and head and neck cancers. Cancers 2014, 6, 1793–1820. [Google Scholar] [CrossRef] [PubMed]
- Chai, R.C.; Lambie, D.; Verma, M.; Punyadeera, C. Current trends in the etiology and diagnosis of HPV-related head and neck cancers. Cancer Med. 2015, 4, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Kreimer, A.R.; Johansson, M.; Waterboer, T.; Kaaks, R.; Chang-Claude, J.; Drogen, D.; Tjønneland, A.; Overvad, K.; Quirós, J.R.; González, C.A.; et al. Evaluation of human papillomavirus antibodies and risk of subsequent head and neck cancer. J. Clin. Oncol. 2013, 31, 2708–2715. [Google Scholar] [CrossRef] [PubMed]
- Wallace, N.A.; Galloway, D.A. Novel Functions of the Human Papillomavirus E6 Oncoproteins. Annu. Rev. Virol. 2015, 2, 403–423. [Google Scholar] [CrossRef] [PubMed]
- Cornet, I.; Bouvard, V.; Campo, M.S.; Thomas, M.; Banks, L.; Gissmann, L.; Lamartine, J.; Sylla, B.S.; Accardi, R.; Tommasino, M. Comparative analysis of transforming properties of E6 and E7 from different beta human papillomavirus types. J. Virol. 2012, 86, 2366–2370. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Tomaić, V.; Pim, D.; Myers, M.P.; Tommasino, M.; Banks, L. Interactions between E6AP and E6 proteins from alpha and beta HPV types. Virology 2013, 435, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Brimer, N.; Lyons, C.; Wallberg, A.E.; Vande Pol, S.B. Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene 2012, 31, 4639–4646. [Google Scholar] [CrossRef] [PubMed]
- Meyers, J.M.; Spangle, J.M.; Munger, K. The human papillomavirus type 8 E6 protein interferes with NOTCH activation during keratinocyte differentiation. J. Virol. 2013, 87, 4762–4767. [Google Scholar] [CrossRef] [PubMed]
- Lowell, S.; Jones, P.; Le Roux, I.; Dunne, J.; Watt, F.M. Stimulation of human epidermal differentiation by delta-notch signalling at the boundaries of stem-cell clusters. Curr. Biol. 2000, 10, 491–500. [Google Scholar] [CrossRef]
- Rangarajan, A.; Talora, C.; Okuyama, R.; Nicolas, M.; Mammucari, C.; Oh, H.; Aster, J.C.; Krishna, S.; Metzger, D.; Chambon, P.; et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001, 20, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Muench, P.; Probst, S.; Schuetz, J.; Leiprecht, N.; Busch, M.; Wesselborg, S.; Stubenrauch, F.; Iftner, T. Cutaneous papillomavirus E6 proteins must interact with p300 and block p53-mediated apoptosis for cellular immortalization and tumorigenesis. Cancer Res. 2010, 70, 6913–6924. [Google Scholar] [CrossRef] [PubMed]
- Muschik, D.; Braspenning-Wesch, I.; Stockfleth, E.; Rösl, F.; Hofmann, T.G.; Nindl, I. Cutaneous HPV23 E6 prevents p53 phosphorylation through interaction with HIPK2. PLoS ONE 2011, 6, e27655. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.M.; Underbrink, M.P.; Howie, H.L.; Galloway, D.A. The E6 oncoproteins from human betapapillomaviruses differentially activate telomerase through an E6AP-dependent mechanism and prolong the lifespan of primary keratinocytes. J. Virol. 2008, 82, 3894–3902. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, S.; Zehbe, I.; Accardi, R.; Malanchi, I.; Dong, W.; Giarrè, M.; de Villiers, E.M.; Filotico, R.; Boukamp, P.; Tommasino, M. The E6 and E7 proteins of the cutaneous human papillomavirus type 38 display transforming properties. J. Virol. 2003, 77, 2195–2206. [Google Scholar] [CrossRef] [PubMed]
- Gabet, A.S.; Accardi, R.; Bellopede, A.; Popp, S.; Boukamp, P.; Sylla, B.S.; Londoño-Vallejo, J.A.; Tommasino, M. Impairment of the telomere/telomerase system and genomic instability are associated with keratinocyte immortalization induced by the skin human papillomavirus type 38. FASEB J. 2008, 22, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Holloway, A.; Simmonds, M.; Azad, A.; Fox, J.L.; Storey, A. Resistance to UV-induced apoptosis by β-HPV5 E6 involves targeting of activated BAK for proteolysis by recruitment of the HERC1 ubiquitin ligase. Int. J. Cancer 2015, 136, 2831–2843. [Google Scholar] [CrossRef] [PubMed]
