
Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth

 ,
,
Abstract
:1. Introduction
2. Characteristics of CAFs
2.1. Markers of CAFs

{kind=link}
{kind=link}
{kind=link}
| Positive Marker | Negative Marker |
|---|---|
| α-SMA | Cytokeratin |
| Fibroblast activation protein | CD31 |
| tenascin-C | |
| periostin | |
| Neuron glial antigen-2 | |
| Vimentin | |
| Desmin | |
| Platelet derived growth factor receptor | |
| Fibroblast specific protein-1 |
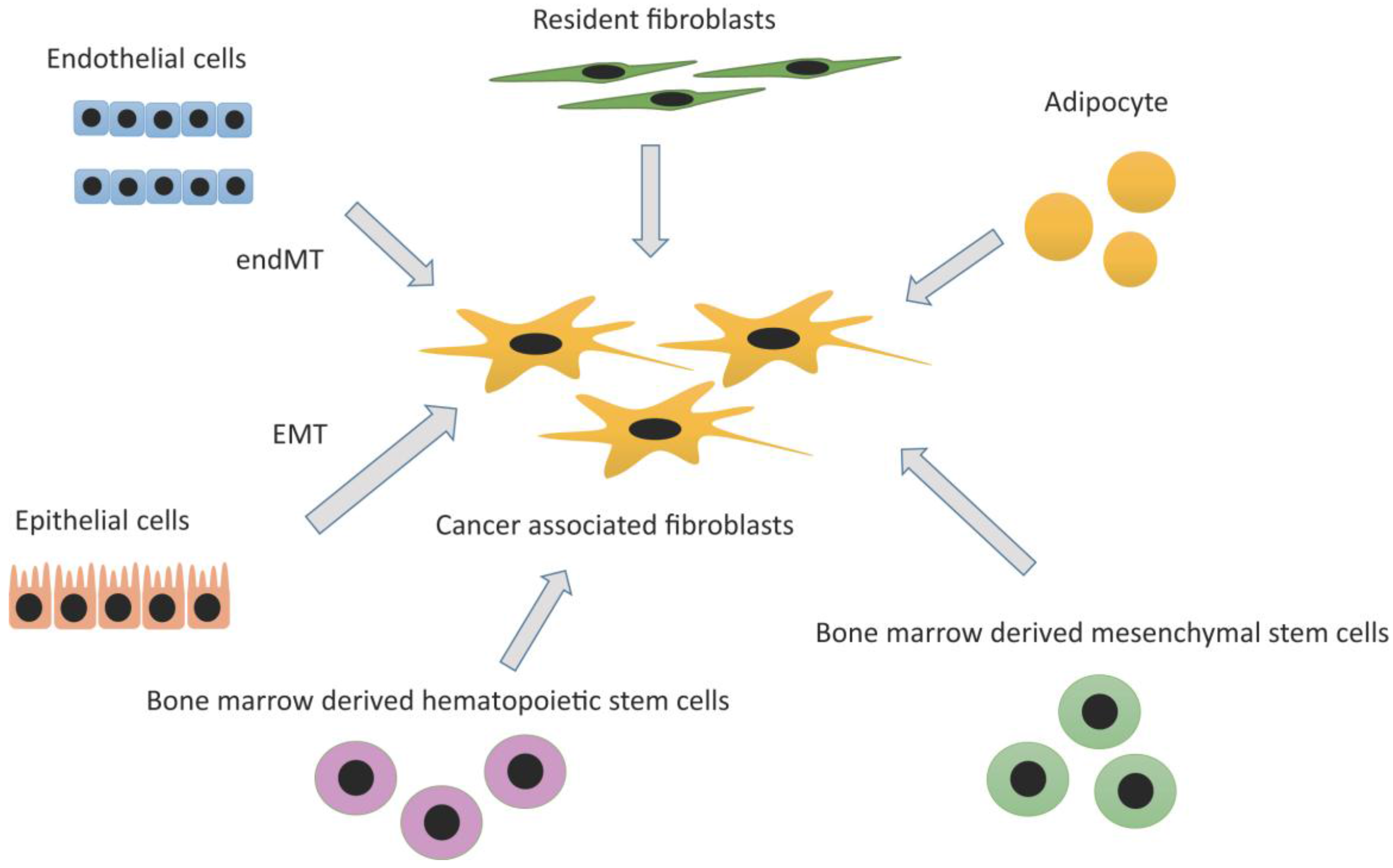
2.2. Heterogeneity and Origins of CAFs
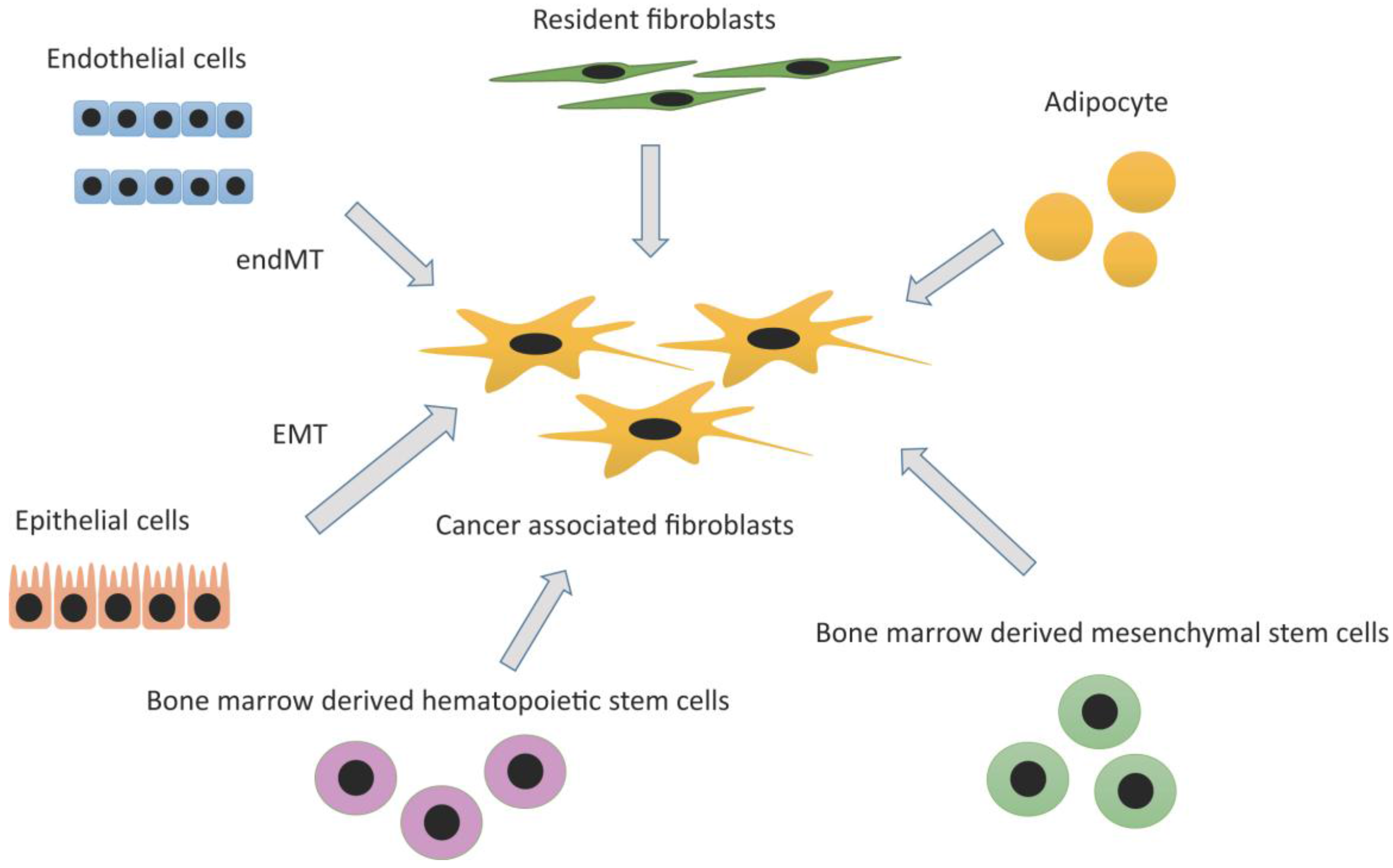
2.2.1. Resident Fibroblasts
2.2.2. Adipocytes
2.2.3. Bone Marrow-Derived Mesenchymal Stem Cell (MSC) and Hematopoietic Stem Cell (HSC)
2.2.4. Epithelial Cells: Epithelial Mesenchymal Transition (EMT)
2.2.5. Endothelial Cells: Endothelial-Mesenchymal Transition (EndMT)
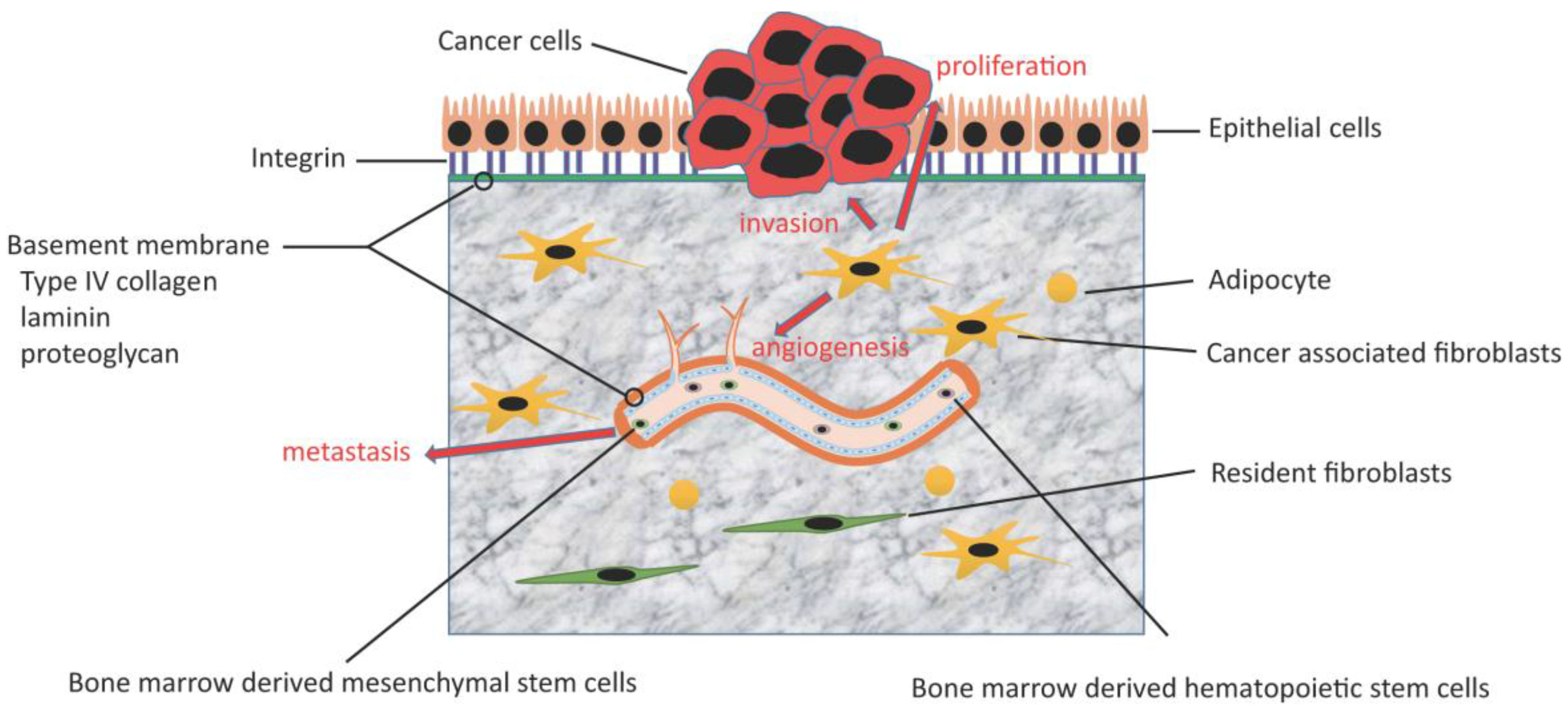
3. The Role of CAFs
3.1. Tumor-Stroma Interaction
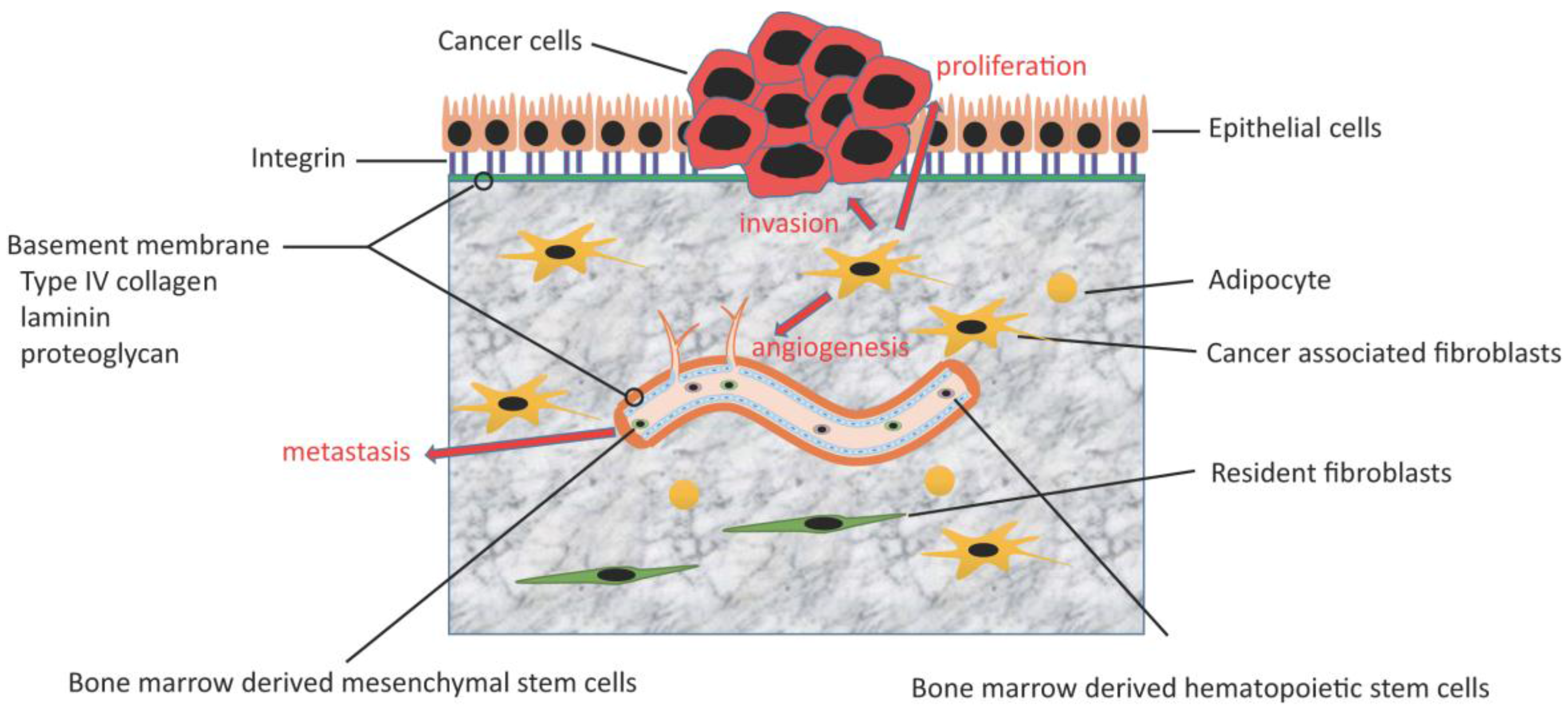
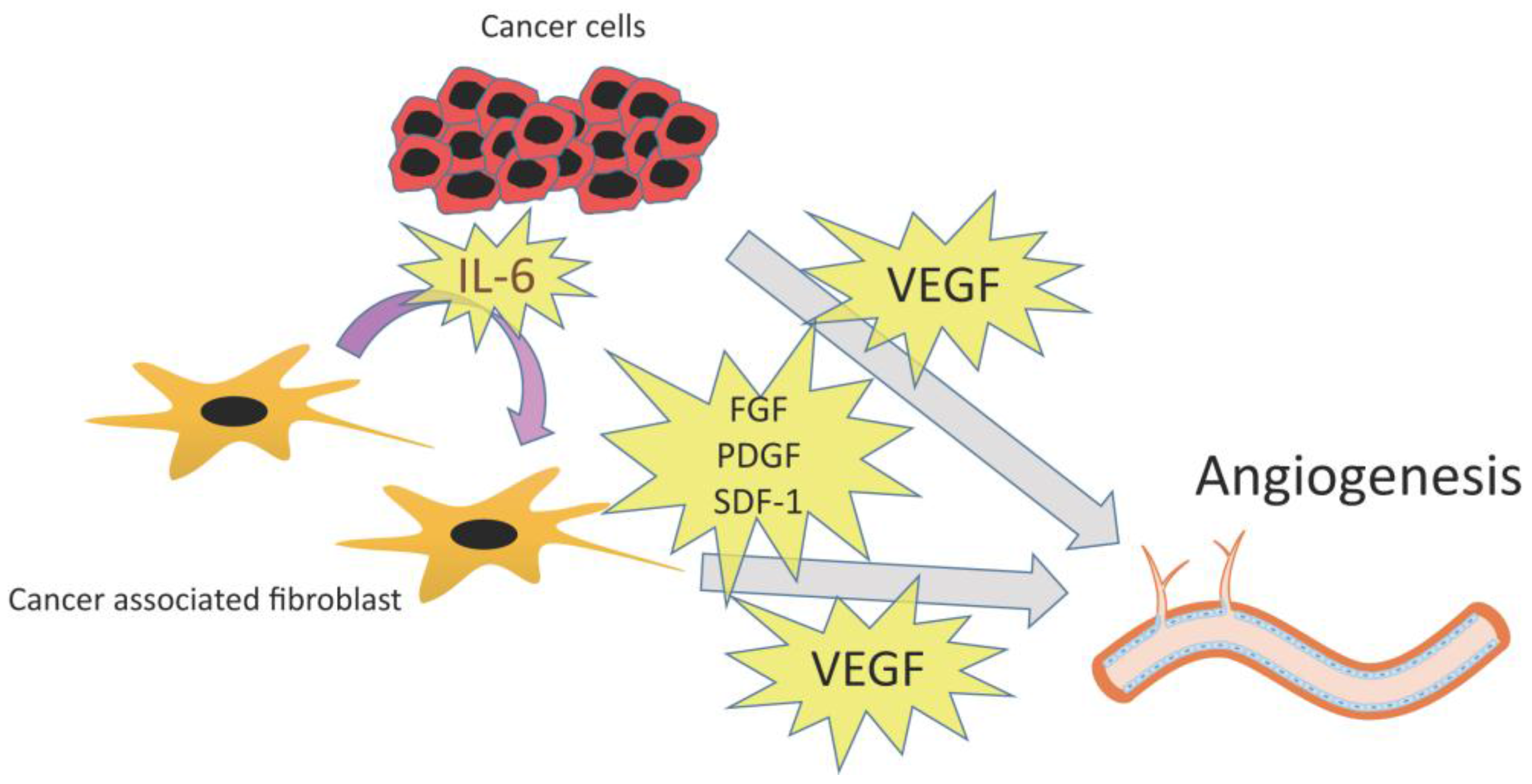
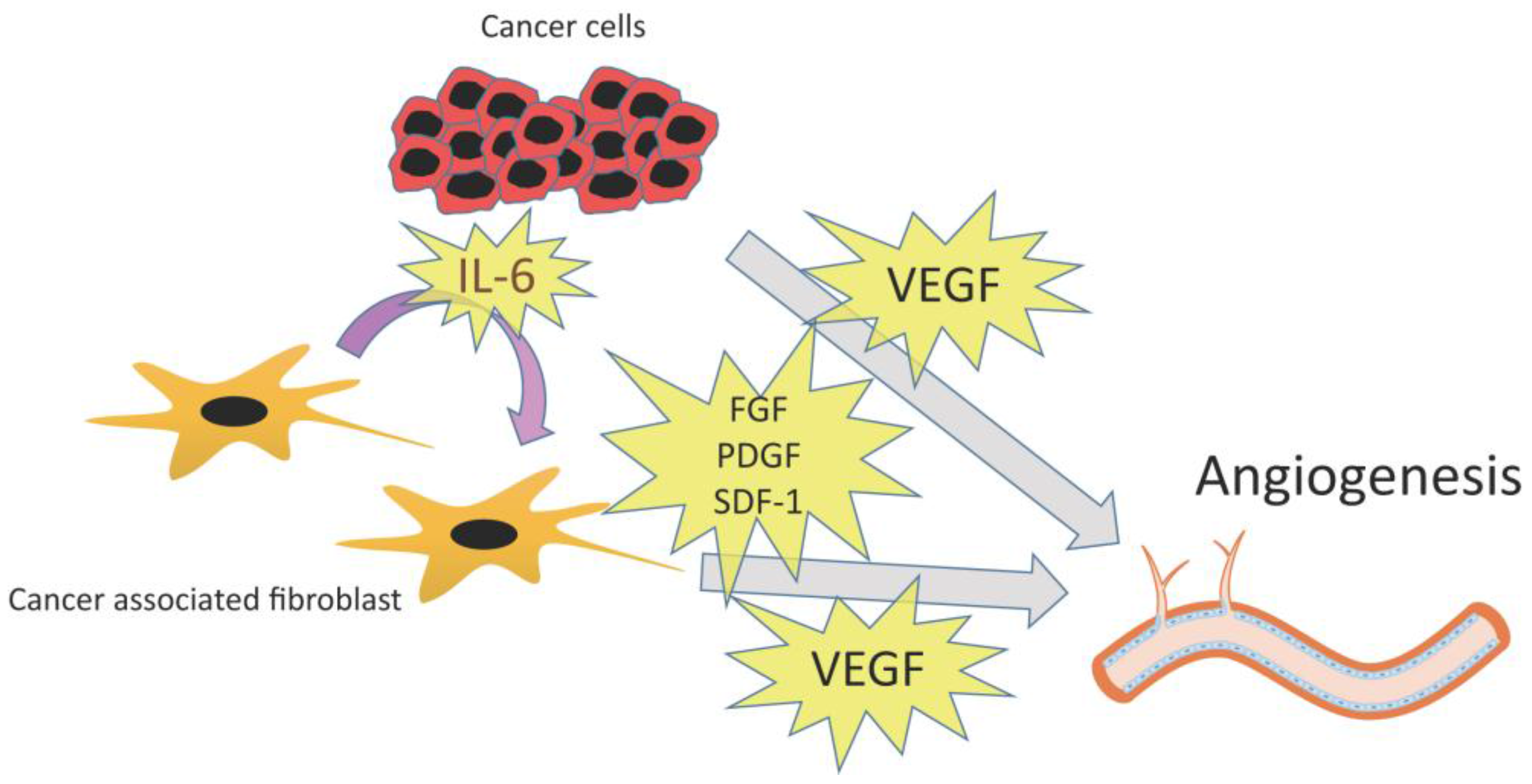
3.2. Angiogenesis
3.2.1. VEGF
3.2.2. Other Angiogenic Factors: PDGF, FGF, and SDF-1
3.3. Metabolism of CAFs: the Warburg Effect and Reverse Warburg Effect
3.4. Chemoresistance
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CAFs | cancer-associated fibroblasts |
| IL-6 | interleukin-6 |
| α-SMA | α-smooth muscle actin |
| FAP | fibroblast activation protein |
| NG2 | neuron glial antigen-2 |
| PDGFR | platelet derived growth factor receptor |
| FSP-1 | fibroblast specific protein-1 |
| EMT | epithelial- mesenchymal transition |
| EndMT | endothelial-mesenchymal transition |
| ROS | reactive oxygen species |
| HIF | hypoxia-inducible factor |
| BM-MSCs | bone marrow derived mesenchymal stem cells |
| TGFβ-1 | transforming growth factor-β1 |
| LPS | lipopolysaccharide |
| ASCs | adipose tissue-derived stem cells |
| CAAs | cancer associated adipocytes |
| MSC | mesenchymal stem cell |
| HSC | hematopoietic stem cell |
| OPN | osteopontin |
| MZF1 | myeloid zinc finger 1 |
| EGFP | enhanced green fluorescent protein |
| ECM | extracellular matrix |
| SDF-1 | stromal-cell-derived factor 1 |
| MMP | matrix metalloproteinase |
| TNF-α | Tumor necrosis factor-α |
| VEGF | vascular endothelial growth factor |
| FGF | fibroblast growth factor |
| PDGF | platelet-derived growth factor |
| IGF | insulin-like growth factor |
| STC-1 | glycoprotein stanniocalcin-1 |
| mTOR | mammalian target of rapamycin |
| SFM-DR | soluble factor-mediated drug resistance |
| CAM-DR | cell adhesion-mediated drug resistance |
| HGF | hepatocyte growth factor |
| EGFR | epidermal growth factor receptor |
| CSCs | cancer stem cells |
| CICs | cancer-initiating cells |
| IFP | interstitial fluid pressure |
| HMGB1 | high mobility group box 1 |
References
- Paget, S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989, 8, 98–101. [Google Scholar] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- De Wever, O.; Demetter, P.; Mareel, M.; Bracke, M. Stromal myofibroblasts are drivers of invasive cancer growth. Int. J. Cancer 2008, 123, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Sappino, A.P.; Skalli, O.; Jackson, B.; Schürch, W.; Gabbiani, G. Smooth-muscle differentiation in stromal cells of malignant and non-malignant breast tissues. Int. J. Cancer 1988, 41, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Serini, G.; Bochaton-Piallat, M.L.; Ropraz, P.; Geinoz, A.; Borsi, L.; Zardi, L.; Gabbiani, G. The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-β1. J. Cell Biol. 1998, 142, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Weinberg, R.A. Heterogeneity of stromal fibroblasts in tumors. Cancer Biol. Ther. 2007, 6, 618–619. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Lenter, M.C.; Zimmermann, R.N.; Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J. Biol. Chem. 1999, 274, 36505–36512. [Google Scholar] [CrossRef] [PubMed]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Wikberg, M.L.; Edin, S.; Lundberg, I.V.; van Guelpen, B.; Dahlin, A.M.; Rutegard, J.; Stenling, R.; Oberg, A.; Palmqvist, R. High intratumoral expression of fibroblast activation protein (FAP) in colon cancer is associated with poorer patient prognosis. Tumour Biol. 2013, 34, 1013–1020. [Google Scholar] [CrossRef] [PubMed]
- Dohi, O.; Ohtani, H.; Hatori, M.; Sato, E.; Hosaka, M.; Nagura, H.; Itoi, E.; Kokubun, S. Histogenesis-specific expression of fibroblast activation protein and dipeptidylpeptidase-iv in human bone and soft tissue tumours. Histopathology 2009, 55, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Mentlein, R.; Hattermann, K.; Hemion, C.; Jungbluth, A.A.; Held-Feindt, J. Expression and role of the cell surface protease seprase/fibroblast activation protein-α (FAP-α) in astroglial tumors. Biol. Chem. 2011, 392, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Akatsuka, T.; Imanaka-Yoshida, K. Tenascin-c and integrins in cancer. Cell Adh. Migr. 2015, 9, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, Y.; Kashima, T.G.; Nishiyama, T.; Shimazu, K.; Morishita, Y.; Shimazaki, M.; Kii, I.; Horie, H.; Nagai, H.; Kudo, A.; et al. Periostin is expressed in pericryptal fibroblasts and cancer-associated fibroblasts in the colon. J. Histochem. Cytochem. 2008, 56, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Togo, S.; Polanska, U.M.; Horimoto, Y.; Orimo, A. Carcinoma-associated fibroblasts are a promising therapeutic target. Cancers 2013, 5, 149–169. [Google Scholar] [CrossRef] [PubMed]
- Xing, F.; Saidou, J.; Watabe, K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci. (Landmark Ed.) 2010, 15, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Sukowati, C.H.; Anfuso, B.; Croce, L.S.; Tiribelli, C. The role of multipotent cancer associated fibroblasts in hepatocarcinogenesis. BMC Cancer 2015, 15, 188. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Livingstone, J.; Buchanan, M.; Reid, J.F.; Hallett, M.; Basik, M. A functional in vitro model of heterotypic interactions reveals a role for interferon-positive carcinoma associated fibroblasts in breast cancer. BMC Cancer 2015, 15, 130. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Giannoni, E.; Segura, C.; Barcellos-de-Souza, P.; Raspollini, M.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Qu, C.; Chen, H.; Xu, L.; Qi, Q.; Luo, J.; Wang, K.; Meng, Z.; Chen, Z.; Wang, P.; et al. Chinese herbal medicine suppresses invasion-promoting capacity of cancer-associated fibroblasts in pancreatic cancer. PLoS ONE 2014, 9, e96177. [Google Scholar] [CrossRef] [PubMed]
- Massani, M.; Stecca, T.; Fabris, L.; Caratozzolo, E.; Ruffolo, C.; Furlanetto, A.; Morton, S.; Cadamuro, M.; Strazzabosco, M.; Bassi, N. Isolation and characterization of biliary epithelial and stromal cells from resected human cholangiocarcinoma: A novel in vitro model to study tumor-stroma interactions. Oncol. Rep. 2013, 30, 1143–1148. [Google Scholar] [CrossRef] [PubMed]
