
Voltage-Gated Ion Channels in Cancer Cell Proliferation

Abstract
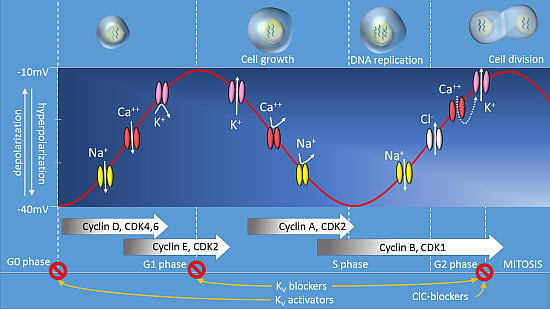
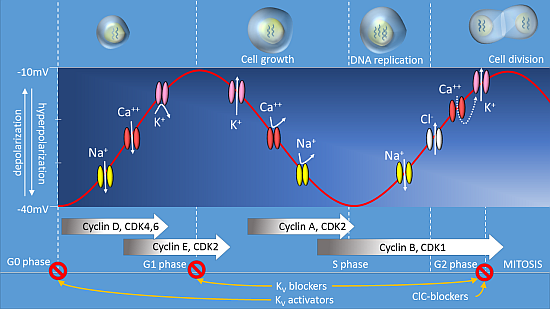
:
1. Introduction
Membrane Potential and Cell Proliferation
2. Voltage-Gated Potassium Channels
2.1. The EAG Superfamily of Potassium Channels in Cancer Biology
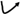 = no ionic flux.
= no ionic flux.
= no ionic flux.
= no ionic flux.
2.2. Other Kv Channels
3. Voltage-Gated Calcium Channels
3.1. CaV1 Channels
3.2. CaV3 Channels
3.3. Auxiliary CaV-α2δ Subunits
3.4. Mechanistic Considerations
4. Voltage-Gated Na+ Channels
5. Voltage-Gated Chloride Ion Channels
6. VGICs as Therapeutic Targets for Cancer
VGIC Activators for Cancer Therapy

{kind=link}
{kind=link}
| VGIC | Expression | Therapeutic Approach | Effect | Ref |
|---|---|---|---|---|
| Voltage gated K+ channel Kv 10.1 Kv 11.1 | Cervical cancer, Breast cancer Ovarian Cancer Osteosarcoma Glioma Melanoma small cell Lung cancer Myeloid leukemia | Small molecule channel inhibitors Kv10.1; Astemizole, Imipramine Kv11.1; Way123398, E4031 Small molecule channel activators NS1643 Antibody Kv10.1 Toxins Kv11.1; Ergotoxin siRNA/shRNA Kv10.1, Kv11.1 | Reduced proliferation by increasing apoptosis or cell cycle arrest at G0/G1, G1/S or G2/M phase | [36,39,48,133,143,151,152,164,169] |
| Voltage gated Ca++ channel Cav 1/Cav 1.3 Cav 3/Cav 3.1 | Adrenal Adenomas Prostate cancer Melanoma Glioblastoma | Small molecule Inhibitors Cav; mibefradil, TTL1177, endostatin siRNA/shRNA Cav3.1, Cav1.3 | Reduced cell proliferation by inducing apoptosis | [88,94,95] |
| Voltage gated Na+ channel Nav 1.5, 1.6, 1.7, 1.9 | Prostate cancer Ovarian cancer Cervical cancer Breast cancer | Small molecule Inhibitors Phenytoin, novel hydroxyl amide Toxins Tetradotoxin | Reduced cell proliferation via cell cycle arrest observed with the small molecule inhibitors. | [156,157,158] |
| Voltage gated Cl− channel | Gliomas | Small molecule inhibitors 5-nitro-2-(3-phenylpropyl-amino) benzoic acid (NPPB)NPPB Small molecule activators Bufadienolides Toxin Chlorotoxin siRNA CLC-3 | Reduced proliferation by cell cycle inhibition at G1, G2/M phase via cell volume regulation
Channel activators induce apoptosis | [131,159,160,161,162,163,164,165,166,171] |
7. Conclusions
Acknowledgments
Author Contributions
- -
- Vidhya R. Rao has contributed to the collection of literature and writing of the manuscript.
- -
- Mathew Perez-Neut has contributed to the collection of literature and writing of the manuscript.
- -
- Simon Kaja has contributed to the collection of literature and writing of the manuscript.
- -
- Saverio Gentile is the principal investigator and corresponding author who was responsible for the layout, design and organization with critical inputs.
