
Not All Next Generation Sequencing Diagnostics are Created Equal: Understanding the Nuances of Solid Tumor Assay Design for Somatic Mutation Detection

Abstract
:1. Introduction
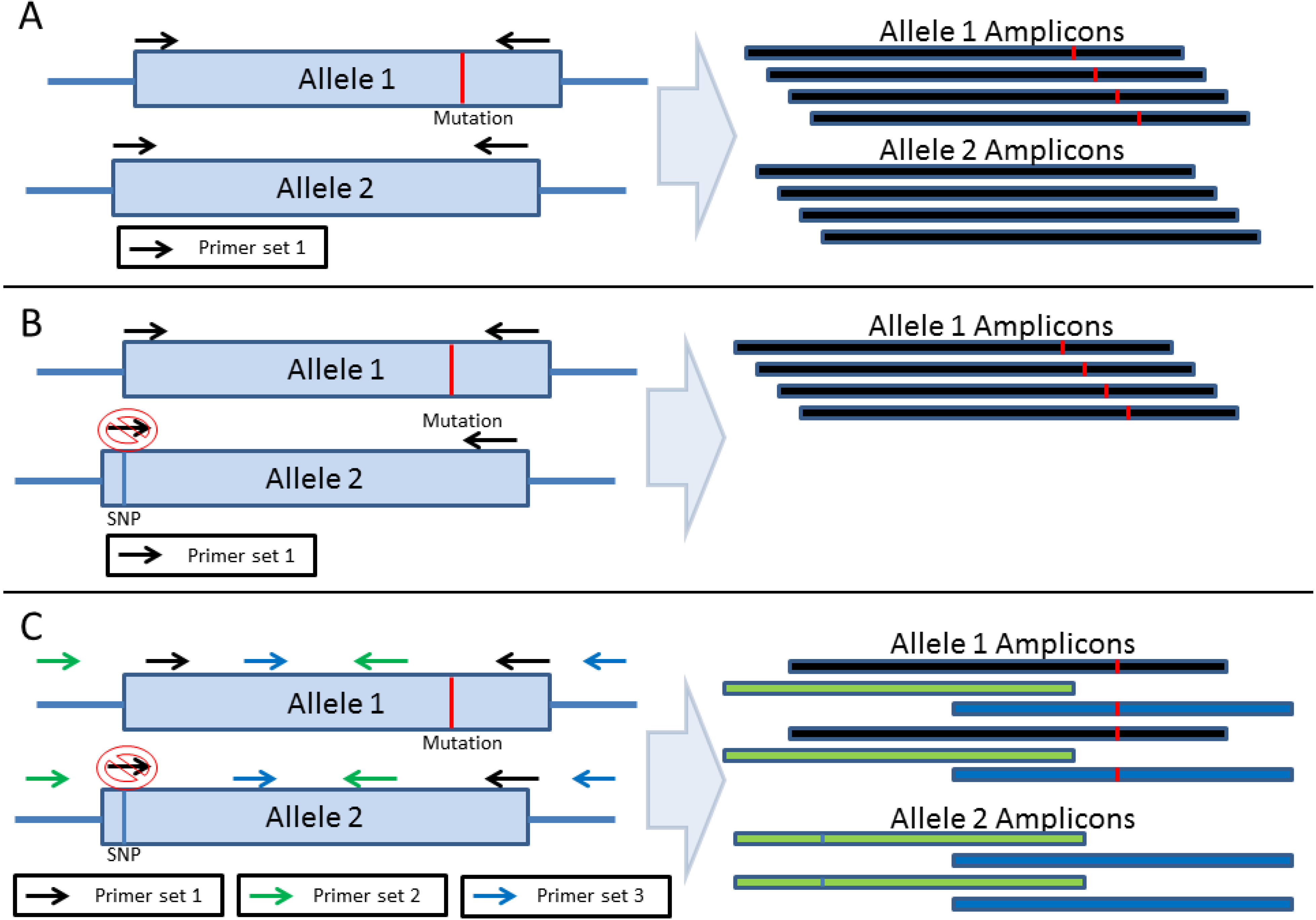
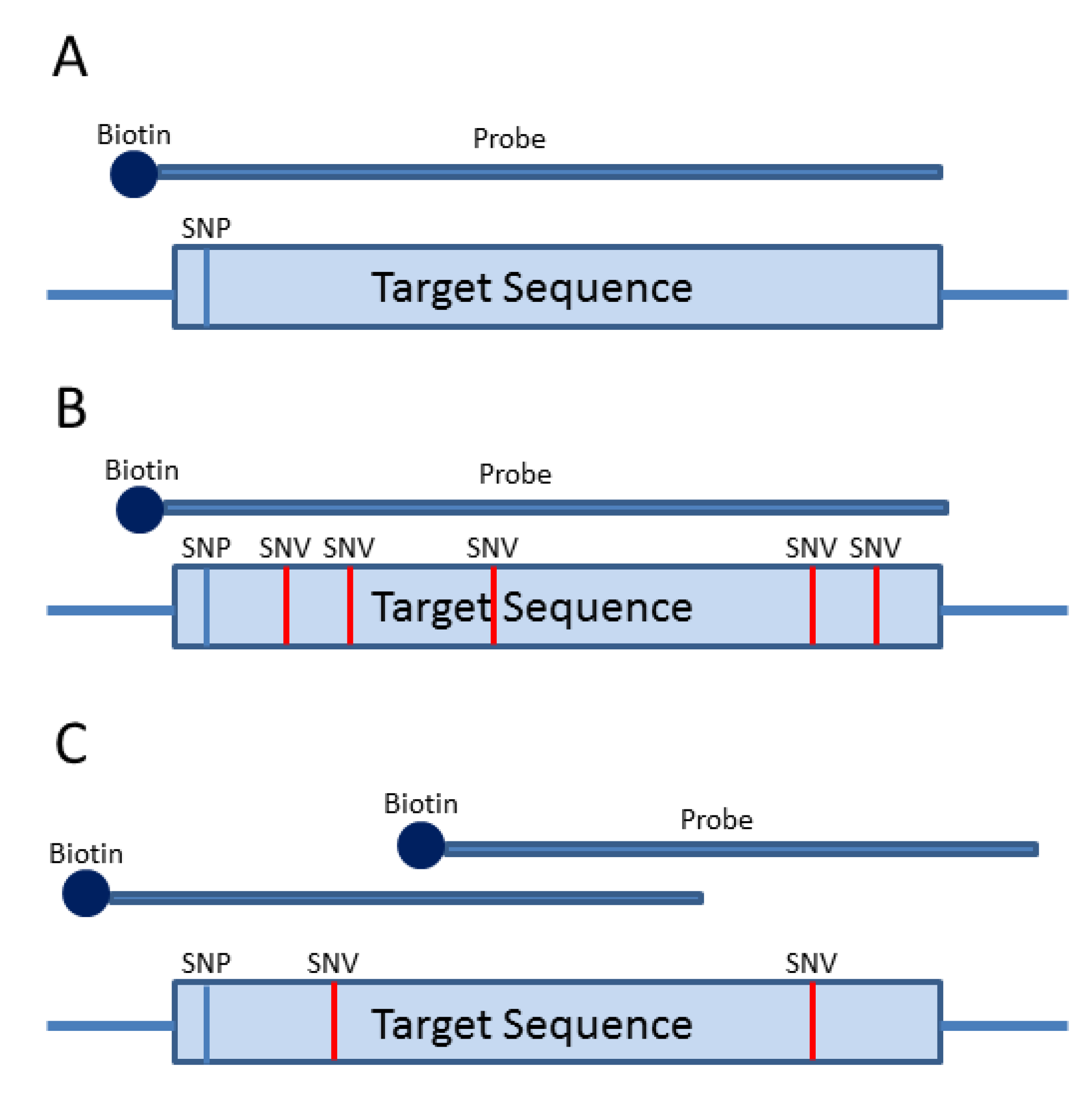
2. Amplification-Based Enrichment Methods


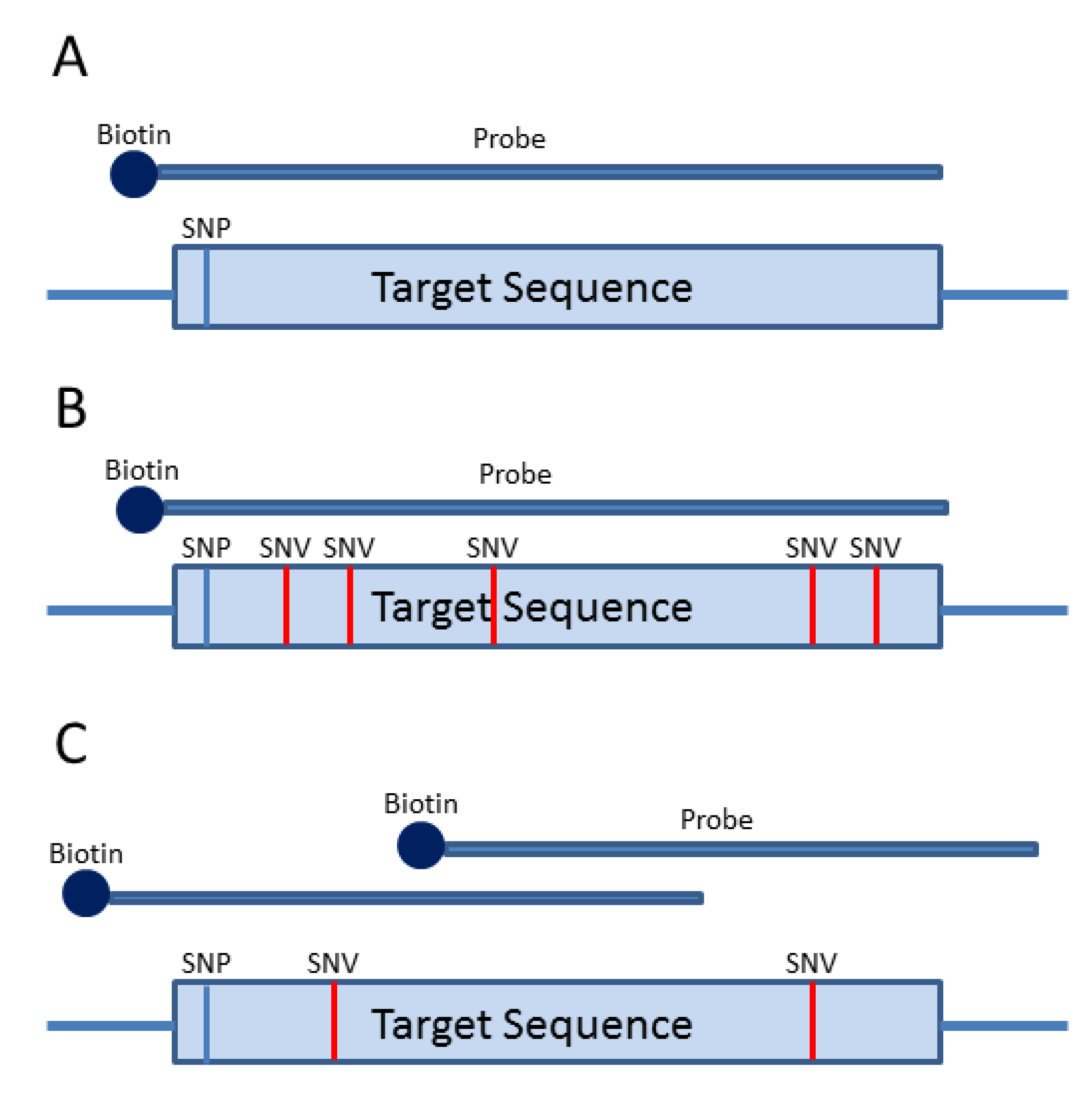
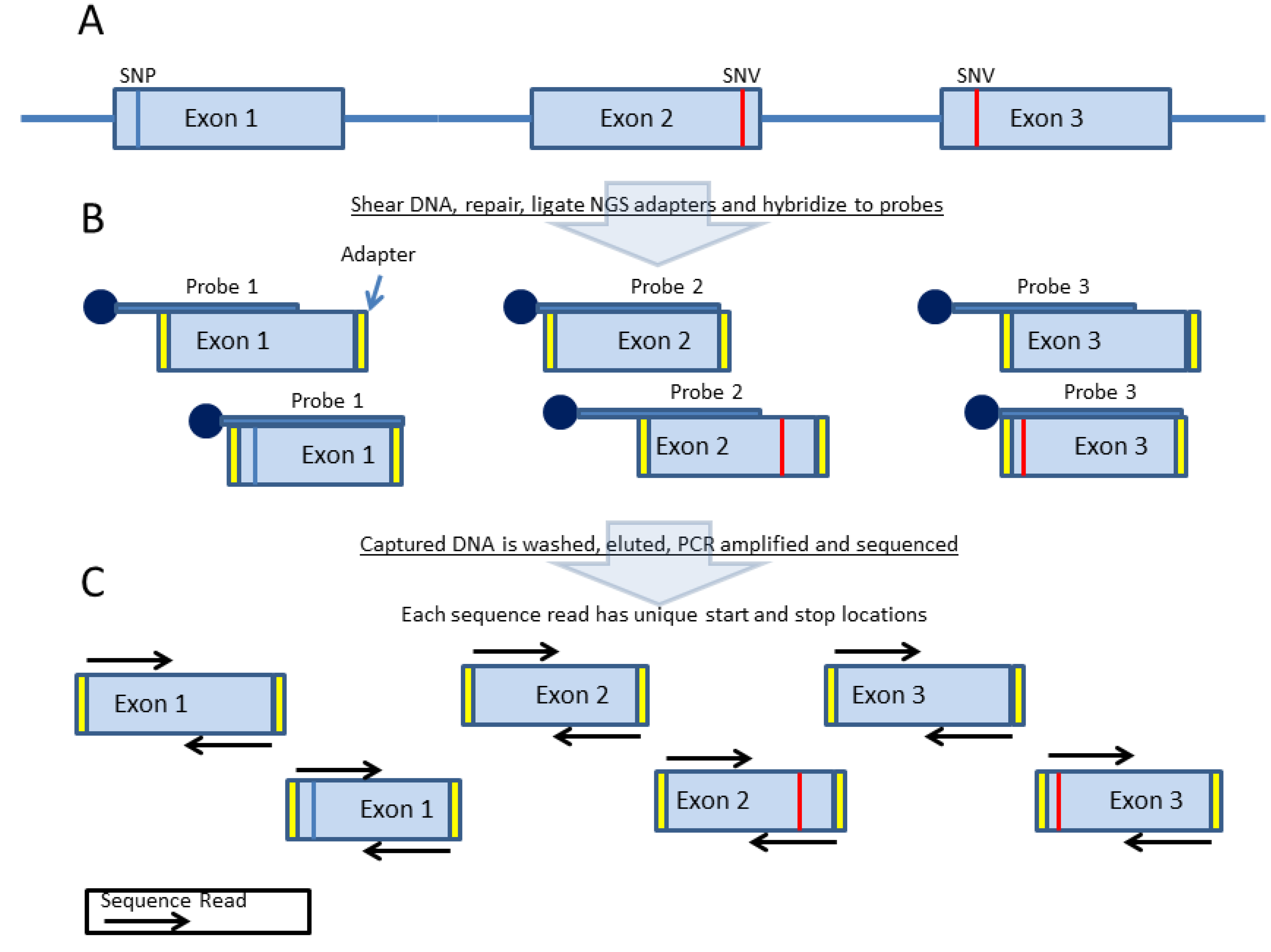
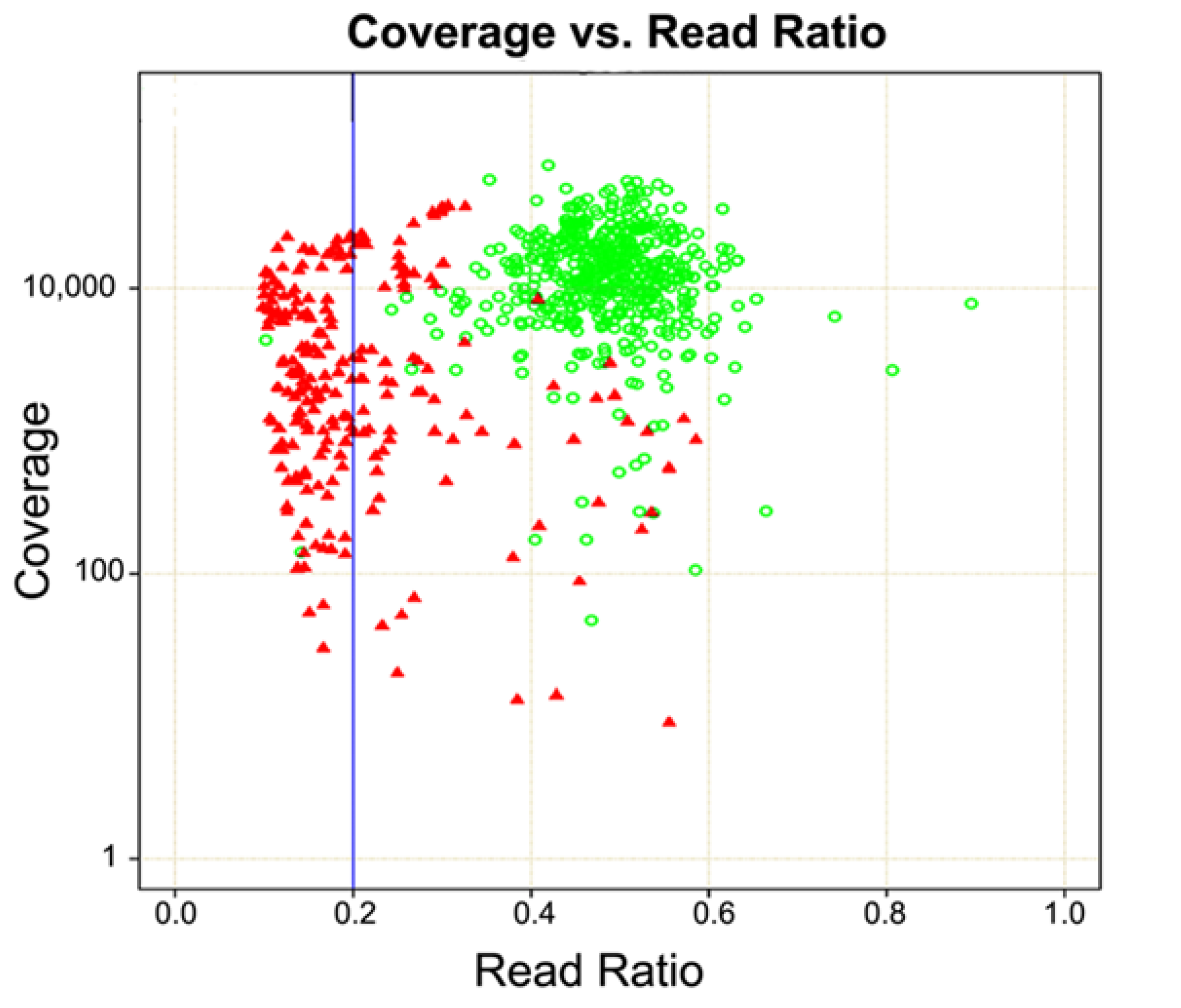
3. Probe-Based Enrichment Methods

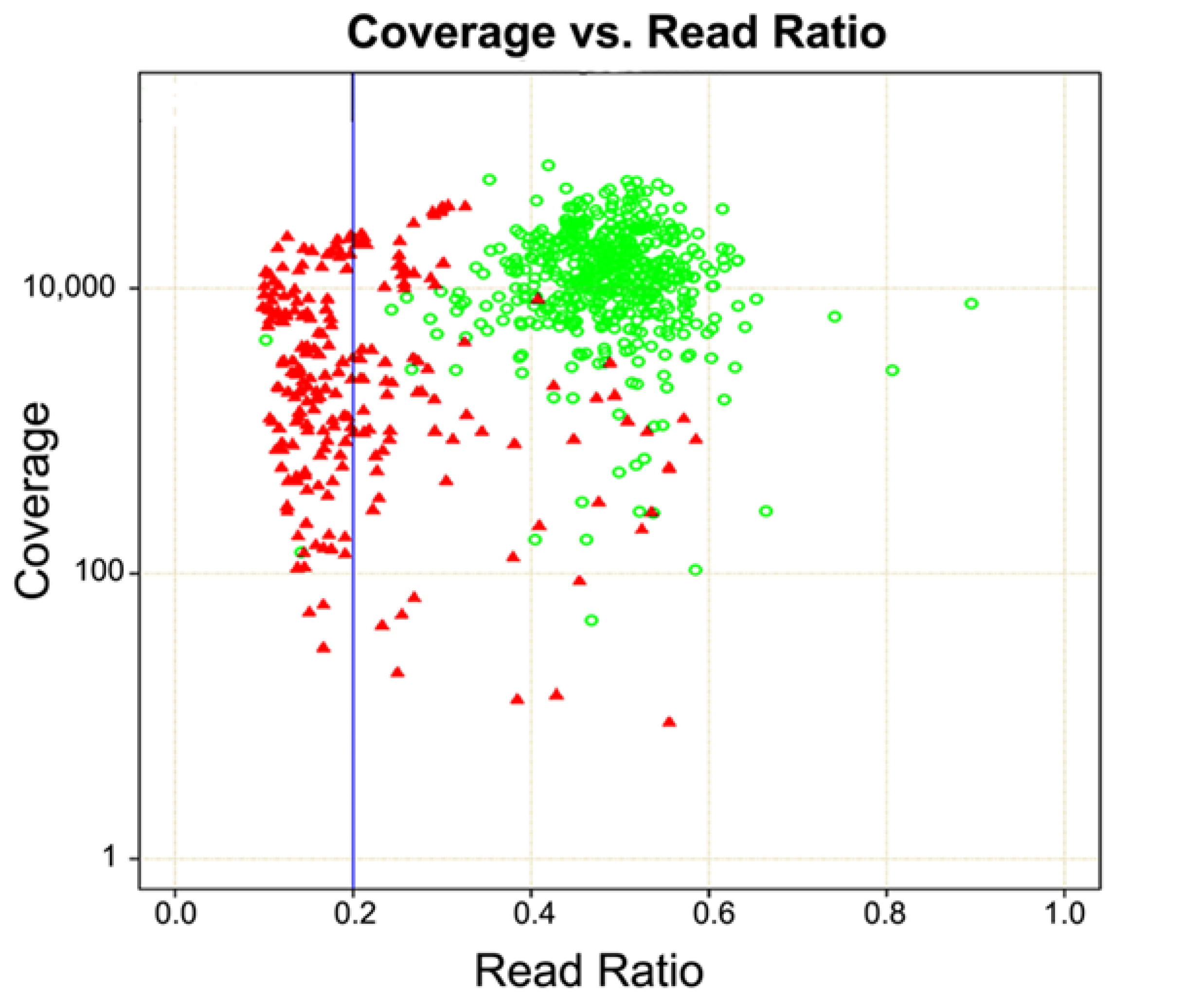
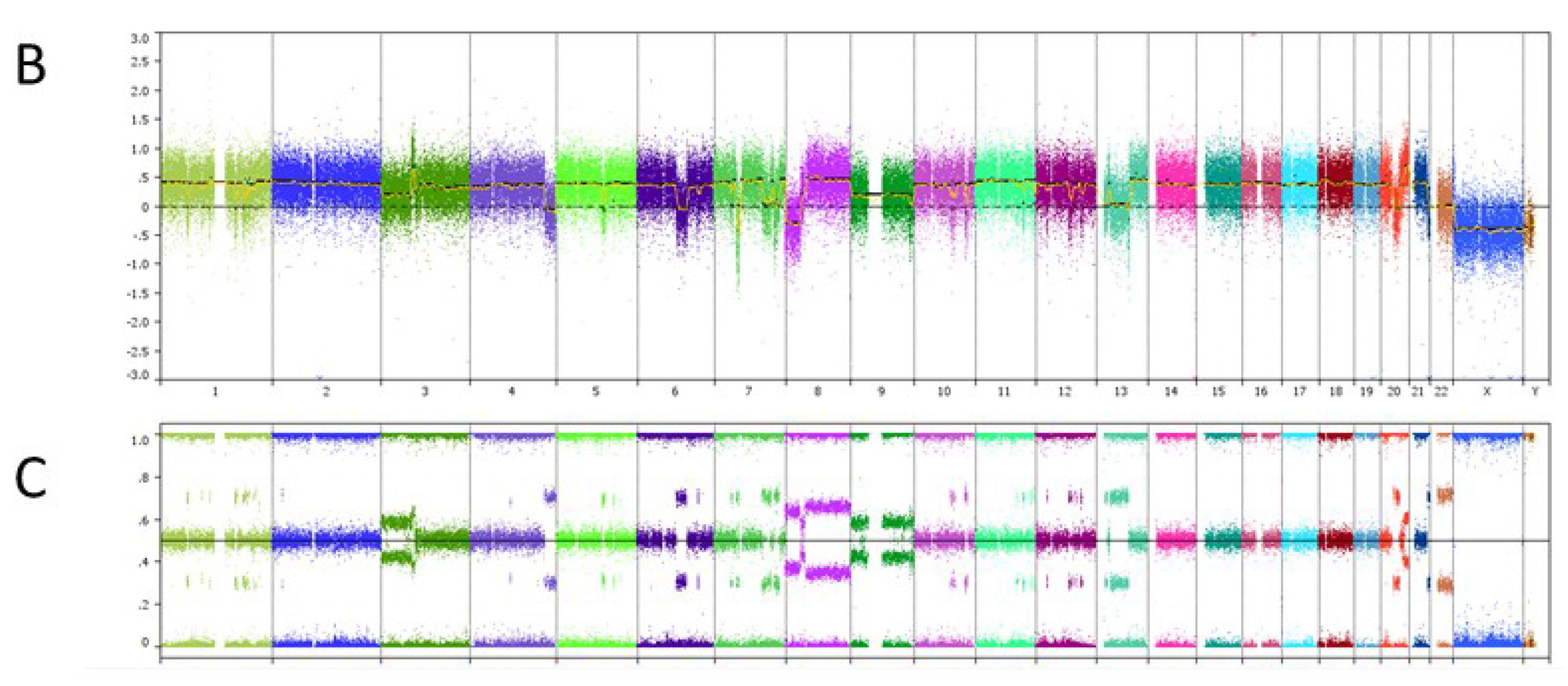
4. Copy Number Analysis
5. Germline Status Incorporated into Bioinformatic Analysis
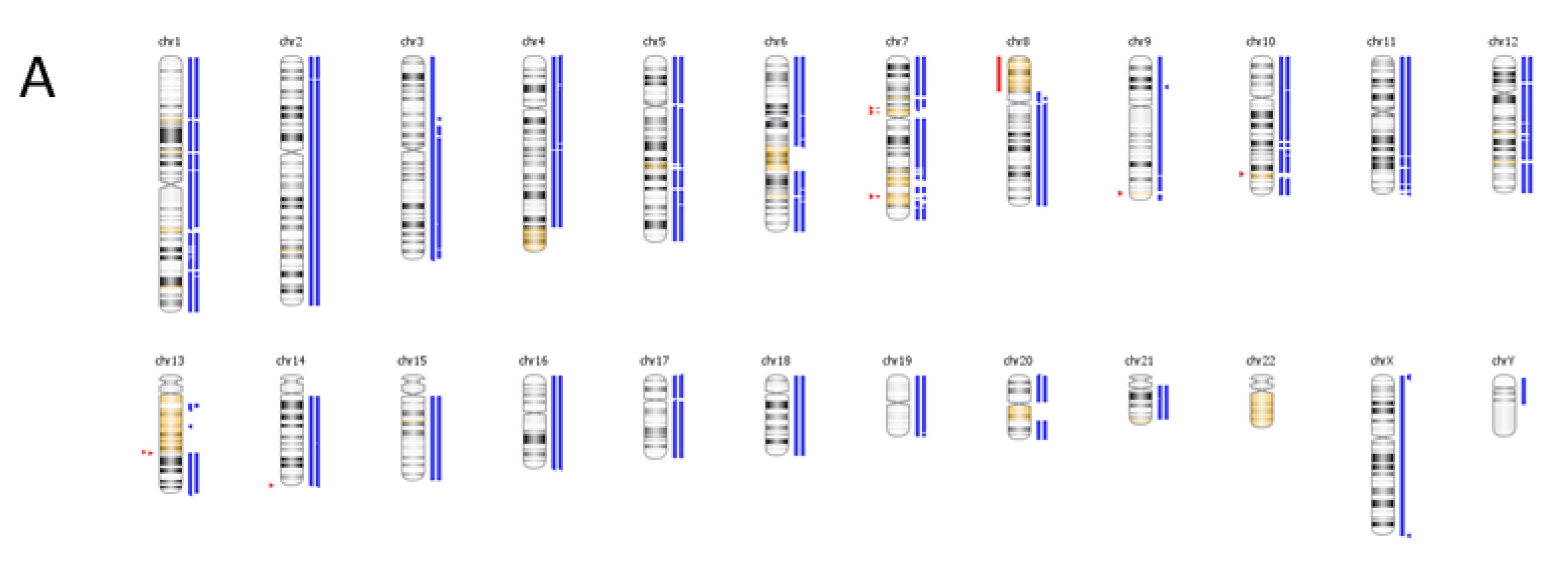
6. Case Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Alterations Identified | |||
|---|---|---|---|
| MITF amplification-equivocal | FOXP1 amplification-equivocal | IKZF1 loss | TMPRSS2 rearrangement intron 2 |
| Variants of Unknown Significance | |||
| ATM V2424G | BCOR K1260R | CHD4 K775R | EGFR loss |
| FAT1 N4492K | INHBA R229Q | PREX2 M1298L | RPTOR A862T |
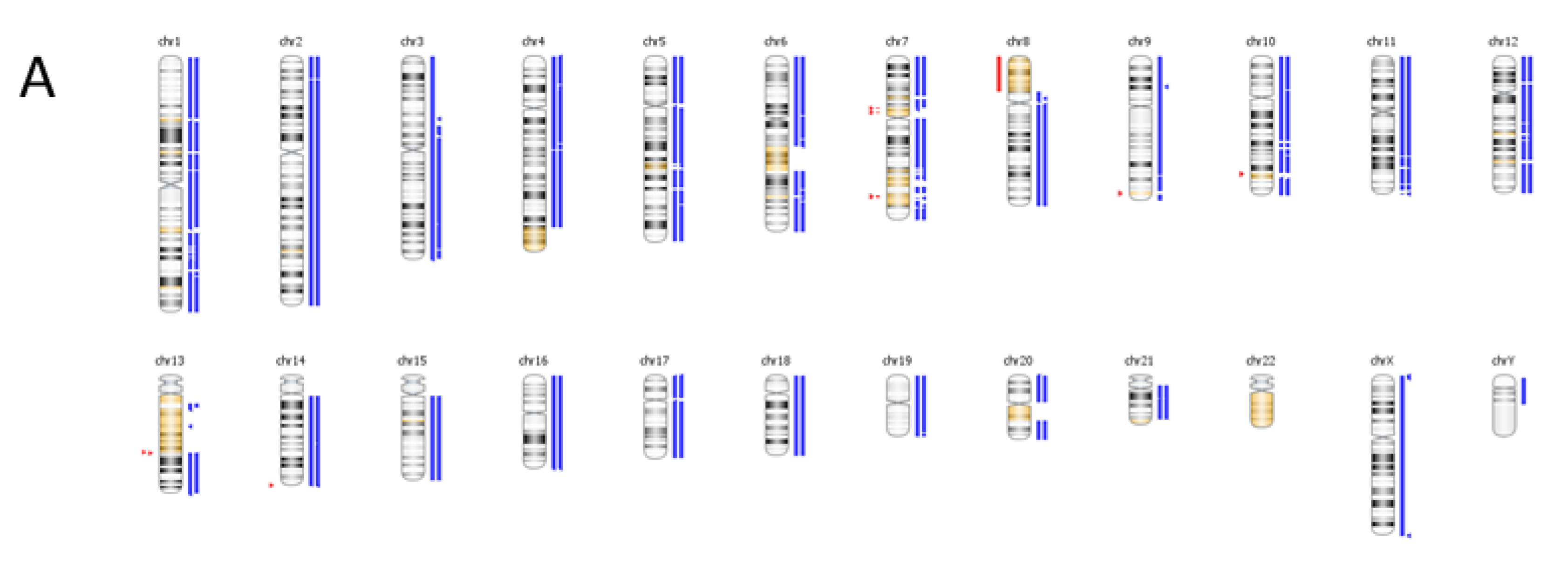
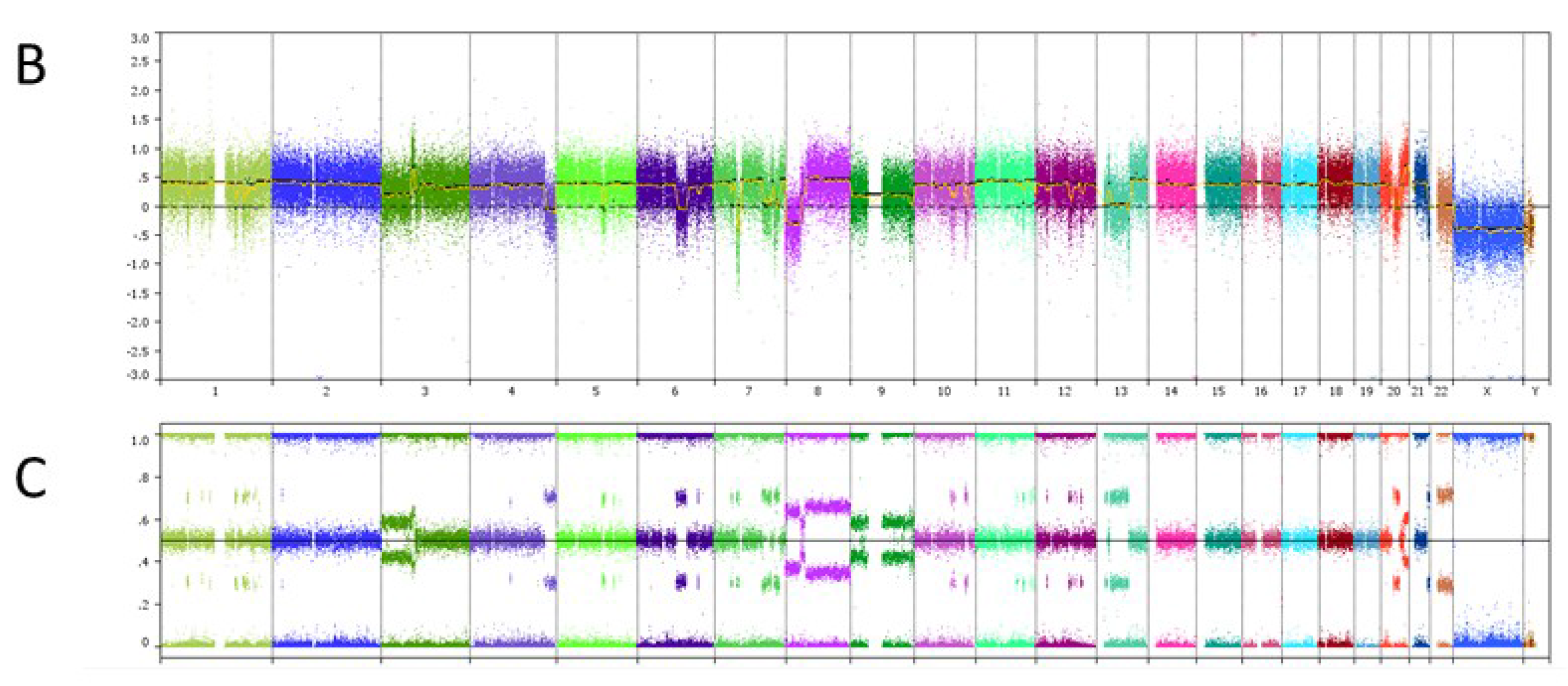
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Igbokwe, A.; Lopez-Terrada, D.H. Molecular testing of solid tumors. Arch. Pathol. Lab. Med. 2011, 135, 67–82. [Google Scholar] [PubMed]
