
Stress Response Leading to Resistance in Glioblastoma—The Need for Innovative Radiotherapy (iRT) Concepts


{kind=link}
Abstract
:1. Introduction
Improvements in Technical Equipment Made Further Dose-Increases Possible
2. Exploitation of Hypoxia in Human Glioblastomas
3. Targeting of Heat Shock Proteins (HSPs) and Other Related Factors
4. Dose Escalation: Particle Radiotherapy for the Treatment of Gliomas—Promise or Not?
5. Vaccination Strategies for Glioblastoma
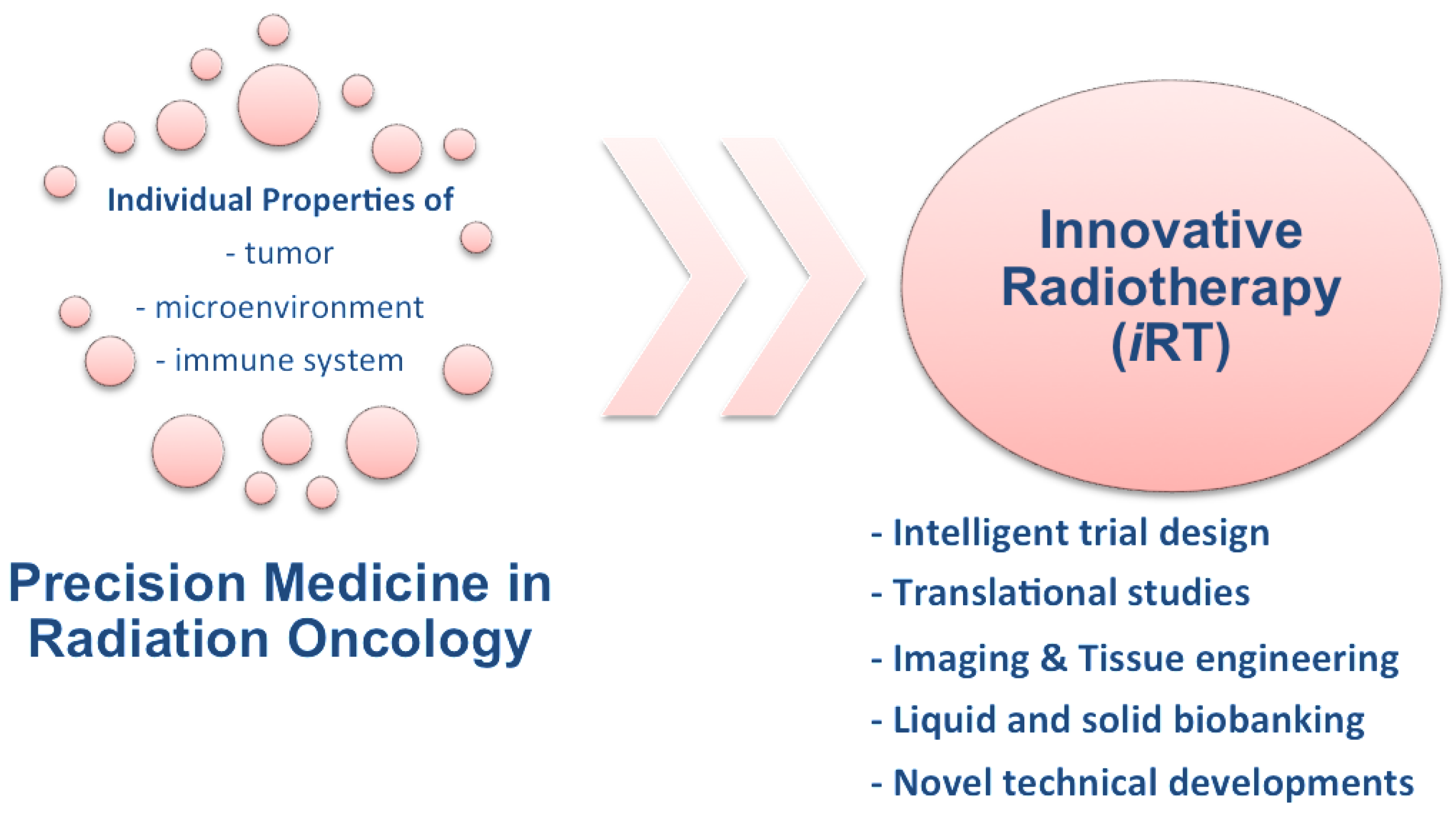
6. Innovative Radiotherapy (iRT)
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [PubMed]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes. Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Roila, F.; Group, E.G.W. Malignant glioma: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann. Oncol. 2009, 20, S126–S128. [Google Scholar] [CrossRef] [PubMed]
- Minniti, G.; Amelio, D.; Amichetti, M.; Salvati, M.; Muni, R.; Bozzao, A.; Lanzetta, G.; Scarpino, S.; Arcella, A.; Enrici, R.M. Patterns of failure and comparison of different target volume delineations in patients with glioblastoma treated with conformal radiotherapy plus concomitant and adjuvant temozolomide. Radiother. Oncol. 2010, 97, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef]
- Jaber, M.; Wolfer, J.; Ewelt, C.; Holling, M.; Hasselblatt, M.; Niederstadt, T.; Zoubi, T.; Weckesser, M.; Stummer, W. The value of 5-ALA in low-grade gliomas and high-grade gliomas lacking glioblastoma imaging features: An analysis based on fluorescence, mri, 18F-FET PET, and tumor molecular factors. Neurosurgery 2015. [Google Scholar] [CrossRef] [PubMed]
- Ringel, F.; Pape, H.; Sabel, M.; Krex, D.; Bock, H.C.; Misch, M.; Weyerbrock, A.; Westermaier, T.; Senft, C.; Schucht, P.; et al. Clinical benefit from resection of recurrent glioblastomas: Results of a multicenter study including 503 patients with recurrent glioblastomas undergoing surgical resection. Neuro-oncology 2015, 18, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; van den Bent, M.J.; Westphal, M. Cytoreductive surgery of glioblastoma as the key to successful adjuvant therapies: New arguments in an old discussion. Acta Neurochir. (Wien) 2011, 153, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.D.; Strike, T.A.; Sheline, G.E. An analysis of dose-effect relationship in the radiotherapy of malignant gliomas. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 1725–1731. [Google Scholar] [CrossRef]
- Walker, M.D.; Green, S.B.; Byar, D.P.; Alexander, E., Jr.; Batzdorf, U.; Brooks, W.H.; Hunt, W.E.; MacCarty, C.S.; Mahaley, M.S., Jr.; Mealey, J., Jr.; et al. Randomized comparisons of radiotherapy and nitrosoureas for the treatment of malignant glioma after surgery. N. Engl. J. Med. 1980, 303, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, F.J.; Herscher, L.L.; Cook, J.A.; Smith, J.; Steinberg, S.M.; Epstein, A.H.; Oldfield, E.H.; Goffman, T.E.; Kinsella, T.J.; Mitchell, J.B.; et al. National cancer institute (phase II) study of high-grade glioma treated with accelerated hyperfractionated radiation and iododeoxyuridine: Results in anaplastic astrocytoma. Int. J. Radiat. Oncol. Biol. Phys. 1994, 30, 583–590. [Google Scholar] [CrossRef]
- Curran, W.J., Jr.; Scott, C.B.; Nelson, J.S.; Weinstein, A.S.; Phillips, T.L.; Murray, K.; Fischbach, A.J.; Yakar, D.; Schwade, J.G.; Powlis, W.D.; et al. A randomized trial of accelerated hyperfractionated radiation therapy and bis-chloroethyl nitrosourea for malignant glioma. A preliminary report of radiation therapy oncology group 83–02. Cancer 1992, 70, 2909–2917. [Google Scholar] [CrossRef]