- Gorchs, L.; Hellevik, T.; Bruun, J.A.; Camilio, K.A.; Al-Saad, S.; Stuge, T.B.; Martinez-Zubiaurre, I. Cancer-associated fibroblasts from lung tumors maintain their immunosuppressive abilities after high-dose irradiation. Front. Oncol. 2015, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Wang, Z.; Zhai, L.; Yang, M.; Shan, L.; Chai, C.; Liu, M.; Wang, L. Effects of cancer-associated fibroblasts on the migration and invasion abilities of SGC-7901 gastric cancer cells. Oncol. Lett. 2013, 5, 609–612. [Google Scholar] [PubMed]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.R.; Baker, D.; Farren, M.; Pommier, A.; Swann, R.; Wang, X.; Mistry, S.; McDaid, K.; Kendrew, J.; Womack, C.; et al. Tumor stromal architecture can define the intrinsic tumor response to VEGF-targeted therapy. Clin. Cancer Res. 2013, 19, 6943–6956. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. Micrornas reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef] [PubMed]
- Toullec, A.; Gerald, D.; Despouy, G.; Bourachot, B.; Cardon, M.; Lefort, S.; Richardson, M.; Rigaill, G.; Parrini, M.C.; Lucchesi, C.; et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol. Med. 2010, 2, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Niu, Y.; Yeh, S.; Chang, C. BM-MSCS promote prostate cancer progression via the conversion of normal fibroblasts to cancer-associated fibroblasts. Int. J. Oncol. 2015, 47, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, K.; Murase, N.; Stolz, D.B.; Toyokawa, H.; O’Donnell, D.R.; Smith, D.M.; Dudas, J.R.; Rubin, J.P.; Marra, K.G. Characterization of transplanted green fluorescent protein+ bone marrow cells into adipose tissue. Stem Cells 2008, 26, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; McDonald, L.T.; Russell, D.L.; Kelly, R.R.; Wilson, K.R.; Mehrotra, M.; Soloff, A.C.; LaRue, A.C. Hematopoietic stem cell-derived adipocytes and fibroblasts in the tumor microenvironment. World J. Stem Cells 2015, 7, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Buache, E.; Chenard, M.P.; Dali-Youcef, N.; Rio, M.C. Adipocyte is a non-trivial, dynamic partner of breast cancer cells. Int. J. Dev. Biol. 2011, 55, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Motrescu, E.R.; Rio, M.C. Cancer cells, adipocytes and matrix metalloproteinase 11: A vicious tumor progression cycle. Biol. Chem. 2008, 389, 1037–1041. [Google Scholar] [CrossRef] [PubMed]
- Jotzu, C.; Alt, E.; Welte, G.; Li, J.; Hennessy, B.T.; Devarajan, E.; Krishnappa, S.; Pinilla, S.; Droll, L.; Song, Y.H. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Anal. Cell. Pathol. 2010, 33, 61–79. [Google Scholar] [CrossRef]
- Ishii, G.; Sangai, T.; Oda, T.; Aoyagi, Y.; Hasebe, T.; Kanomata, N.; Endoh, Y.; Okumura, C.; Okuhara, Y.; Magae, J.; et al. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem. Biophys. Res. Commun. 2003, 309, 232–240. [Google Scholar] [CrossRef]
- Direkze, N.C.; Hodivala-Dilke, K.; Jeffery, R.; Hunt, T.; Poulsom, R.; Oukrif, D.; Alison, M.R.; Wright, N.A. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004, 64, 8492–8495. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.E.; Kothari, A.N.; Wai, P.Y.; Li, N.Y.; Driver, J.; Zapf, M.A.; Franzen, C.A.; Gupta, G.N.; Osipo, C.; Zlobin, A.; et al. Osteopontin mediates an MZF1-TGF-β1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene 2015, 34, 4821–4833. [Google Scholar] [CrossRef] [PubMed]