Conflicts of Interest
References
- Stewart, B.; Wild, C. World Cancer Report; IARC Press: Lyon, France, 2014. [Google Scholar]
- Cone, C.D., Jr.; Tongier, M., Jr. Mitotic synchronization of L-strain fibroblasts with 5-aminouracil as determined by time-lapse cinephotography. NASA TN D-5021. Tech. Note U.S. Natl. Aeronaut. Space Adm. 1969. [Google Scholar] [PubMed]
- DeSantis, C.E.; Lin, C.C.; Mariotto, A.B.; Siegel, R.L.; Stein, K.D.; Kramer, J.L.; Alteri, R.; Robbins, A.S.; Jemal, A. Cancer treatment and survivorship statistics, 2014. CA Cancer J. Clin. 2014, 64, 252–271. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R. The Biology of Cancer; Garland Science: New York, NY, USA, 2007. [Google Scholar]
- Pecorino, L. Molecular Biology of Cancer: Mechanisms, Targets, and Therapeutics; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed Cell Death Pathways in Cancer: A Review of Apoptosis, Autophagy and Programmed Necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xiong, Z.-G. Ion Channels as Targets for Cancer Therapy. Int. J. Physiol. Pathophysiol. Pharmacol. 2011, 3, 156. [Google Scholar] [PubMed]
- Pedersen, S.F.; Stock, C. Ion channels and transporters in cancer: Pathophysiology, regulation, and clinical potential. Cancer Res. 2013, 73, 1658–1661. [Google Scholar] [CrossRef] [PubMed]
- Djamgoz, M.B.; Coombes, R.C.; Schwab, A. Ion transport and cancer: From initiation to metastasis. Philos. Trans. R Soc. Lond. B Biol. Sci. 2014, 369, 20130092. [Google Scholar] [CrossRef] [PubMed]
- Golias, C.H.; Charalabopoulos, A.; Charalabopoulos, K. Cell proliferation and cell cycle control: A mini review. Int. J. Clin. Pract. 2004, 58, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, K.; van Bockstaele, D.R.; Berneman, Z.N. The cell cycle: A review of regulation, deregulation and therapeutic targets in cancer. Cell. Prolif. 2003, 36, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Brackenbury, W.J. Membrane potential and cancer progression. Front. Physiol. 2013, 4, 185. [Google Scholar] [CrossRef] [PubMed]
- Blackiston, D.J.; McLaughlin, K.A.; Levin, M. Bioelectric controls of cell proliferation: ion channels, membrane voltage and the cell cycle. Cell Cycle 2009, 8, 3527–3536. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.H. Generation of resting membrane potential. Adv. Physiol. Educ. 2004, 28, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Cone, C.D., Jr. Electroosmotic interactions accompanying mitosis initation in sarcoma cells in vitro. Trans. NY Acad. Sci. 1969, 31, 404–427. [Google Scholar] [CrossRef]
- Cone, C.D., Jr.; Tongier, M., Jr. Control of somatic cell mitosis by simulated changes in the transmembrane potential level. Oncology 1971, 25, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Stillwell, E.F.; Cone, C.M.; Cone, C.D., Jr. Stimulation of DNA synthesis in CNS neurones by sustained depolarisation. Nat. New Biol. 1973, 246, 110–111. [Google Scholar] [CrossRef] [PubMed]
- Wonderlin, W.F.; Woodfork, K.A.; Strobl, J.S. Changes in membrane potential during the progression of MCF-7 human mammary tumor cells through the cell cycle. J. Cell. Physiol. 1995, 165, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, J.; Mummery, C.L.; Tertoolen, L.G.; van der Saag, P.T.; de Laat, S.W. Cation transport and growth regulation in neuroblastoma cells. Modulations of K+ transport and electrical membrane properties during the cell cycle. J. Cell. Physiol. 1981, 107, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Cone, C.D., Jr.; Cone, C.M. Induction of mitosis in mature neurons in central nervous system by sustained depolarization. Science 1976, 192, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Sachs, H.G.; Stambrook, P.J.; Ebert, J.D. Changes in membrane potential during the cell cycle. Exp. Cell. Res. 1974, 83, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Sundelacruz, S.; Levin, M.; Kaplan, D.L. Role of membrane potential in the regulation of cell proliferation and differentiation. Stem Cell Rev. 2009, 5, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Jan, L.Y. Targeting potassium channels in cancer. J. Cell Biol. 2014, 206, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Bezanilla, F. Voltage-gated ion channels. IEEE Trans. Nanobioscience 2005, 4, 34–48. [Google Scholar]