- Fitzek, M.M.; Thornton, A.F.; Rabinov, J.D.; Lev, M.H.; Pardo, F.S.; Munzenrider, J.E.; Okunieff, P.; Bussiere, M.; Braun, I.; Hochberg, F.H.; et al. Accelerated fractionated proton/photon irradiation to 90 cobalt gray equivalent for glioblastoma multiforme: Results of a phase II prospective trial. J. Neurosurg. 1999, 91, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Baumert, B.G.; Lutterbach, J.; Bernays, R.; Davis, J.B.; Heppner, F.L. Fractionated stereotactic radiotherapy boost after post-operative radiotherapy in patients with high-grade gliomas. Radiother. Oncol. 2003, 67, 183–190. [Google Scholar] [CrossRef]
- Vaupel, P.; Höckel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal. 2007, 9, 1221–1235. [Google Scholar] [CrossRef] [PubMed]
- Maftei, C.A.; Shi, K.; Bayer, C.; Astner, S.T.; Vaupel, P. Comparison of (immuno-)fluorescence data with serial [18F]fmiso PET/CT imaging for assessment of chronic and acute hypoxia in head and neck cancers. Radiother. Oncol. 2011, 99, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Evans, S.M.; Jenkins, K.W.; Jenkins, W.T.; Dilling, T.; Judy, K.D.; Schrlau, A.; Judkins, A.; Hahn, S.M.; Koch, C.J. Imaging and analytical methods as applied to the evaluation of vasculature and hypoxia in human brain tumors. Radiat. Res. 2008, 170, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P. Tumor microenvironmental physiology and its implications for radiation oncology. Semin. Radiat. Oncol. 2004, 14, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P. Hypoxia and aggressive tumor phenotype: Implications for therapy and prognosis. Oncologist 2008, 13, S21–S26. [Google Scholar] [CrossRef] [PubMed]
- Höckel, M.; Vaupel, P. Tumor hypoxia: Definitions and current clinical, biologic, and molecular aspects. J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Park, M.; Lee, D.; Mintz, A.; Androutsellis-Theotokis, A.; McKay, R.D.; Engh, J.; Iwama, T.; Kunisada, T.; Kassam, A.B.; et al. Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1α. Oncogene 2009, 28, 3949–3959. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhou, C.; Xu, L.; Xiao, H. Hypoxia enhances stemness of cancer stem cells in glioblastoma: An in vitro study. Int. J. Med. Sci. 2013, 10, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Kayama, T.; Yoshimoto, T.; Fujimoto, S.; Sakurai, Y. Intratumoral oxygen pressure in malignant brain tumor. J. Neurosurg. 1991, 74, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Rampling, R.; Cruickshank, G.; Lewis, A.D.; Fitzsimmons, S.A.; Workman, P. Direct measurement of pO2 distribution and bioreductive enzymes in human malignant brain tumors. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 427–431. [Google Scholar] [CrossRef]
- Vaupel, P. Blood flow and metabolic microenvironment of brain tumors. J. Neurooncol. 1994, 22, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, G.S.; Rampling, R.P.; Cowans, W. Direct measurement of the pO2 distribution in human malignant brain tumours. Adv. Exp. Med. Biol. 1994, 345, 465–470. [Google Scholar] [PubMed]
- Moringlane, J.R. Measurement of oxygen partial pressure in brain tumors under stereotactic conditions. Adv. Exp. Med. Biol. 1994, 345, 471–477. [Google Scholar] [PubMed]
- Collingridge, D.R.; Piepmeier, J.M.; Rockwell, S.; Knisely, J.P. Polarographic measurements of oxygen tension in human glioma and surrounding peritumoural brain tissue. Radiother. Oncol. 1999, 53, 127–131. [Google Scholar] [CrossRef]