- McDonald, L.T.; LaRue, A.C. Hematopoietic stem cell derived carcinoma-associated fibroblasts: A novel origin. Int. J. Clin. Exp. Pathol. 2012, 5, 863–873. [Google Scholar] [PubMed]
- Ogawa, M.; LaRue, A.C.; Drake, C.J. Hematopoietic origin of fibroblasts/myofibroblasts: Its pathophysiologic implications. Blood 2006, 108, 2893–2896. [Google Scholar] [CrossRef] [PubMed]
- Greenburg, G.; Hay, E.D. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Tse, J.C.; Kalluri, R. Mechanisms of metastasis: Epithelial-to-mesenchymal transition and contribution of tumor microenvironment. J. Cell. Biochem. 2007, 101, 816–829. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Petersen, O.W.; Lind Nielsen, H.; Gudjonsson, T.; Villadsen, R.; Ronnov-Jessen, L.; Bissell, M.J. The plasticity of human breast carcinoma cells is more than epithelial to mesenchymal conversion. Breast Cancer Res. 2001, 3, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, C.P.; Pan, J.Y.; Zheng, W.W.; Cao, X.J.; Fan, G.K. Cancer-associated fibroblasts in a human hep-2 established laryngeal xenografted tumor are not derived from cancer cells through epithelial-mesenchymal transition, phenotypically activated but karyotypically normal. PLoS ONE 2015, 10, e0117405. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Watanabe-Takano, H.; Takano, K.; Hatano, M.; Tokuhisa, T.; Endo, T. DA-Raf-mediated suppression of the Ras-ERK pathway is essential for TGF-β1-induced epithelial-mesenchymal transition in alveolar epithelial type 2 cells. PLoS ONE 2015, 10, e0127888. [Google Scholar] [CrossRef] [PubMed]
- Markwald, R.R.; Fitzharris, T.P.; Smith, W.N. Sturctural analysis of endocardial cytodifferentiation. Dev. Biol. 1975, 42, 160–180. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Potenta, S.; Xie, L.; Zeisberg, M.; Kalluri, R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007, 67, 10123–10128. [Google Scholar] [CrossRef] [PubMed]
- Potenta, S.; Zeisberg, E.; Kalluri, R. The role of endothelial-to-mesenchymal transition in cancer progression. Br. J. Cancer 2008, 99, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Wang, N.; Zhang, T.C. The role of endothelial-mesenchymal transition in development and pathological process. IUBMB Life 2012, 64, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, Y.; Asai, N.; Enomoto, A.; Kato, T.; Mii, S.; Kondo, Y.; Ushida, K.; Niimi, K.; Tsunoda, N.; Nagino, M.; et al. Akt-girdin signaling in cancer-associated fibroblasts contributes to tumor progression. Cancer Res. 2015, 75, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Garcia-Palmero, I.; Herrera, M.; Bartolome, R.A.; Pena, C.; Fernandez-Acenero, M.J.; Padilla, G.; Pelaez-Garcia, A.; Lopez-Lucendo, M.; Rodriguez-Merlo, R.; et al. LOXL2 is highly expressed in cancer-associated fibroblasts and associates to poor colon cancer survival. Clin. Cancer Res. 2015, 21, 4892–4902. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Miles, F.L.; Sikes, R.A. Insidious changes in stromal matrix fuel cancer progression. Mol. Cancer Res. 2014, 12, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Dimanche-Boitrel, M.T.; Vakaet, L., Jr.; Pujuguet, P.; Chauffert, B.; Martin, M.S.; Hammann, A.; van Roy, F.; Mareel, M.; Martin, F. In vivo and in vitro invasiveness of a rat colon-cancer cell line maintaining e-cadherin expression: An enhancing role of tumor-associated myofibroblasts. Int. J. Cancer 1994, 56, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Angelucci, C.; Maulucci, G.; Lama, G.; Proietti, G.; Colabianchi, A.; Papi, M.; Maiorana, A.; de Spirito, M.; Micera, A.; Balzamino, O.B.; et al. Epithelial-stromal interactions in human breast cancer: Effects on adhesion, plasma membrane fluidity and migration speed and directness. PLoS ONE 2012, 7, e50804. [Google Scholar] [CrossRef] [PubMed]