- McCourt, C.M.; Boyle, D.; James, J.; Salto-Tellez, M. Immunohistochemistry in the era of personalised medicine. J. Clin. Pathol. 2013, 66, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Pauletti, G.; Dandekar, S.; Rong, H.; Ramos, L.; Peng, H.; Seshadri, R.; Slamon, D.J. Assessment of methods for tissue-based detection of the her-2/neu alteration in human breast cancer: A direct comparison of fluorescence in situ hybridization and immunohistochemistry. J. Clin. Oncol. 2000, 18, 3651–3664. [Google Scholar] [PubMed]
- Lebeau, A.; Deimling, D.; Kaltz, C.; Sendelhofert, A.; Iff, A.; Luthardt, B.; Untch, M.; Lohrs, U. Her-2/neu analysis in archival tissue samples of human breast cancer: Comparison of immunohistochemistry and fluorescence in situ hybridization. J. Clin. Oncol. 2001, 19, 354–363. [Google Scholar] [PubMed]
- Angulo, B.; Garcia-Garcia, E.; Martinez, R.; Suarez-Gauthier, A.; Conde, E.; Hidalgo, M.; Lopez-Rios, F. A commercial real-time pcr kit provides greater sensitivity than direct sequencing to detect kras mutations: A morphology-based approach in colorectal carcinoma. J. Mol. Diagn. 2010, 12, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez de Castro, D.; Angulo, B.; Gomez, B.; Mair, D.; Martinez, R.; Suarez-Gauthier, A.; Shieh, F.; Velez, M.; Brophy, V.H.; Lawrence, H.J.; et al. A comparison of three methods for detecting kras mutations