- Catterall, W.A. Structure and function of voltage-gated ion channels. Annu. Rev. Biochem. 1995, 64, 493–531. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.M.; Hille, B. Voltage-gated ion channels and electrical excitability. Neuron 1998, 20, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Miller, C. An overview of the potassium channel family. Genome Biol. 2000, 1, 1–5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pardo, L.A.; del Camino, D.; Sanchez, A.; Alves, F.; Bruggemann, A.; Beckh, S.; Stuhmer, W. Oncogenic potential of EAG K(+) channels. EMBO J. 1999, 18, 5540–5547. [Google Scholar] [CrossRef] [PubMed]
- Hemmerlein, B.; Weseloh, R.M.; Mello de Queiroz, F.; Knotgen, H.; Sanchez, A.; Rubio, M.E.; Martin, S.; Schliephacke, T.; Jenke, M.; Heinz Joachim, R.; et al. Overexpression of Eag1 potassium channels in clinical tumours. Mol. Cancer 2006, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Patt, S.; Preussat, K.; Beetz, C.; Kraft, R.; Schrey, M.; Kalff, R.; Schonherr, K.; Heinemann, S.H. Expression of ether a go-go potassium channels in human gliomas. Neurosci. Lett. 2004, 368, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Mello de Queiroz, F.; Suarez-Kurtz, G.; Stuhmer, W.; Pardo, L.A. Ether a go-go potassium channel expression in soft tissue sarcoma patients. Mol. Cancer 2006, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.W.; Luo, H.S.; Jin, X.; Yan, J.J.; Ai, Y.W. Aberrant expression of Eag1 potassium channels in gastric cancer patients and cell lines. Med. Oncol. 2007, 24, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.W.; Yan, J.J.; An, P.; Lu, P.; Luo, H.S. Aberrant expression of ether a go-go potassium channel in colorectal cancer patients and cell lines. World J. Gastroenterol. 2007, 13, 1257–1261. [Google Scholar] [CrossRef] [PubMed]
- Ousingsawat, J.; Spitzner, M.; Puntheeranurak, S.; Terracciano, L.; Tornillo, L.; Bubendorf, L.; Kunzelmann, K.; Schreiber, R. Expression of voltage-gated potassium channels in human and mouse colonic carcinoma. Clin. Cancer Res. 2007, 13, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Varela, D.; Zwick-Wallasch, E.; Knotgen, H.; Sanchez, A.; Hettmann, T.; Ossipov, D.; Weseloh, R.; Contreras-Jurado, C.; Rothe, M.; Stuhmer, W.; et al. Monoclonal antibody blockade of the human Eag1 potassium channel function exerts antitumor activity. Cancer Res. 2007, 67, 7343–7349. [Google Scholar] [CrossRef] [PubMed]
- Hartung, F.; Stuhmer, W.; Pardo, L.A. Tumor cell-selective apoptosis induction through targeting of K(V)10.1 via bifunctional TRAIL antibody. Mol. Cancer 2011, 10, 109. [Google Scholar] [CrossRef] [PubMed]
- Asher, V.; Sowter, H.; Shaw, R.; Bali, A.; Khan, R. Eag and HERG potassium channels as novel therapeutic targets in cancer. World J. Surg. Oncol. 2010, 8, 113. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Mello de Queiroz, F.; Downie, B.R.; Suckow, A.; Stuhmer, W.; Pardo, L.A. Silencing the activity and proliferative properties of the human EagI Potassium Channel by RNA Interference. J. Biol. Chem. 2006, 281, 13030–13037. [Google Scholar] [CrossRef] [PubMed]
- Warmke, J.W.; Ganetzky, B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. USA 1994, 91, 3438–3442. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, J.I.; Perry, M.D.; Perrin, M.J.; Mann, S.A.; Ke, Y.; Hill, A.P. hERG K(+) channels: structure, function, and clinical significance. Physiol. Rev. 2012, 92, 1393–1478. [Google Scholar] [CrossRef] [PubMed]
- Babcock, J.J.; Li, M. hERG channel function: Beyond long QT. Acta Pharmacol. Sin. 2013, 34, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Arcangeli, A. Expression and role of hERG channels in cancer cells. Novartis Found. Symp. 2005, 266, 225–234. [Google Scholar]
- Wang, H.; Zhang, Y.; Cao, L.; Han, H.; Wang, J.; Yang, B.; Nattel, S.; Wang, Z. HERG K+ channel, a regulator of tumor cell apoptosis and proliferation. Cancer Res. 2002, 62, 4843–4848. [Google Scholar] [PubMed]