- Knauper, V.; Smith, B.; Lopez-Otin, C.; Murphy, G. Activation of progelatinase b (proMMP-9) by active collagenase-3 (MMP-13). Eur. J. Biochem. 1997, 248, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Stuelten, C.H.; DaCosta Byfield, S.; Arany, P.R.; Karpova, T.S.; Stetler-Stevenson, W.G.; Roberts, A.B. Breast cancer cells induce stromal fibroblasts to express MMP-9 via secretion of TNF-α and TGF-β. J. Cell Sci. 2005, 118, 2143–2153. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, A.; Kawana, K.; Tomio, K.; Yamashita, A.; Isobe, Y.; Nagasaka, K.; Koga, K.; Inoue, T.; Nishida, H.; Kojima, S. Matrix metalloproteinase (mmp)-9 in cancer-associated fibroblasts (CAFs) is suppressed by omega-3 polyunsaturated fatty acids in vitro and in vivo. PLoS ONE 2014, 9, e89605. [Google Scholar] [CrossRef] [PubMed]
- Vong, S.; Kalluri, R. The role of stromal myofibroblast and extracellular matrix in tumor angiogenesis. Genes Cancer 2011, 2, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; D’Amore, P.A. Blood vessel formation: What is its molecular basis? Cell 1996, 87, 1153–1155. [Google Scholar] [CrossRef]
- Ferrara, N.; Davis-Smyth, T. The biology of vascular endothelial growth factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Claesson-Welsh, L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 2006, 312, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Hlatky, L.; Tsionou, C.; Hahnfeldt, P.; Coleman, C.N. Mammary fibroblasts may influence breast tumor angiogenesis via hypoxia-induced vascular endothelial growth factor up-regulation and protein expression. Cancer Res. 1994, 54, 6083–6086. [Google Scholar] [PubMed]
- Sakurai, T.; Kudo, M. Signaling pathways governing tumor angiogenesis. Oncology 2011, 81, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, S.; Arii, S.; Furutani, M.; Niwano, M.; Harada, T.; Mizumoto, M.; Mori, A.; Onodera, H.; Imamura, M. Predictive value of vascular endothelial growth factor (VEGF) in metastasis and prognosis of human colorectal cancer. Br. J. Cancer 1998, 78, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Lin, H.Z.; Ma, S.P.; Ji, P.; Xie, D.; Yu, J.X. Vascular endothelial growth factor-a and -c: Expression and correlations with lymphatic metastasis and prognosis in colorectal cancer. Med. Oncol. 2011, 28, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Q.; Fang, J.M.; Xiao, Y.Y.; Zhao, Y.; Cui, R.; Hu, F.; Xu, Q. Prognostic role of vascular endothelial growth factor in prostate cancer: A systematic review and meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 2289–2298. [Google Scholar] [PubMed]
- Nagasaki, T.; Hara, M.; Shiga, K.; Takeyama, H. Relationship between inflammation and cancer progression: Recent advances in interleukin-6 signaling and its blockage in cancer therapy. Recept. Clin. Investig. 2014, 1. [Google Scholar] [CrossRef]