in formalin-fixed colorectal cancer specimens. Br. J. Cancer 2012, 107, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Rios, F.; Angulo, B.; Gomez, B.; Mair, D.; Martinez, R.; Conde, E.; Shieh, F.; Vaks, J.; Langland, R.; Lawrence, H.J.; et al. Comparison of testing methods for the detection of braf v600e mutations in malignant melanoma: Pre-approval validation study of the companion diagnostic test for vemurafenib. PLoS ONE 2013, 8, e53733. [Google Scholar] [CrossRef] [PubMed]
- Solin, L.J.; Gray, R.; Baehner, F.L.; Butler, S.M.; Hughes, L.L.; Yoshizawa, C.; Cherbavaz, D.B.; Shak, S.; Page, D.L.; Sledge, G.W., Jr.; et al. A multigene expression assay to predict local recurrence risk for ductal carcinoma in situ of the breast. J. Natl. Cancer Inst. 2013, 105, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Cobas 4800 braf v600 mutation test. Available online: http://molecular.roche.com/assays/Pages/cobas4800BRAFV600MutationTest.aspx (accessed on 15 May 2015).
- Cobas egfr mutation test. Available online: http://molecular.roche.com/assays/Pages/cobasEGFRMutationTest.aspx (accessed on 15 May 2015).
- Lambros, M.B.; Natrajan, R.; Reis-Filho, J.S. Chromogenic and fluorescent in situ hybridization in breast cancer. Hum. Pathol. 2007, 38, 1105–1122. [Google Scholar] [CrossRef] [PubMed]