- Glassmeier, G.; Hempel, K.; Wulfsen, I.; Bauer, C.K.; Schumacher, U.; Schwarz, J.R. Inhibition of HERG1 K+ channel protein expression decreases cell proliferation of human small cell lung cancer cells. Pflugers. Arch. 2012, 463, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Staudacher, I.; Jehle, J.; Staudacher, K.; Pledl, H.W.; Lemke, D.; Schweizer, P.A.; Becker, R.; Katus, H.A.; Thomas, D. HERG K+ channel-dependent apoptosis and cell cycle arrest in human glioblastoma cells. PLoS ONE 2014, 9, e88164. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.; Schweizer, P.A.; Katus, H.A.; Thomas, D. Novel roles for hERG K(+) channels in cell proliferation and apoptosis. Cell Death Dis. 2011, 2, e193. [Google Scholar] [CrossRef] [PubMed]
- Perez-Neut, M.; Shum, A.; Cuevas, B.D.; Miller, R.; Gentile, S. Stimulation of hERG1 channel activity promotes a calcium-dependent degradation of cyclin E2, but not cyclin E1, in breast cancer cells. Oncotarget 2015, 6, 1631–1639. [Google Scholar] [PubMed]
- Lee, Y.S.; Sayeed, M.M.; Wurster, R.D. Inhibition of cell growth by K+ channel modulators is due to interference with agonist-induced Ca2+ release. Cell. Signal. 1993, 5, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Hegle, A.P.; Marble, D.D.; Wilson, G.F. A voltage-driven switch for ion-independent signaling by ether-a-go-go K+ channels. Proc. Natl. Acad. Sci. USA 2006, 103, 2886–2891. [Google Scholar] [CrossRef] [PubMed]
- Downie, B.R.; Sanchez, A.; Knotgen, H.; Contreras-Jurado, C.; Gymnopoulos, M.; Weber, C.; Stuhmer, W.; Pardo, L.A. Eag1 expression interferes with hypoxia homeostasis and induces angiogenesis in tumors. J. Biol. Chem. 2008, 283, 36234–36240. [Google Scholar] [CrossRef] [PubMed]
- Cidad, P.; Jimenez-Perez, L.; Garcia-Arribas, D.; Miguel-Velado, E.; Tajada, S.; Ruiz-McDavitt, C.; Lopez-Lopez, J.R.; Perez-Garcia, M.T. Kv1.3 channels can modulate cell proliferation during phenotypic switch by an ion-flux independent mechanism. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Wonderlin, W.F.; Strobl, J.S. Potassium channels, proliferation and G1 progression. J. Membr. Biol. 1996, 154, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Woodfork, K.A.; Wonderlin, W.F.; Peterson, V.A.; Strobl, J.S. Inhibition of ATP-sensitive potassium channels causes reversible cell-cycle arrest of human breast cancer cells in tissue culture. J. Cell. Physiol. 1995, 162, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Fan, Y.; Wang, S.; Wang, L.; He, W.; Zhang, Q.; Li, X. Effects of voltage-gated K+ channel on cell proliferation in multiple myeloma. Sci. World J. 2014. [Google Scholar] [CrossRef]
- Nilius, B.; Schwarz, G.; Droogmans, G. Control of intracellular calcium by membrane potential in human melanoma cells. Am. J. Physiol. 1993, 265, C1501–C1510. [Google Scholar] [PubMed]
- Gelfand, E.W.; Cheung, R.K.; Grinstein, S. Role of membrane potential in the regulation of lectin-induced calcium uptake. J. Cell. Physiol. 1984, 121, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Lepple-Wienhues, A.; Berweck, S.; Bohmig, M.; Leo, C.P.; Meyling, B.; Garbe, C.; Wiederholt, M. K+ channels and the intracellular calcium signal in human melanoma cell proliferation. J. Membr. Biol. 1996, 151, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Menendez, S.T.; Villaronga, M.A.; Rodrigo, J.P.; Alvarez-Teijeiro, S.; Garcia-Carracedo, D.; Urdinguio, R.G.; Fraga, M.F.; Pardo, L.A.; Viloria, C.G.; Suarez, C.; et al. Frequent aberrant expression of the human ether a go-go (hEAG1) potassium channel in head and neck cancer: Pathobiological mechanisms and clinical implications. J. Mol. Med. (Berl.) 2012, 90, 1173–1184. [Google Scholar] [CrossRef]
- Ouadid-Ahidouch, H.; Rodat-Despoix, L.; Matifat, F.; Morin, G.; Ahidouch, A. DNA methylation of channel-related genes in cancers. Biochim. Biophys. Acta 2015. [Google Scholar] [CrossRef]