- Beppu, T.; Kamada, K.; Yoshida, Y.; Arai, H.; Ogasawara, K.; Ogawa, A. Change of oxygen pressure in glioblastoma tissue under various conditions. J. Neurooncol. 2002, 58, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Clavo, B.; Robaina, F.; Morera, J.; Ruiz-Egea, E.; Perez, J.L.; Macias, D.; Carames, M.A.; Catala, L.; Hernandez, M.A.; Günderoth, M. Increase of brain tumor oxygenation during cervical spinal cord stimulation. Report of three cases. J. Neurosurg. 2002, 96, 94–100. [Google Scholar] [PubMed]
- Knisely, J.P.; Rockwell, S. Importance of hypoxia in the biology and treatment of brain tumors. Neuroimaging Clin. N. Am. 2002, 12, 525–536. [Google Scholar] [CrossRef]
- Evans, S.M.; Judy, K.D.; Dunphy, I.; Jenkins, W.T.; Hwang, W.T.; Nelson, P.T.; Lustig, R.A.; Jenkins, K.; Magarelli, D.P.; Hahn, S.M.; et al. Hypoxia is important in the biology and aggression of human glial brain tumors. Clin. Cancer Res. 2004, 10, 8177–8184. [Google Scholar] [CrossRef] [PubMed]
- Lally, B.E.; Colasanto, J.M.; Fischer, J.J.; Knisely, J.P. Is there an optimal hemoglobin level for patients with glioblastoma multiforme? Cancer J. 2004, 10, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Graffman, S.; Bjork, P.; Ederoth, P.; Ihse, I. Polarographic pO2 measurements of intra-abdominal adenocarcinoma in connection with intraoperative radiotherapy before and after change of oxygen concentration of anaesthetic gases. Acta Oncol. 2001, 40, 105–107. [Google Scholar] [PubMed]
- Evans, S.M.; Jenkins, K.W.; Chen, H.I.; Jenkins, W.T.; Judy, K.D.; Hwang, W.T.; Lustig, R.A.; Judkins, A.R.; Grady, M.S.; Hahn, S.M.; et al. The relationship among hypoxia, proliferation, and outcome in patients with de novo glioblastoma: A pilot study. Transl. Oncol. 2010, 3, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Said, H.M.; Hagemann, C.; Staab, A.; Stojic, J.; Kuhnel, S.; Vince, G.H.; Flentje, M.; Roosen, K.; Vordermark, D. Expression patterns of the hypoxia-related genes osteopontin, CA9, erythropoietin, VEGF and HIF-1α in human glioma in vitro and in vivo. Radiother. Oncol. 2007, 83, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Reszec, J.; Rutkowski, R.; Chyczewski, L. The expression of hypoxia-inducible factor-1 in primary brain tumors. Int. J. Neurosci. 2013, 123, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Erpolat, O.P.; Gocun, P.U.; Akmansu, M.; Ozgun, G.; Akyol, G. Hypoxia-related molecules HIF-1α, CA9, and osteopontin : Predictors of survival in patients with high-grade glioma. Strahlenther. Onkol. 2013, 189, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.; Schneider, F.; Vaupel, P.; Sommer, C.; Schmidberger, H. Differential expression of HIF-1 in glioblastoma multiforme and anaplastic astrocytoma. Int. J. Oncol. 2012, 41, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, G.A.; Karamoutsios, A.; Lallas, G.; Ragos, V.; Goussia, A.; Kyritsis, A.P.; Voulgaris, S.; Vartholomatos, G. Expression of heat shock proteins in brain tumors. Turk. Neurosurg. 2014, 24, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Rerole, A.L.; Jego, G.; Garrido, C. Hsp70: Anti-apoptotic and tumorigenic protein. Methods Mol. Biol. 2011, 787, 205–230. [Google Scholar] [PubMed]
- Liu, Y.; Zheng, T.; Zhao, S.; Liu, H.; Han, D.; Zhen, Y.; Xu, D.; Wang, Y.; Yang, H.; Zhang, G.; et al. Inhibition of heat shock protein response enhances ps-341-mediated glioma cell death. Ann. Surg. Oncol. 2012, 19, S421–S429. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, K.; Yokota, S.; Takahashi, A.; Ohnishi, T. Induction of radiation resistance by a heat shock protein inhibitor, knk437, in human glioblastoma cells. Int. J. Radiat. Biol. 2006, 82, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Neckers, K. Heat-shock protein 90 inhibitors as novel cancer chemotherapeutic agents. Expert. Opin. Emerg. Drugs 2002, 7, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lin, Y.; Guo, Z.; Cheng, J.; Huang, J.; Deng, L.; Liao, W.; Chen, Z.; Liu, Z.; Su, B. The essential role of MEKK3 in TNF-induced NF-κB activation. Nat. Immunol. 