- Gruver, A.M.; Peerwani, Z.; Tubbs, R.R. Out of the darkness and into the light: Bright field in situ hybridisation for delineation of erbb2 (her2) status in breast carcinoma. J. Clin. Pathol. 2010, 63, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Korbel, J.O.; Urban, A.E.; Affourtit, J.P.; Godwin, B.; Grubert, F.; Simons, J.F.; Kim, P.M.; Palejev, D.; Carriero, N.J.; Du, L.; et al. Paired-end mapping reveals extensive structural variation in the human genome. Science 2007, 318, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.L.; Ordonez, G.R.; Bignell, G.R.; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010, 463, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. Breakdancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Chiang, D.Y.; Getz, G.; Jaffe, D.B.; O'Kelly, M.J.; Zhao, X.; Carter, S.L.; Russ, C.; Nusbaum, C.; Meyerson, M.; Lander, E.S. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat. Methods 2009, 6, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Berger, M.F.; Davis, M.J.; Blumenstiel, B.; Defelice, M.; Pochanard, P.; Ducar, M.; van Hummelen, P.; Macconaill, L.E.; Hahn, W.C.; et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2011, 2, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, C.E.; Al-Kateb, H.; Bredemeyer, A.J.; Duncavage, E.J.; Spencer, D.H.; Abel, H.J.; Lockwood, C.M.; Hagemann, I.S.; O’Guin, S.M.; Burcea, L.C.; et al. Validation of a next-generation sequencing assay for clinical molecular oncology. J. Mol. Diagn. 