- Feng, Q.; Hawes, S.E.; Stern, J.E.; Wiens, L.; Lu, H.; Dong, Z.M.; Jordan, C.D.; Kiviat, N.B.; Vesselle, H. DNA methylation in tumor and matched normal tissues from non-small cell lung cancer patients. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Cicek, M.S.; Koestler, D.C.; Fridley, B.L.; Kalli, K.R.; Armasu, S.M.; Larson, M.C.; Wang, C.; Winham, S.J.; Vierkant, R.A.; Rider, D.N.; et al. Epigenome-wide ovarian cancer analysis identifies a methylation profile differentiating clear-cell histology with epigenetic silencing of the HERG K+ channel. Hum. Mol. Genet. 2013, 22, 3038–3047. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Li, Z.; Chen, C.; Luo, X.; Xiao, J.; Dong, D.; Lu, Y.; Yang, B.; Wang, Z. Transcriptional and post-transcriptional mechanisms for oncogenic overexpression of ether a go-go K+ channel. PLoS ONE 2011, 6, e20362. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.P.; Marron Fernandez de Velasco, E.; Mizuno, F.; Scappini, E.L.; Gloss, B.; Erxleben, C.; Williams, J.G.; Stapleton, H.M.; Gentile, S.; Armstrong, D.L. A rapid cytoplasmic mechanism for PI3 kinase regulation by the nuclear thyroid hormone receptor, TRbeta, and genetic evidence for its role in the maturation of mouse hippocampal synapses in vivo. Endocrinology 2014, 155, 3713–3724. [Google Scholar] [CrossRef] [PubMed]
- Gentile, S.; Darden, T.; Erxleben, C.; Romeo, C.; Russo, A.; Martin, N.; Rossie, S.; Armstrong, D.L. Rac GTPase signaling through the PP5 protein phosphatase. Proc. Natl. Acad. Sci. USA 2006, 103, 5202–5206. [Google Scholar] [CrossRef] [PubMed]
- Storey, N.M.; Gentile, S.; Ullah, H.; Russo, A.; Muessel, M.; Erxleben, C.; Armstrong, D.L. Rapid signaling at the plasma membrane by a nuclear receptor for thyroid hormone. Proc. Natl. Acad. Sci. USA 2006, 103, 5197–5201. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, T.; Genazzani, A.R. Non-genomic actions of sex steroid hormones. Eur. J. Endocrinol. 2003, 148, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Kurokawa, J. Non-genomic regulation of cardiac ion channels by sex hormones. Cardiovasc. Hematol. Disord. Drug Targets 2008, 8, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Arcangeli, A.; Yuan, J.X. American Journal of Physiology-Cell Physiology theme: Ion channels and transporters in cancer. Am. J. Physiol. Cell Physiol. 2011, 301, C253–C254. [Google Scholar] [CrossRef]
- Ouadid-Ahidouch, H.; Roudbaraki, M.; Delcourt, P.; Ahidouch, A.; Joury, N.; Prevarskaya, N. Functional and molecular identification of intermediate-conductance Ca2+-activated K+ channels in breast cancer cells: Association with cell cycle progression. Am. J. Physiol. Cell Physiol. 2004, 287, C125–C134. [Google Scholar] [CrossRef] [PubMed]
- Arcangeli, A.; Bianchi, L.; Becchetti, A.; Faravelli, L.; Coronnello, M.; Mini, E.; Olivotto, M.; Wanke, E. A novel inward-rectifying K+ current with a cell-cycle dependence governs the resting potential of mammalian neuroblastoma cells. J. Physiol. 1995, 489, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Villalonga, N.; Martinez-Marmol, R.; Roura-Ferrer, M.; David, M.; Valenzuela, C.; Soler, C.; Felipe, A. Cell cycle-dependent expression of Kv1.5 is involved in myoblast proliferation. Biochim. Biophys. Acta 2008, 1783, 728–736. [Google Scholar] [CrossRef]
- Huang, X.; Dubuc, A.M.; Hashizume, R.; Berg, J.; He, Y.; Wang, J.; Chiang, C.; Cooper, M.K.; Northcott, P.A.; Taylor, M.D.; et al. Voltage-gated potassium channel EAG2 controls mitotic entry and tumor growth in medulloblastoma via regulating cell volume dynamics. Genes Dev. 2012, 26, 1780–1796. [Google Scholar] [CrossRef] [PubMed]
- MacLean, J.N.; Zhang, Y.; Johnson, B.R.; Harris-Warrick, R.M. Activity-independent homeostasis in rhythmically active neurons. Neuron 2003, 37, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Holmes, T.C.; Berman, K.; Swartz, J.E.; Dagan, D.; Levitan, I.B. Expression of voltage-gated potassium channels decreases cellular protein tyrosine phosphorylation. J. Neurosci. 1997, 17, 8964–8974. [Google Scholar] [PubMed]