- De Boeck, A.; Hendrix, A.; Maynard, D.; van Bockstal, M.; Daniels, A.; Pauwels, P.; Gespach, C.; Bracke, M.; de Wever, O. Differential secretome analysis of cancer-associated fibroblasts and bone marrow-derived precursors to identify microenvironmental regulators of colon cancer progression. Proteomics 2013, 13, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Tommelein, J.; Verset, L.; Boterberg, T.; Demetter, P.; Bracke, M.; de Wever, O. Cancer-associated fibroblasts connect metastasis-promoting communication in colorectal cancer. Front. Oncol. 2015, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Huang, H.J.; Kazlauskas, A.; Cavenee, W.K. Induction of vascular endothelial growth factor expression in endothelial cells by platelet-derived growth factor through the activation of phosphatidylinositol 3-kinase. Cancer Res. 1999, 59, 1464–1472. [Google Scholar] [PubMed]
- Kitadai, Y.; Sasaki, T.; Kuwai, T.; Nakamura, T.; Bucana, C.D.; Hamilton, S.R.; Fidler, I.J. Expression of activated platelet-derived growth factor receptor in stromal cells of human colon carcinomas is associated with metastatic potential. Int. J. Cancer 2006, 119, 2567–2574. [Google Scholar] [CrossRef] [PubMed]
- Pena, C.; Cespedes, M.V.; Lindh, M.B.; Kiflemariam, S.; Mezheyeuski, A.; Edqvist, P.H.; Hagglof, C.; Birgisson, H.; Bojmar, L.; Jirstrom, K.; et al. STC1 expression by cancer-associated fibroblasts drives metastasis of colorectal cancer. Cancer Res. 2013, 73, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Cao, R.; Hedlund, E.M. R regulation of tumor angiogenesis and metastasis by fgf and pdgf signaling pathways. J. Mol. Med. 2008, 86, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Giulianelli, S.; Cerliani, J.P.; Lamb, C.A.; Fabris, V.T.; Bottino, M.C.; Gorostiaga, M.A.; Novaro, V.; Gongora, A.; Baldi, A.; Molinolo, A.; et al. Carcinoma-associated fibroblasts activate progesterone receptors and induce hormone independent mammary tumor growth: A role for the FGF-2/FGFR-2 axis. Int. J. Cancer 2008, 123, 2518–2531. [Google Scholar] [CrossRef] [PubMed]
- Fabris, V.T.; Sahores, A.; Vanzulli, S.I.; Colombo, L.; Molinolo, A.A.; Lanari, C.; Lamb, C.A. Inoculated mammary carcinoma-associated fibroblasts: Contribution to hormone independent tumor growth. BMC Cancer 2010, 10, 293. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Whitaker-Menezes, D.; Daumer, K.M.; Milliman, J.N.; Chiavarina, B.; Migneco, G.; Witkiewicz, A.K.; Martinez-Cantarin, M.P.; Flomenberg, N.; et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010, 9, 2423–2433. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, C.D.; Alvarez, S.; Ropolo, A.; Rosenzvit, C.; Bagnes, M.F.; Vaccaro, M.I. Autophagy, warburg, and warburg reverse effects in human cancer. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed]
- Shan-Wei, W.; Kan-Lun, X.; Shu-Qin, R.; Li-Li, Z.; Li-Rong, C. Overexpression of caveolin-1 in cancer-associated fibroblasts predicts good outcome in breast cancer. Breast Care (Basel) 2012, 7, 477–483. [Google Scholar] [PubMed]
- Cohen, R.; Neuzillet, C.; Tijeras-Raballand, A.; Faivre, S.; de Gramont, A.; Raymond, E. Targeting cancer cell metabolism in pancreatic adenocarcinoma. Oncotarget 2015, 6, 16832–16847. [Google Scholar] [CrossRef] [PubMed]
- Sahra, I.B.; le Marchand-Brustel, Y.; Tanti, J.-F.; Bost, F. Metformin in cancer therapy: A new perspective for an old antidiabetic drug? Mol. Cancer Ther. 2010, 9, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Al-Ansari, M.M.; Aboussekhra, A. Caffeine mediates sustained inactivation of breast cancer-associated myofibroblasts via up-regulation of tumor suppressor genes. PLoS ONE 2014, 9, e90907. [Google Scholar] [CrossRef] [PubMed]