2014, 16, 89–105. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Salipante, S.J.; Koehler, K.; Smith, C.; Scroggins, S.; Wood, B.; Wu, D.; Lee, M.K.; Dintzis, S.; Adey, A.; et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J. Mol. Diagn. 2014, 16, 56–67. [Google Scholar] [CrossRef] [PubMed]
- “Next generation” sequencing (ngs) guidelines for somatic genetic variant detection. Available online: http://www.wadsworth.org/labcert/TestApproval/forms/NextGenSeq_ONCO_Guidelines.pdf (accessed on 15 May 2015).
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Wang, L.; Arcila, M.E.; Balasubramanian, S.; Greenbowe, J.R.; Ross, J.S.; Stephens, P.; Lipson, D.; Miller, V.A.; Kris, M.G.; et al. Broad, hybrid capture-based next-generation sequencing identifies actionable genomic alterations in lung adenocarcinomas otherwise negative for such alterations by other genomic testing approaches. Clin. Cancer Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ikegawa, S.; Mabuchi, A.; Ogawa, M.; Ikeda, T. Allele-specific pcr amplification due to sequence identity between a pcr primer and an amplicon: Is direct sequencing so reliable? Hum. Genet. 2002, 110, 606–608. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.; Garvin, A.M.; Di Naro, E.; Holzgreve, W. Allele drop-out can occur in alleles differing by a single nucleotide and is not alleviated by preamplification or minor template increments. Genet. Test 1998, 2, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Barnard, R.; Futo, V.; Pecheniuk, N.; Slattery, M.; Walsh, T. Pcr bias toward the wild-type k-ras and p53 sequences: Implications for pcr detection of mutations and cancer diagnosis. BioTechniques 1998, 25, 684–691. [Google Scholar] [PubMed]
- Chong, H.K.; Wang, T.; Lu, H.M.; Seidler, S.; Lu, H.; Keiles, S.; Chao, E.C.; Stuenkel, A.J.; Li, X.; Elliott, A.M. The validation and clinical implementation of brcaplus: A comprehensive high-risk breast cancer diagnostic assay. PLoS ONE 2014, 9, e97408. [Google Scholar] [CrossRef] [PubMed]
- Kozarewa, I.; Ning, Z.; Quail, M.A.; Sanders, M.J.; Berriman, M.; Turner, D.J. Amplification-free illumina sequencing-library preparation facilitates improved mapping and assembly of (g+c)-biased genomes. Nat. Methods 2009, 6, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Pireddu, L.; Leo, S.; Zanetti, G. Seal: A distributed short read mapping and duplicate removal tool. Bioinformatics 2011, 27, 2159–2160. [Google Scholar] [CrossRef] [PubMed]
- Picard. Available online: http://broadinstitute.github.io/picard/ (accessed on 15 May 2015).