2001, 2, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. Hsp90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Morimoto, R.I. Role of the heat shock response and molecular chaperones in oncogenesis and cell death. J. Natl. Cancer Inst. 2000, 92, 1564–1572. [Google Scholar] [CrossRef] [PubMed]
- Jakubowicz-Gil, J.; Langner, E.; Badziul, D.; Wertel, I.; Rzeski, W. Silencing of Hsp27 and Hsp72 in glioma cells as a tool for programmed cell death induction upon temozolomide and quercetin treatment. Toxicol. Appl. Pharmacol. 2013, 273, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Belkacemi, L.; Hebb, M.O. Hsp27 knockdown produces synergistic induction of apoptosis by hsp90 and kinase inhibitors in glioblastoma multiforme. Anticancer. Res. 2014, 34, 4915–4927. [Google Scholar] [PubMed]
- Matokanovic, M.; Barisic, K.; Filipovic-Grcic, J.; Maysinger, D. Hsp70 silencing with sirna in nanocarriers enhances cancer cell death induced by the inhibitor of hsp90. Eur. J. Pharm. Sci. 2013, 50, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Shervington, L.; Patil, H.; Shervington, A. Could the anti-chaperone VER155008 replace temozolomide for glioma treatment. J. Cancer 2015, 6, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, Y.; Ishiwata, T.; Yoshimura, H.; Hagio, M.; Arai, T. Inhibition of nestin suppresses stem cell phenotype of glioblastomas through the alteration of post-translational modification of heat shock protein HSPA8/HSC71. Cancer Lett. 2015, 357, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.G.; Lopes, M.H.; Martins, V.R. Targeting prion protein interactions in cancer. Prion 2015, 9, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.H.; Santos, T.G.; Rodrigues, B.R.; Queiroz-Hazarbassanov, N.; Cunha, I.W.; Wasilewska-Sampaio, A.P.; Costa-Silva, B.; Marchi, F.A.; Bleggi-Torres, L.F.; Sanematsu, P.I.; et al. Disruption of prion protein-hop engagement impairs glioblastoma growth and cognitive decline and improves overall survival. Oncogene 2015, 34, 3305–3314. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.R. Heat shock proteins and protection of the nervous system. Ann. N. Y. Acad. Sci. 2007, 1113, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Khalouei, S.; Chow, A.M.; Brown, I.R. Localization of heat shock protein HSPA6 (HSP70B’) to sites of transcription in cultured differentiated human neuronal cells following thermal stress. J. Neurochem. 2014, 131, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Khalouei, S.; Chow, A.M.; Brown, I.R. Stress-induced localization of HSPA6 (HSP70B′) and HSPA1A (HSP70–1) proteins to centrioles in human neuronal cells. Cell Stress Chaperones 2014, 19, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.A.; Kim, A.V.; Samochernych, K.A.; Romanova, I.V.; Margulis, B.A.; Guzhova, I.V.; Yakovenko, I.V.; Ischenko, A.M.; Khachatryan, W.A. Pilot study of intratumoral injection of recombinant heat shock protein 70 in the treatment of malignant brain tumors in children. Onco Targets Ther. 2014, 7, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Tosti, G.; Cocorocchio, E.; Pennacchioli, E.; Ferrucci, P.F.; Testori, A.; Martinoli, C. Heat-shock proteins-based immunotherapy for advanced melanoma in the era of target therapies and immunomodulating agents. Expert. Opin. Biol. Ther. 2014, 14, 955–967. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.A.; Han, S.J.; Ahn, B.; Oehlke, J.; Kivett, V.; Fedoroff, A.; Butowski, N.; Chang, S.M.; Clarke, J.; Berger, M.S.; et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin. Cancer Res. 2013, 19, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Botzler, C.; Jennen, L.; Schmidt, J.; Ellwart, J.; Issels, R. Heat shock protein 72 on tumor cells: A recognition structure for natural killer cells. J. Immunol. 1997, 158, 4341–4350. [Google Scholar] [PubMed]