- Felipe, A.; Bielanska, J.; Comes, N.; Vallejo, A.; Roig, S.; Ramon, Y.C.S.; Condom, E.; Hernandez-Losa, J.; Ferreres, J.C. Targeting the voltage-dependent K+ channels Kv1.3 and Kv1.5 as tumor biomarkers for cancer detection and prevention. Curr. Med. Chem. 2012, 19, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Bock, J.; Jekle, A.; Soddemann, M.; Adams, C.; Lang, F.; Zoratti, M.; Gulbins, E. A novel potassium channel in lymphocyte mitochondria. J. Biol. Chem. 2005, 280, 12790–12798. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Bock, J.; Grassme, H.; Soddemann, M.; Wilker, B.; Lang, F.; Zoratti, M.; Gulbins, E. Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc. Natl Acad. Sci. USA 2008, 105, 14861–14866. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Henry, B.; Sassi, N.; Zoratti, M.; Chandy, K.G.; Gulbins, E.; Szabo, I. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol. Med. 2012, 4, 577–593. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Harry, G.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Ransom, C.B.; Liu, X.; Sontheimer, H. Current transients associated with BK channels in human glioma cells. J. Membr. Biol. 2003, 193, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Ransom, C.B.; Liu, X.; Sontheimer, H. BK channels in human glioma cells have enhanced calcium sensitivity. Glia 2002, 38, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chang, Y.; Reinhart, P.H.; Sontheimer, H. Cloning and characterization of glioma BK, a novel BK channel isoform highly expressed in human glioma cells. J. Neurosci. 2002, 22, 1840–1849. [Google Scholar] [PubMed]
- Simms, B.A.; Zamponi, G.W. Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron 2014, 82, 24–45. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Neuronal calcium signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef]
- Guéguinou, M.; Chantôme, A.; Fromont, G.; Bougnoux, P.; Vandier, C.; Potier-Cartereau, M. KCa and Ca2+ channels: The complex thought. Biochim. Biophys. Acta 2014, 1843, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I.; Goh, G.; Stolting, G.; de Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.W.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zeng, X.; Zhang, R.; Huang, J.; Kuang, X.; Yang, J.; Liu, J.; Tawfik, O.; Thrasher, J.B.; Li, B. Cav1.3 channel alpha1D protein is overexpressed and modulates androgen receptor transactivation in prostate cancers. Urol. Oncol. 2014, 32, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, E.; Shiota, M.; Yokomizo, A.; Inokuchi, J.; Uchiumi, T.; Naito, S. EP2 signaling mediates suppressive effects of celecoxib on androgen receptor expression and cell proliferation in prostate cancer. Prostate Cancer Prostatic Dis. 2014, 17, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tao, J.; Huang, H.; Ding, G.; Cheng, Y.; Sun, W. Effects of celecoxib on voltage-gated calcium channel currents in rat pheochromocytoma (PC12) cells. Pharmacol. Res. 2007, 56, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.Q.; Zhang, H.; Tan, S.J.; Gu, Y.C. Nifedipine promotes the proliferation and migration of breast cancer cells. PLoS ONE 2014, 9, e113649. [Google Scholar] [CrossRef] [PubMed]
- Gray, L.S.; Perez-Reyes, E.; Gomora, J.C.; Haverstick, D.M.; Shattock, M.; McLatchie, L.; Harper, J.; Brooks, G.; Heady, T.; Macdonald, T.L. The role of voltage gated T-type Ca2+ channel isoforms in mediating “capacitative” Ca2+ entry in cancer cells. Cell Calcium 2004, 36, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Ohkubo, T.; Yamazaki, J. T-type voltage-activated calcium channel Cav3.1, but not Cav3.2, is involved in the inhibition of proliferation and apoptosis in MCF-7 human breast cancer cells. Int. J. Oncol. 2012, 41, 267–725. [Google Scholar] [PubMed]
- Yu, W.; Wang, P.; Ma, H.; Zhang, G.; Yulin, Z.; Lu, B.; Wang, H.; Dong, M. Suppression of T-type Ca2+ channels inhibited human laryngeal squamous cell carcinoma cell proliferation running title: roles of T-type Ca2+ channels in LSCC cell proliferation. Clin. Lab. 2014, 60, 621–628. [Google Scholar] [PubMed]
- Das, A.; Pushparaj, C.; Bahi, N.; Sorolla, A.; Herreros, J.; Pamplona, R.; Vilella, R.; Matias-Guiu, X.; Marti, R.M.; Canti, C. Functional expression of voltage-gated calcium channels in human melanoma. Pigment. Cell. Melanoma Res. 2012, 25, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Pushparaj, C.; Herreros, J.; Nager, M.; Vilella, R.; Portero, M.; Pamplona, R.; Matias-Guiu, X.; Marti, R.M.; Canti, C. T-type calcium channel blockers inhibit autophagy and promote apoptosis of malignant melanoma cells. Pigment. Cell. Melanoma Res. 2013, 26, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Valerie, N.C.; Dziegielewska, B.; Hosing, A.S.; Augustin, E.; Gray, L.S.; Brautigan, D.L.; Larner, J.M.; Dziegielewski, J. Inhibition of T-type calcium channels disrupts Akt signaling and promotes apoptosis in glioblastoma cells. Biochem. Pharmacol. 2013, 85, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, J.; Jiang, D.; Zhang, D.; Qian, Z.; Liu, C.; Tao, J. Inhibition of T-type Ca2+ channels by endostatin attenuates human glioblastoma cell proliferation and migration. Br. J. Pharmacol. 2012, 166, 1247–1260. [Google Scholar] [CrossRef] [PubMed]