- Smith, E.N.; Jepsen, K.; Khosroheidari, M.; Rassenti, L.Z.; D'Antonio, M.; Ghia, E.M.; Carson, D.A.; Jamieson, C.H.; Kipps, T.J.; Frazer, K.A. Biased estimates of clonal evolution and subclonal heterogeneity can arise from pcr duplicates in deep sequencing experiments. Genome Biol. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Kanagawa, T. Bias and artifacts in multitemplate polymerase chain reactions (pcr). J. Biosci. Bioeng. 2003, 96, 317–323. [Google Scholar] [CrossRef]
- Rait, V.K.; Zhang, Q.; Fabris, D.; Mason, J.T.; O’Leary, T.J. Conversions of formaldehyde-modified 2'-deoxyadenosine 5'-monophosphate in conditions modeling formalin-fixed tissue dehydration. J. Histochem. Cytochem. 2006, 54, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Miner, B.E.; Stoger, R.J.; Burden, A.F.; Laird, C.D.; Hansen, R.S. Molecular barcodes detect redundancy and contamination in hairpin-bisulfite pcr. Nucleic Acids Res. 2004, 32, e135. [Google Scholar] [CrossRef] [PubMed]
- Casbon, J.A.; Osborne, R.J.; Brenner, S.; Lichtenstein, C.P. A method for counting pcr template molecules with application to next-generation sequencing. Nucleic Acids Res. 2011, 39, e81. [Google Scholar] [CrossRef] [PubMed]
- Hiatt, J.B.; Pritchard, C.C.; Salipante, S.J.; O’Roak, B.J.; Shendure, J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013, 23, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Zeisel, A.; Joost, S.; La Manno, G.; Zajac, P.; Kasper, M.; Lonnerberg, P.; Linnarsson, S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 2014, 11, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Hodges, E.; Xuan, Z.; Balija, V.; Kramer, M.; Molla, M.N.; Smith, S.W.; Middle, C.M.; Rodesch, M.J.; Albert, T.J.; Hannon, G.J.; et al. Genome-wide in situ exon capture for selective resequencing. Nat. Genet. 2007, 39, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Okou, D.T.; Steinberg, K.M.; Middle, C.; Cutler, D.J.; Albert, T.J.; Zwick, M.E. Microarray-based genomic selection for high-throughput resequencing. Nat. Methods 2007, 4, 907–909. [Google Scholar] [CrossRef] [PubMed]
- Albert, T.J.; Molla, M.N.; Muzny, D.M.; Nazareth, L.; Wheeler, D.; Song, X.; Richmond, T.A.; Middle, C.M.; Rodesch, M.J.; Packard, C.J.; et al. Direct selection of human genomic loci by microarray hybridization. Nat. Methods 2007, 4, 903–905. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Morrison, C.D.; Johnson, C.S.; Trump, D.L.; Qin, M.; Conroy, J.C.; Wang, J.; Liu, S. Computational methods for detecting copy number variations in cancer genome using next generation sequencing: Principles and challenges. Oncotarget 2013, 4, 1868–1881. [Google Scholar] [PubMed]
- Zhao, M.; Wang, Q.; Wang, Q.; Jia, P.; Zhao, Z. Computational tools for copy number variation (cnv) detection using next-generation sequencing data: Features and perspectives. BMC Bioinformatics 2013, 14, S1. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.; Biricik, A.; Bono, S.; Spizzichino, L.; Cotroneo, E.; Cottone, G.; Kokocinski, F.; Michel, C.E. Development and validation of a next-generation sequencing-based protocol for 24-chromosome aneuploidy screening of embryos. Fertil. Steril. 2014, 101, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, F.; Bono, S.; Biricik, A.; Nuccitelli, A.; Cotroneo, E.; Cottone, G.; Kokocinski, F.; Michel, C.E.; Minasi, M.G.; Greco, E. Application of next-generation sequencing technology for comprehensive aneuploidy screening of blastocysts in clinical preimplantation genetic screening cycles. Hum. Reprod. 2014, 29, 2802–2813. [Google Scholar] [CrossRef] [PubMed]
- Pinkel, D.; Segraves, R.; Sudar, D.; Clark, S.; Poole, I.; Kowbel, D.; Collins, C.; Kuo, W.L.; Chen, C.; Zhai, Y.; et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 1998, 20, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Pollack, J.R.; Perou, C.M.; Alizadeh, A.A.; Eisen, M.B.; Pergamenschikov, A.; Williams, C.F.; Jeffrey, S.S.; Botstein, D.; Brown, P.O. Genome-wide analysis of DNA copy-number changes using cdna microarrays. Nat. Genet. 1999, 23, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Lo, J.; Kerns, B.J.; Amling, C.L.; Robertson, C.N.; Layfield, L.J. Correlation of DNA ploidy and histologic diagnosis from prostate core-needle biopsies: Is DNA ploidy more sensitive than histology for the diagnosis of carcinoma in small specimens? J. Surg. Oncol. 1996, 63, 41–45. [Google Scholar] [CrossRef]
- Koivisto, P. Aneuploidy and rapid cell proliferation in recurrent prostate cancers with androgen receptor gene amplification. Prostate Cancer Prostat. Dis. 1997, 1, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Reinholz, M.M.; Bruzek, A.K.; Visscher, D.W.; Lingle, W.L.; Schroeder, M.J.; Perez, E.A.; Jenkins, R.B. Breast cancer and aneusomy 17: Implications for carcinogenesis and therapeutic response. Lancet 2009, 10, 267–277. [Google Scholar] [CrossRef]
- Krishnamurti, U.; Hammers, J.L.; Atem, F.D.; Storto, P.D.; Silverman, J.F. Poor prognostic significance of unamplified chromosome 17 polysomy in invasive breast carcinoma. Mod. Pathol. 2009, 22, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Watters, A.D.; Going, J.J.; Cooke, T.G.; Bartlett, J.M. Chromosome 17 aneusomy is associated with poor prognostic factors in invasive breast carcinoma. Breast Cancer Res. Treat. 2003, 77, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Hagenkord, J.M.; Chang, C.C. The rewards and challenges of array-based karyotyping for clinical oncology applications. Leukemia 2009, 23, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Gunn, S.R.; Mohammed, M.S.; Gorre, M.E.; Cotter, P.D.; Kim, J.; Bahler, D.W.; Preobrazhensky, S.N.; Higgins, R.A.; Bolla, A.R.; Ismail, S.H.; et al. Whole-genome scanning by array comparative genomic hybridization as a clinical tool for risk assessment in chronic lymphocytic leukemia. J. Mol. Diagn. 2008, 10, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Lyons-Weiler, M.; Hagenkord, J.; Sciulli, C.; Dhir, R.; Monzon, F.A. Optimization of the affymetrix genechip mapping 10k 2.0 assay for routine clinical use on formalin-fixed paraffin-embedded tissues. Diagn. Mol. Pathol. 2008, 17, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Kim, C.; Wolmark, N. Her2 status and benefit from adjuvant trastuzumab in breast cancer. N. Engl. J. Med. 2008, 358, 1409–1411. [Google Scholar] [CrossRef] [PubMed]
- Wolff, A.C.; Hammond, M.E.; Hicks, D.G.; Dowsett, M.; McShane, L.M.; Allison, K.H.; Allred, D.C.; Bartlett, J.M.; Bilous, M.; Fitzgibbons, P.; et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American society of clinical oncology/college of american pathologists clinical practice guideline update. J. Clin. Oncol. 2013, 31, 3997–4013. [Google Scholar] [CrossRef] [PubMed]