- Hantschel, M.; Pfister, K.; Jordan, A.; Scholz, R.; Andreesen, R.; Schmitz, G.; Schmetzer, H.; Hiddemann, W.; Multhoff, G. Hsp70 plasma membrane expression on primary tumor biopsy material and bone marrow of leukemic patients. Cell Stress Chaperones 2000, 5, 438–442. [Google Scholar] [CrossRef]
- Gehrmann, M.; Schmetzer, H.; Eissner, G.; Haferlach, T.; Hiddemann, W.; Multhoff, G. Membrane-bound heat shock protein 70 (Hsp70) in acute myeloid leukemia: A tumor specific recognition structure for the cytolytic activity of autologous nk cells. Haematologica 2003, 88, 474–476. [Google Scholar] [PubMed]
- Gross, C.; Hansch, D.; Gastpar, R.; Multhoff, G. Interaction of heat shock protein 70 peptide with NK cells involves the NK receptor CD94. Biol. Chem. 2003, 384, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Pfister, K.; Botzler, C.; Jordan, A.; Scholz, R.; Schmetzer, H.; Burgstahler, R.; Hiddemann, W. Adoptive transfer of human natural killer cells in mice with severe combined immunodeficiency inhibits growth of hsp70-expressing tumors. Int. J. Cancer 2000, 88, 791–797. [Google Scholar] [CrossRef]
- Multhoff, G.; Pfister, K.; Gehrmann, M.; Hantschel, M.; Gross, C.; Hafner, M.; Hiddemann, W. A 14-mer Hsp70 peptide stimulates natural killer (NK) cell activity. Cell Stress Chaperones 2001, 6, 337–344. [Google Scholar] [CrossRef]
- Krause, S.W.; Gastpar, R.; Andreesen, R.; Gross, C.; Ullrich, H.; Thonigs, G.; Pfister, K.; Multhoff, G. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous natural killer cells: A clinical phase i trial. Clin. Cancer Res. 2004, 10, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G. Heat shock protein 72 (HSP72), a hyperthermia-inducible immunogenic determinant on leukemic K562 and ewing’s sarcoma cells. Int. J. Hyperthermia 1997, 13, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, M.; Pfister, K.; Hutzler, P.; Gastpar, R.; Margulis, B.; Multhoff, G. Effects of antineoplastic agents on cytoplasmic and membrane-bound heat shock protein 70 (HSP70) levels. Biol. Chem. 2002, 383, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, M.; Brunner, M.; Pfister, K.; Reichle, A.; Kremmer, E.; Multhoff, G. Differential up-regulation of cytosolic and membrane-bound heat shock protein 70 in tumor cells by anti-inflammatory drugs. Clin. Cancer Res. 2004, 10, 3354–3364. [Google Scholar] [CrossRef] [PubMed]
- Gastpar, R.; Gross, C.; Rossbacher, L.; Ellwart, J.; Riegger, J.; Multhoff, G. The cell surface-localized heat shock protein 70 epitope TKD induces migration and cytolytic activity selectively in human NK cells. J. Immunol. 2004, 172, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Pasi, F.; Paolini, A.; Nano, R.; Di Liberto, R.; Capelli, E. Effects of single or combined treatments with radiation and chemotherapy on survival and danger signals expression in glioblastoma cell lines. Biomed. Res. Int. 2014, 2014, 453497. [Google Scholar] [CrossRef] [PubMed]