- Weaver, E.M.; Zamora, F.J.; Puplampu-Dove, Y.A.; Kiessu, E.; Hearne, J.L.; Martin-Caraballo, M. Regulation of T-type calcium channel expression by sodium butyrate in prostate cancer cells. Eur. J. Pharmacol. 2015, 749, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Killary, A.M.; Wolf, M.E.; Giambernardi, T.A.; Naylor, S.L. Definition of a tumor suppressor locus within human chromosome 3p21-p22. Proc. Natl. Acad. Sci. USA 1992, 89, 10877–10881. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Sekido, Y.; Maximov, A.; Saad, M.; Forgacs, E.; Latif, F.; Wei, M.H.; Lerman, M.; Lee, J.H.; Perez-Reyes, E.; et al. Functional properties of a new voltage-dependent calcium channel alpha(2)delta auxiliary subunit gene (CACNA2D2). J. Biol. Chem. 2000, 275, 12237–12242. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, D.; Duh, F.M.; Wei, M.F.; Johnson, B.E.; Lerman, M.I. A G-to-A single nucleotide polymorphism in intron 2 of the human CACNA2D2 gene that maps at 3p21.3. Mol. Cell. Probes 2001, 15, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Lerman, M.I.; Minna, J.D. The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: Identification and evaluation of the resident candidate tumor suppressor genes. The International Lung Cancer Chromosome 3p21.3 Tumor Suppressor Gene Consortium. Cancer Res. 2000, 60, 6116–6133. [Google Scholar] [PubMed]
- Carboni, G.L.; Gao, B.; Nishizaki, M.; Xu, K.; Minna, J.D.; Roth, J.A.; Ji, L. CACNA2D2-mediated apoptosis in NSCLC cells is associated with alterations of the intracellular calcium signaling and disruption of mitochondria membrane integrity. Oncogene 2003, 22, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Warnier, M.; Roudbaraki, M.; Derouiche, S.; Delcourt, P.; Bokhobza, A.; Prevarskaya, N.; Mariot, P. CACNA2D2 promotes tumorigenesis by stimulating cell proliferation and angiogenesis. Oncogene 2015. [Google Scholar] [CrossRef]
- Wanajo, A.; Sasaki, A.; Nagasaki, H.; Shimada, S.; Otsubo, T.; Owaki, S.; Shimizu, Y.; Eishi, Y.; Kojima, K.; Nakajima, Y.; et al. Methylation of the calcium channel-related gene, CACNA2D3, is frequent and a poor prognostic factor in gastric cancer. Gastroenterology 2008, 135, 580–590. [Google Scholar] [CrossRef]
- Palmieri, C.; Rudraraju, B.; Monteverde, M.; Lattanzio, L.; Gojis, O.; Brizio, R.; Garrone, O.; Merlano, M.; Syed, N.; Lo Nigro, C.; et al. Methylation of the calcium channel regulatory subunit alpha2delta-3 (CACNA2D3) predicts site-specific relapse in oestrogen receptor-positive primary breast carcinomas. Br. J. Cancer 2012, 107, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.M.; Kong, K.L.; Chen, L.; Liu, M.; Wong, A.M.; Zhu, C.; Tsang, J.W.; Guan, X.Y. Characterization of CACNA2D3 as a putative tumor suppressor gene in the development and progression of nasopharyngeal carcinoma. Int. J. Cancer 2013, 133, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, C.L.; Nie, C.J.; Li, J.C.; Zeng, T.T.; Zhou, J.; Chen, J.; Chen, K.; Fu, L.; Liu, H.; et al. Investigation of tumor suppressing function of CACNA2D3 in esophageal squamous cell carcinoma. PLoS ONE 2013, 8, e60027. [Google Scholar] [CrossRef] [PubMed]
- Gackiere, F.; Warnier, M.; Katsogiannou, M.; Derouiche, S.; Delcourt, P.; Dewailly, E.; Slomianny, C.; Humez, S.; Prevarskaya, N.; Roudbaraki, M.; et al. Functional coupling between large-conductance potassium channels and Cav3.2 voltage-dependent calcium channels participates in prostate cancer cell growth. Biol. Open 2013, 2, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Catterall, W.A. Overview of the voltage-gated sodium channel family. Genome Biol. 2003, 4, 207. [Google Scholar] [CrossRef] [PubMed]