- Ballou, L.M.; Lin, R.Z. Rapamycin and mtor kinase inhibitors. J. Chem. Biol. 2008, 1, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Vascular and interstitial barriers to delivery of therapeutic agents in tumors. Cancer Metastasis Rev. 1990, 9, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Herman, T.S.; Holden, S.A.; Wang, Y.Y.; Pfeffer, M.R.; Crawford, J.W.; Frei, E., 3rd. Tumor resistance to alkylating agents conferred by mechanisms operative only in vivo. Science 1990, 247, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Hu, S.Q.; Xiao, L. The cancer-associated fibroblasts and drug resistance. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 2112–2119. [Google Scholar] [PubMed]
- Kharaishvili, G.; Simkova, D.; Bouchalova, K.; Gachechiladze, M.; Narsia, N.; Bouchal, J. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer Cell Int. 2014, 14, 41. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Keller, E.T.; Garfield, D.H.; Shen, K.; Wang, J. Stromal cells in tumor microenvironment and breast cancer. Cancer Metastasis Rev. 2013, 32, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to raf inhibitors through hgf secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.C.; Ansell, A.; Jerhammar, F.; Lindh, M.B.; Grenman, R.; Munck-Wikland, E.; Ostman, A.; Roberg, K. Cancer-associated fibroblasts induce matrix metalloproteinase-mediated cetuximab resistance in head and neck squamous cell carcinoma cells. Mol. Cancer Res. 2012, 10, 1158–1168. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Giannoni, E.; Taddei, M.L.; Cirri, P.; Marini, A.; Pintus, G.; Nativi, C.; Richichi, B.; Scozzafava, A.; Carta, F.; et al. Carbonic anhydrase ix from cancer-associated fibroblasts drives epithelial-mesenchymal transition in prostate carcinoma cells. Cell Cycle 2013, 12, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Kharaziha, P.; Rodriguez, P.; Li, Q.; Rundqvist, H.; Bjorklund, A.C.; Augsten, M.; Ullen, A.; Egevad, L.; Wiklund, P.; Nilsson, S.; et al. Targeting of distinct signaling cascades and cancer-associated fibroblasts define the efficacy of sorafenib against prostate cancer cells. Cell Death Dis. 2012, 3, e262. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, M.; Kruger, J.A.; Niethammer, A.G.; Reisfeld, R.A. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Invest. 2006, 116, 1955–1962. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bhatia, R. Stem cell quiescence. Clin. Cancer Res. 2011, 17, 4936–4941. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef] [PubMed]
- Lotti, F.; Jarrar, A.M.; Pai, R.K.; Hitomi, M.; Lathia, J.; Mace, A.; Gantt, G.A., Jr.; Sukhdeo, K.; devecchio, J.; Vasanji, A.; et al. Chemotherapy activates cancer-associated fibroblasts to maintain colorectal cancer-initiating cells by IL-17A. J. Exp. Med. 2013, 210, 2851–2872. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Mao, Y.; Wang, J.; Zu, L.; Hao, M.; Cheng, G.; Qu, Q.; Cui, D.; Keller, E.; Chen, X. Il-6 secreted by cancer-associated fibroblasts induces tamoxifen resistance in luminal breast cancer. Oncogene 2014, 33, 4450. [Google Scholar] [CrossRef] [PubMed]