- Beaman, G.M.; Dennison, S.R.; Chatfield, L.K.; Phoenix, D.A. Reliability of HSP70 (HSPA) expression as a prognostic marker in glioma. Mol. Cell Biochem. 2014, 393, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Breuninger, S.; Erl, J.; Knape, C.; Gunther, S.; Regel, I.; Rödel, F.; Gaipl, U.; Thorsteinsdottir, J.; Giannitrapani, L.; Dickinson, A.; et al. Quantitative analysis of liposomal heat shock protein 70 (HSP70) in the blood of tumor patients using a novel liphsp70 elisa. J. Clin. Cell. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Ramaekers, B.L.; Pijls-Johannesma, M.; Joore, M.A.; van den Ende, P.; Langendijk, J.A.; Lambin, P.; Kessels, A.G.; Grutters, J.P. Systematic review and meta-analysis of radiotherapy in various head and neck cancers: Comparing photons, carbon-ions and protons. Cancer Treat. Rev. 2011, 37, 185–201. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Suzuki, M.; Abe, A.; Suzuki, Y.; Morita, S.; Mizoe, J.; Sato, S.; Miyamoto, T.; Kamada, T.; Kato, H.; et al. The phase I/II clinical study of carbon ion therapy for cancer of the uterine cervix. Cancer J. Sci. Am. 1999, 5, 362–369. [Google Scholar] [PubMed]
- Weyrather, W.K.; Kraft, G. Rbe of carbon ions: Experimental data and the strategy of rbe calculation for treatment planning. Radiother. Oncol. 2004, 73, S161–S169. [Google Scholar] [CrossRef]
- Allen, A.M.; Pawlicki, T.; Dong, L.; Fourkal, E.; Buyyounouski, M.; Cengel, K.; Plastaras, J.; Bucci, M.K.; Yock, T.I.; Bonilla, L.; et al. An evidence based review of proton beam therapy: The report of astro’s emerging technology committee. Radiother. Oncol. 2012, 103, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Alan Mitteer, R.; Wang, Y.; Shah, J.; Gordon, S.; Fager, M.; Butter, P.P.; Jun Kim, H.; Guardiola-Salmeron, C.; Carabe-Fernandez, A.; Fan, Y. Proton beam radiation induces DNA damage and cell apoptosis in glioma stem cells through reactive oxygen species. Sci. Rep. 2015, 5, 13961. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, C.; Piro, S.; Tabbi, G.; Ragusa, M.; Di Pietro, V.; Zimmitti, V.; Cuda, F.; Anello, M.; Consoli, U.; Salinaro, E.T.; et al. Cellular and molecular effects of protons: Apoptosis induction and potential implications for cancer therapy. Apoptosis 2006, 11, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.C.; Lomax, A.J.; Rutz, H.P.; Stadelmann, O.; Egger, E.; Timmermann, B.; Pedroni, E.S.; Verwey, J.; Miralbell, R.; Goitein, G.; et al. Spot-scanning proton radiation therapy for recurrent, residual or untreated intracranial meningiomas. Radiother. Oncol. 2004, 71, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, M.; Tsuboi, K.; Igaki, H.; Yamamoto, T.; Takano, S.; Oshiro, Y.; Hayashi, Y.; Hashii, H.; Kanemoto, A.; Nakayama, H.; et al. Phase I/II trial of hyperfractionated concomitant boost proton radiotherapy for supratentorial glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, M.; Yamamoto, T.; Takano, S.; Ishikawa, E.; Matsumura, A.; Ishikawa, H.; Okumura, T.; Sakurai, H.; Miyatake, S.; Tsuboi, K. Long-term survival after treatment of glioblastoma multiforme with hyperfractionated concomitant boost proton beam therapy. Pract. Radiat. Oncol. 2015, 5, e9–e16. [Google Scholar] [CrossRef] [PubMed]
- Akino, Y.; Teshima, T.; Kihara, A.; Kodera-Suzumoto, Y.; Inaoka, M.; Higashiyama, S.; Furusawa, Y.; Matsuura, N. Carbon-ion beam irradiation effectively suppresses migration and invasion of human non-small-cell lung cancer cells. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Teshima, T.; Kagawa, K.; Hishikawa, Y.; Takahashi, Y.; Kawaguchi, A.; Suzumoto, Y.; Nojima, K.; Furusawa, Y.; Matsuura, N. Particle irradiation suppresses metastatic potential of cancer cells. Cancer Res. 2005, 65, 113–120. [Google Scholar] [PubMed]