- Underbrink, M.P.; Howie, H.L.; Bedard, K.M.; Koop, J.I.; Galloway, D.A. E6 proteins from multiple human betapapillomavirus types degrade Bak and protect keratinocytes from apoptosis after UVB irradiation. J. Virol. 2008, 82, 10408–10417. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.S.; Vass, W.C.; Lowy, D.R.; Schiller, J.T. In vitro biological activities of the E6 and E7 genes vary among human papillomaviruses of different oncogenic potential. J. Virol. 1991, 65, 292–298. [Google Scholar] [PubMed]
- Münger, K.; Werness, B.A.; Dyson, N.; Phelps, W.C.; Harlow, E.; Howley, P.M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989, 8, 4099–4105. [Google Scholar] [PubMed]
- Accardi, R.; Dong, W.; Smet, A.; Cui, R.; Hautefeuille, A.; Gabet, A.S.; Sylla, B.S.; Gissmann, L.; Hainaut, P.; Tommasino, M. Skin human papillomavirus type 38 alters p53 functions by accumulation of deltaNp73. EMBO Rep. 2006, 7, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Accardi, R.; Scalise, M.; Gheit, T.; Hussain, I.; Yue, J.; Carreira, C.; Collino, A.; Indiveri, C.; Gissmann, L.; Sylla, B.S.; et al. IkappaB kinase beta promotes cell survival by antagonizing p53 functions through DeltaNp73alpha phosphorylation and stabilization. Mol. Cell. Biol. 2011, 31, 2210–2226. [Google Scholar] [CrossRef] [PubMed]
- Saidj, D.; Cros, M.P.; Hernandez-Vargas, H.; Guarino, F.; Sylla, B.S.; Tommasino, M.; Accardi, R. Oncoprotein E7 from beta human papillomavirus 38 induces formation of an inhibitory complex for a subset of p53-regulated promoters. J. Virol. 2013, 87, 12139–12150. [Google Scholar] [CrossRef] [PubMed]
- Tommasino, M. The human papillomavirus family and its role in carcinogenesis. Semin. Cancer Biol. 2014, 26, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Schaper, I.D.; Marcuzzi, G.P.; Weissenborn, S.J.; Kasper, H.U.; Dries, V.; Smyth, N.; Fuchs, P.; Pfister, H. Development of skin tumors in mice transgenic for early genes of human papillomavirus type 8. Cancer Res. 2005, 65, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Viarisio, D.; Mueller-Decker, K.; Kloz, U.; Aengeneyndt, B.; Kopp-Schneider, A.; Gröne, H.J.; Gheit, T.; Flechtenmacher, C.; Gissmann, L.; Tommasino, M. E6 and E7 from beta HPV38 cooperate with ultraviolet light in the development of actinic keratosis-like lesions and squamous cell carcinoma in mice. PLoS Pathog. 2011, 7, e1002125. [Google Scholar] [CrossRef] [PubMed]
- Cotsarelis, G.; Sun, T.T.; Lavker, R.M. Label-retaining cells reside in the bulge area of pilosebaceous unit: Implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell 1990, 61, 1329–1337. [Google Scholar] [CrossRef]
- Cotsarelis, G. Epithelial stem cells: A folliculocentric view. J. Investig. Dermatol. 2006, 126, 1459–1468. [Google Scholar] [CrossRef] [PubMed]
- Ghazizadeh, S.; Taichman, L.B. Organization of stem cells and their progeny in human epidermis. J. Investig. Dermatol. 2005, 124, 367–372. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomaić, V. Functional Roles of E6 and E7 Oncoproteins in HPV-Induced Malignancies at Diverse Anatomical Sites. Cancers 2016, 8, 95. https://doi.org/10.3390/cancers8100095
Tomaić V. Functional Roles of E6 and E7 Oncoproteins in HPV-Induced Malignancies at Diverse Anatomical Sites. Cancers. 2016; 8(10):95. https://doi.org/10.3390/cancers8100095
Chicago/Turabian StyleTomaić, Vjekoslav. 2016. "Functional Roles of E6 and E7 Oncoproteins in HPV-Induced Malignancies at Diverse Anatomical Sites" Cancers 8, no. 10: 95. https://doi.org/10.3390/cancers8100095
APA StyleTomaić, V. (2016). Functional Roles of E6 and E7 Oncoproteins in HPV-Induced Malignancies at Diverse Anatomical Sites. Cancers, 8(10), 95. https://doi.org/10.3390/cancers8100095