- Paraiso, K.H.; Smalley, K.S. Fibroblast-mediated drug resistance in cancer. Biochem. Pharmacol. 2013, 85, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, L.A.; Dalton, W.S. Mechanisms associated with cell adhesion mediated drug resistance (CAM-DR) in hematopoietic malignancies. Cancer Metastasis Rev. 2001, 20, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Sherman-Baust, C.A.; Weeraratna, A.T.; Rangel, L.B.; Pizer, E.S.; Cho, K.R.; Schwartz, D.R.; Shock, T.; Morin, P.J. Remodeling of the extracellular matrix through overexpression of collagen vi contributes to cisplatin resistance in ovarian cancer cells. Cancer Cell 2003, 3, 377–386. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, M.; Yang, L.; Tu, G.; Zhu, Q.; Chen, M.; Cheng, H.; Luo, H.; Fu, W.; Li, Z.; et al. Acquisition of epithelial-mesenchymal transition phenotype in the tamoxifen-resistant breast cancer cell: A new role for g protein-coupled estrogen receptor in mediating tamoxifen resistance through cancer-associated fibroblast-derived fibronectin and beta1-integrin signaling pathway in tumor cells. Breast Cancer Res. 2015, 17, 69. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Ghatak, S.; Zoltan-Jones, A.; Toole, B.P. Regulation of multidrug resistance in cancer cells by hyaluronan. J. Biol. Chem. 2003, 278, 25285–25288. [Google Scholar] [CrossRef] [PubMed]
- Curti, B.D. Physical barriers to drug delivery in tumors. Crit. Rev. Oncol. Hematol. 1993, 14, 29–39. [Google Scholar] [CrossRef]
- Pietras, K.; Ostman, A.; Sjoquist, M.; Buchdunger, E.; Reed, R.K.; Heldin, C.H.; Rubin, K. Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and increases transcapillary transport in tumors. Cancer Res. 2001, 61, 2929–2934. [Google Scholar] [PubMed]
- Pietras, K.; Rubin, K.; Sjoblom, T.; Buchdunger, E.; Sjoquist, M.; Heldin, C.H.; Ostman, A. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002, 62, 5476–5484. [Google Scholar] [PubMed]
- Sumida, T.; Kitadai, Y.; Shinagawa, K.; Tanaka, M.; Kodama, M.; Ohnishi, M.; Ohara, E.; Tanaka, S.; Yasui, W.; Chayama, K. Anti-stromal therapy with imatinib inhibits growth and metastasis of gastric carcinoma in an orthotopic nude mouse model. Int. J. Cancer 2011, 128, 2050–2062. [Google Scholar] [CrossRef] [PubMed]
- Vlahovic, G.; Rabbani, Z.N.; Herndon, J.E., II; Dewhirst, M.W.; Vujaskovic, Z. Treatment with imatinib in NSCLC is associated with decrease of phosphorylated PDGFR-β and vegf expression, decrease in interstitial fluid pressure and improvement of oxygenation. Br. J. Cancer 2006, 95, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Amornsupuk, K.; Insawang, T.; Thuwajit, P.; Pornchai, O.; Eccles, S.A.; Thuwajit, C. Cancer-associated fibroblasts induce high mobility group box 1 and contribute to resistance to doxorubicin in breast cancer cells. BMC Cancer 2014, 14, 955. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers 2015, 7, 2443-2458. https://doi.org/10.3390/cancers7040902
Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H. Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers. 2015; 7(4):2443-2458. https://doi.org/10.3390/cancers7040902
Chicago/Turabian StyleShiga, Kazuyoshi, Masayasu Hara, Takaya Nagasaki, Takafumi Sato, Hiroki Takahashi, and Hiromitsu Takeyama. 2015. "Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth" Cancers 7, no. 4: 2443-2458. https://doi.org/10.3390/cancers7040902
APA StyleShiga, K., Hara, M., Nagasaki, T., Sato, T., Takahashi, H., & Takeyama, H. (2015). Cancer-Associated Fibroblasts: Their Characteristics and Their Roles in Tumor Growth. Cancers, 7(4), 2443-2458. https://doi.org/10.3390/cancers7040902