- Dowsett, M.; Hanna, W.M.; Kockx, M.; Penault-Llorca, F.; Ruschoff, J.; Gutjahr, T.; Habben, K.; van de Vijver, M.J. Standardization of her2 testing: Results of an international proficiency-testing ring study. Mod. Pathol. 2007, 20, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Gunn, S.; Yeh, I.T.; Lytvak, I.; Tirtorahardjo, B.; Dzidic, N.; Zadeh, S.; Kim, J.; McCaskill, C.; Lim, L.; Gorre, M.; et al. Clinical array-based karyotyping of breast cancer with equivocal her2 status resolves gene copy number and reveals chromosome 17 complexity. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Sapino, A.; Goia, M.; Recupero, D.; Marchio, C. Current challenges for her2 testing in diagnostic pathology: State of the art and controversial issues. Front. Oncol. 2013, 3, 129. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.V.; Vikesaa, J.; Buhl, S.S.; Rossing, H.H.; Timmermans-Wielenga, V.; Nielsen, F.C. High-density snp arrays improve detection of her2 amplification and polyploidy in breast tumors. BMC Cancer 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Gunn, S.R. The vanguard has arrived in the clinical laboratory: Array-based karyotyping for prognostic markers in chronic lymphocytic leukemia. J. Mol. Diagn. 2010, 12, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Carlton, V.E.; Karlin-Neumann, G.; Sapolsky, R.; Zhang, L.; Moorhead, M.; Wang, Z.C.; Richardson, A.L.; Warren, R.; Walther, A.; et al. High quality copy number and genotype data from ffpe samples using molecular inversion probe (mip) microarrays. BMC Med. Genom. 2009, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jia, P.; Li, F.; Chen, H.; Ji, H.; Hucks, D.; Dahlman, K.B.; Pao, W.; Zhao, Z. Detecting somatic point mutations in cancer genome sequencing data: A comparison of mutation callers. Genome Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Stead, L.F.; Sutton, K.M.; Taylor, G.R.; Quirke, P.; Rabbitts, P. Accurately identifying low-allelic fraction variants in single samples with next-generation sequencing: Applications in tumor subclone resolution. Hum. Mutat. 2013, 34, 1432–1438. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. Varscan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Karow, J. Tumor-only sequencing may misguide therapy but many labs omit matched control. Available online: https://www.genomeweb.com/cancer/tumor-only-sequencing-may-misguide-therapy-many-labs-omit-matched-control (accessed on 15 May 2015).
- Jones, S.; Anagnostou, V.; Lytle, K.; Parpart-Li, S.; Nesselbush, M.; Riley, D.R.; Shukla, M.; Chesnick, B.; Kadan, M.; Papp, E.; et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci. Transl. Med. 2015, 7, 283ra53. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, S. Mechanisms of inherited cancer susceptibility. J. Zhejiang Uni. Sci. B 2008, 9, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Roy, A.; Plon, S.E.; Roychowdhury, S.; Chinnaiyan, A.M. Clinical tumor sequencing: An incidental casualty of the american college of medical genetics and genomics recommendations for reporting of incidental findings. J. Clin. Oncol. 2014, 32, 2203–2205. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Jonnalagadda, J.; Marsh, A.; Grist, S.; Jenkins, M.; Hobson, K.; Taylor, M.; Lindeman, G.J.; Tavtigian, S.V.; Suthers, G.; et al. Characterization of the breast cancer associated atm 7271T>G (V2424Gg) mutation by gene expression profiling. Genes Chromosom. Cancer 2006, 45, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Stankovic, T.; Kidd, A.M.; Sutcliffe, A.; McGuire, G.M.; Robinson, P.; Weber, P.; Bedenham, T.; Bradwell, A.R.; Easton, D.F.; Lennox, G.G.; et al. Atm mutations and phenotypes in ataxia-telangiectasia families in the british isles: Expression of mutant atm and the risk of leukemia, lymphoma, and breast cancer. Am. J. Hum. Genet. 1998, 62, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J.L.; Teraoka, S.; Southey, M.C.; Jenkins, M.A.; Andrulis, I.L.; Knight, J.A.; John, E.M.; Lapinski, R.; Wolitzer, A.L.; Whittemore, A.S.; et al. Population-based estimates of breast cancer risks associated with atm gene variants c.7271t>g and c.1066–6t>g (ivs10–6t>g) from the breast cancer family registry. Hum. Mutat. 2006, 27, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Goldgar, D.E.; Healey, S.; Dowty, J.G.; Da Silva, L.; Chen, X.; Spurdle, A.B.; Terry, M.B.; Daly, M.J.; Buys, S.M.; Southey, M.C.; et al. Rare variants in the atm gene and risk of breast cancer. Breast Cancer Res. 2011, 13, R73. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the atm and atr kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, R.; Kastan, M.B. The ATM-dependent DNA damage signaling pathway. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Yelamos, J.; Farres, J.; Llacuna, L.; Ampurdanes, C.; Martin-Caballero, J. Parp-1 and parp-2: New players in tumour development. Am. J. Cancer Res. 2011, 1, 328–346. [Google Scholar] [PubMed]
- Tangutoori, S.; Baldwin, P.; Sridhar, S. Parp inhibitors: A new era of targeted therapy. Maturitas 2015, 81, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cottman, M.; Schiffman, J.D. Molecular inversion probes: A novel microarray technology and its application in cancer research. Cancer Genet. 2012, 205, 34–55. [Google Scholar] [CrossRef] [PubMed]
- Piha-Paul, S. Phase II study of BMN 673. Available online: https://clinicaltrials.gov/ct2/show/NCT02286687 (accessed on 15 May 2015).
- FDA approves lynparza to treat advanced ovarian cancer. Available online: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm427554.htm (accessed on 15 May 2015).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gray, P.N.; Dunlop, C.L.M.; Elliott, A.M. Not All Next Generation Sequencing Diagnostics are Created Equal: Understanding the Nuances of Solid Tumor Assay Design for Somatic Mutation Detection. Cancers 2015, 7, 1313-1332. https://doi.org/10.3390/cancers7030837
Gray PN, Dunlop CLM, Elliott AM. Not All Next Generation Sequencing Diagnostics are Created Equal: Understanding the Nuances of Solid Tumor Assay Design for Somatic Mutation Detection. Cancers. 2015; 7(3):1313-1332. https://doi.org/10.3390/cancers7030837
Chicago/Turabian StyleGray, Phillip N., Charles L.M. Dunlop, and Aaron M. Elliott. 2015. "Not All Next Generation Sequencing Diagnostics are Created Equal: Understanding the Nuances of Solid Tumor Assay Design for Somatic Mutation Detection" Cancers 7, no. 3: 1313-1332. https://doi.org/10.3390/cancers7030837
APA StyleGray, P. N., Dunlop, C. L. M., & Elliott, A. M. (2015). Not All Next Generation Sequencing Diagnostics are Created Equal: Understanding the Nuances of Solid Tumor Assay Design for Somatic Mutation Detection. Cancers, 7(3), 1313-1332. https://doi.org/10.3390/cancers7030837