- Ohno, T. Particle radiotherapy with carbon ion beams. EPMA J. 2013, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Combs, S.E.; Schulz-Ertner, D.; Debus, J.; von Deimling, A.; Hartmann, C. Improved correlation of the neuropathologic classification according to adapted world health organization classification and outcome after radiotherapy in patients with atypical and anaplastic meningiomas. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Combs, S.E.; Bohl, J.; Elsasser, T.; Weber, K.J.; Schulz-Ertner, D.; Debus, J.; Weyrather, W.K. Radiobiological evaluation and correlation with the local effect model (LEM) of carbon ion radiation therapy and temozolomide in glioblastoma cell lines. Int. J. Radiat. Biol. 2009, 85, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Mizoe, J.E.; Tsujii, H.; Hasegawa, A.; Yanagi, T.; Takagi, R.; Kamada, T.; Tsuji, H.; Takakura, K.; Organizing Committee of the Central Nervous System Tumor Working Group. Phase I/II clinical trial of carbon ion radiotherapy for malignant gliomas: Combined X-ray radiotherapy, chemotherapy, and carbon ion radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Combs, S.E.; Bruckner, T.; Mizoe, J.E.; Kamada, T.; Tsujii, H.; Kieser, M.; Debus, J. Comparison of carbon ion radiotherapy to photon radiation alone or in combination with temozolomide in patients with high-grade gliomas: Explorative hypothesis-generating retrospective analysis. Radiother. Oncol. 2013, 108, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Mitchell, D.A. Vaccination strategies for neuro-oncology. Neuro-Oncology 2015, 17, vii15–vii25. [Google Scholar] [CrossRef] [PubMed]
- Tagliamonte, M.; Petrizzo, A.; Tornesello, M.L.; Buonaguro, F.M.; Buonaguro, L. Antigen-specific vaccines for cancer treatment. Hum. Vaccin. Immunother. 2014, 10, 3332–3346. [Google Scholar] [CrossRef] [PubMed]
- Freitas, M.; Malheiros, S.; Stavale, J.N.; Biassi, T.P.; Zamuner, F.T.; de Souza Begnami, M.; Soares, F.A.; Vettore, A.L. Expression of cancer/testis antigens is correlated with improved survival in glioblastoma. Oncotarget 2013, 4, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.W.; Chow, K.K.; Lim, M.; Li, G. Current vaccine trials in glioblastoma: A review. J. Immunol. Res. 2014, 2014, 796856. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Black, K.L.; Martin, N.A.; Sykes, S.N.; Bronstein, J.M.; Jouben-Steele, L.; Mischel, P.S.; Belldegrun, A.; Cloughesy, T.F. Treatment of a patient by vaccination with autologous dendritic cells pulsed with allogeneic major histocompatibility complex class i-matched tumor peptides. Case report. Neurosurg. Focus 2000, 9, e8. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Prins, R.M.; Kiertscher, S.M.; Odesa, S.K.; Kremen, T.J.; Giovannone, A.J.; Lin, J.W.; Chute, D.J.; Mischel, P.S.; Cloughesy, T.F.; et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin. Cancer Res. 2005, 11, 5515–5525. [Google Scholar] [CrossRef] [PubMed]
- Freije, W.A.; Castro-Vargas, F.E.; Fang, Z.; Horvath, S.; Cloughesy, T.; Liau, L.M.; Mischel, P.S.; Nelson, S.F. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004, 64, 6503–6510. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Prins, R.M.; Soto, H.; Konkankit, V.; Odesa, S.K.; Eskin, A.; Yong, W.H.; Nelson, S.F.; Liau, L.M. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin. Cancer Res. 2011, 17, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current therapeutic advances targeting egfr and egfrviii in glioblastoma. Front. Oncol. 2015, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.J.; Ruppert, J.M.; Bigner, S.H.; Grzeschik, C.H.; Humphrey, P.A.; Bigner, D.S.; Vogelstein, B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc. Natl. Acad. Sci. USA 1992, 89, 2965–2969. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E., 2nd; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant iii peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, C.A.; Li, G.; Wong, A.J. Targeting egf receptor variant III: Tumor-specific peptide vaccination for malignant gliomas. Expert. Rev. Vaccines 2012, 11, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Mitra, S.; Wong, A.J. The epidermal growth factor variant III peptide vaccine for treatment of malignant gliomas. Neurosurg. Clin. N. Am. 2010, 21, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Fenton, T.R.; Nathanson, D.; Ponte de Albuquerque, C.; Kuga, D.; Iwanami, A.; Dang, J.; Yang, H.; Tanaka, K.; Oba-Shinjo, S.M.; Uno, M.; et al. Resistance to EGF receptor inhibitors in glioblastoma mediated by phosphorylation of the pten tumor suppressor at tyrosine 240. Proc. Natl. Acad. Sci. USA 2012, 109, 14164–14169. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Homma, J.; Yajima, N.; Tsuchiya, N.; Sano, M.; Kobayashi, T.; Yoshida, S.; Abe, T.; Narita, M.; Takahashi, M.; et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: Results of a clinical phase I/II trial. Clin. Cancer Res. 2005, 11, 4160–4167. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Anido, J.; Saez-Borderias, A.; Gonzalez-Junca, A.; Rodon, L.; Folch, G.; Carmona, M.A.; Prieto-Sanchez, R.M.; Barba, I.; Martinez-Saez, E.; Prudkin, L.; et al. TGF-β receptor inhibitors target the CD44high/id1high glioma-initiating cell population in human glioblastoma. Cancer Cell 2010, 18, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Dai, Q.; Min, Z.; Zhang, M. Transforming growth factor β1 enhances the invasiveness of human glioma cell line via ERK/MAPK pathway. Nan Fang Yi Ke Da Xue Xue Bao 2013, 33, 1744–1747. [Google Scholar] [PubMed]
- Rich, J.N.; Zhang, M.; Datto, M.B.; Bigner, D.D.; Wang, X.F. Transforming growth factor-β-mediated p15ink4b induction and growth inhibition in astrocytes is SMAD3-dependent and a pathway prominently altered in human glioma cell lines. J. Biol. Chem. 1999, 274, 35053–35058. [Google Scholar] [CrossRef] [PubMed]
- Combs, S.E.; Ernsberger, U.; Krieglstein, K.; Unsicker, K. Reduction of endogenous TGF-β does not affect phenotypic development of sympathoadrenal progenitors into adrenal chromaffin cells. Mech. Dev. 2001, 109, 295–302. [Google Scholar] [CrossRef]
- Combs, S.E.; Krieglstein, K.; Unsicker, K. Reduction of endogenous TGF-β increases proliferation of developing adrenal chromaffin cells in vivo. J. Neurosci. Res. 2000, 59, 379–383. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Combs, S.E.; Schmid, T.E.; Vaupel, P.; Multhoff, G. Stress Response Leading to Resistance in Glioblastoma—The Need for Innovative Radiotherapy (iRT) Concepts. Cancers 2016, 8, 15. https://doi.org/10.3390/cancers8010015
Combs SE, Schmid TE, Vaupel P, Multhoff G. Stress Response Leading to Resistance in Glioblastoma—The Need for Innovative Radiotherapy (iRT) Concepts. Cancers. 2016; 8(1):15. https://doi.org/10.3390/cancers8010015
Chicago/Turabian StyleCombs, Stephanie E., Thomas E. Schmid, Peter Vaupel, and Gabriele Multhoff. 2016. "Stress Response Leading to Resistance in Glioblastoma—The Need for Innovative Radiotherapy (iRT) Concepts" Cancers 8, no. 1: 15. https://doi.org/10.3390/cancers8010015
APA StyleCombs, S. E., Schmid, T. E., Vaupel, P., & Multhoff, G. (2016). Stress Response Leading to Resistance in Glioblastoma—The Need for Innovative Radiotherapy (iRT) Concepts. Cancers, 8(1), 15. https://doi.org/10.3390/cancers8010015