- Roger, S.; Rollin, J.; Barascu, A.; Besson, P.; Raynal, P.I.; Iochmann, S.; Lei, M.; Bougnoux, P.; Gruel, Y.; Le Guennec, J.Y. Voltage-gated sodium channels potentiate the invasive capacities of human non-small-cell lung cancer cell lines. Int. J. Biochem. Cell. Biol. 2007, 39, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Roger, S.; Potier, M.; Vandier, C.; Besson, P.; Le Guennec, J.Y. Voltage-gated sodium channels: new targets in cancer therapy? Curr. Pharm Des. 2006, 12, 3681–3695. [Google Scholar] [CrossRef] [PubMed]
- Fraser, S.P.; Diss, J.K.; Chioni, A.M.; Mycielska, M.E.; Pan, H.; Yamaci, R.F.; Pani, F.; Siwy, Z.; Krasowska, M.; Grzywna, Z.; et al. Voltage-gated sodium channel expression and potentiation of human breast cancer metastasis. Clin. Cancer Res. 2005, 11, 5381–5389. [Google Scholar] [CrossRef] [PubMed]
- Brackenbury, W.J.; Chioni, A.M.; Diss, J.K.; Djamgoz, M.B. The neonatal splice variant of Nav1.5 potentiates in vitro invasive behaviour of MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2007, 101, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Roger, S.; Besson, P.; Le Guennec, J.Y. Involvement of a novel fast inward sodium current in the invasion capacity of a breast cancer cell line. Biochim. Biophys. Acta 2003, 1616, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.S.; Smith, B.A.; Harper, J.M. Voltage-gated Na+ channels confer invasive properties on human prostate cancer cells. Pflugers. Arch. 2004, 447, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Diss, J.K.; Archer, S.N.; Hirano, J.; Fraser, S.P.; Djamgoz, M.B. Expression profiles of voltage-gated Na(+) channel alpha-subunit genes in rat and human prostate cancer cell lines. Prostate 2001, 48, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Diss, J.K.; Stewart, D.; Pani, F.; Foster, C.S.; Walker, M.M.; Patel, A.; Djamgoz, M.B. A potential novel marker for human prostate cancer: Voltage-gated sodium channel expression in vivo. Prostate Cancer Prostatic Dis. 2005, 8, 266–273. [Google Scholar] [CrossRef]
- Anderson, J.D.; Hansen, T.P.; Lenkowski, P.W.; Walls, A.M.; Choudhury, I.M.; Schenck, H.A.; Friehling, M.; Holl, G.M.; Patel, M.K.; Sikes, R.A.; et al. Voltage-gated sodium channel blockers as cytostatic inhibitors of the androgen-independent prostate cancer cell line PC-3. Mol. Cancer Ther. 2003, 2, 1149–1154. [Google Scholar] [PubMed]
- Nakajima, T.; Kubota, N.; Tsutsumi, T.; Oguri, A.; Imuta, H.; Jo, T.; Oonuma, H.; Soma, M.; Meguro, K.; Takano, H.; et al. Eicosapentaenoic acid inhibits voltage-gated sodium channels and invasiveness in prostate cancer cells. Br. J. Pharmacol. 2009, 156, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Andrikopoulos, P.; Fraser, S.P.; Patterson, L.; Ahmad, Z.; Burcu, H.; Ottaviani, D.; Diss, J.K.; Box, C.; Eccles, S.A.; Djamgoz, M.B. Angiogenic functions of voltage-gated Na+ Channels in human endothelial cells: Modulation of vascular endothelial growth factor (VEGF) signaling. J. Biol. Chem. 2011, 286, 16846–16860. [Google Scholar] [CrossRef]
- Davis, G.C.; Kong, Y.; Paige, M.; Li, Z.; Merrick, E.C.; Hansen, T.; Suy, S.; Wang, K.; Dakshanamurthy, S.; Cordova, A.; et al. Asymmetric synthesis and evaluation of a hydroxyphenylamide voltage-gated sodium channel blocker in human prostate cancer xenografts. Bioorg. Med. Chem. 2012, 20, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Mindell, J.A.; Maduke, M. ClC chloride channels. Genome Biol. 2001, 2, 3000–3001. [Google Scholar] [CrossRef]
- Olsen, M.L.; Schade, S.; Lyons, S.A.; Amaral, M.D.; Sontheimer, H. Expression of voltage-gated chloride channels in human glioma cells. J. Neurosci. 2003, 23, 5572–5582. [Google Scholar] [PubMed]
- Hong, S.; Bi, M.; Wang, L.; Kang, Z.; Ling, L.; Zhao, C. CLC-3 channels in cancer (review). Oncol. Rep. 2015, 33, 507–514. [Google Scholar] [PubMed]
- Habela, C.W.; Ernest, N.J.; Swindall, A.F.; Sontheimer, H. Chloride accumulation drives volume dynamics underlying cell proliferation and migration. J. Neurophysiol. 2009, 101, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Habela, C.W.; Sontheimer, H. Cytoplasmic volume condensation is an integral part of mitosis. Cell Cycle 2007, 6, 1613–1620. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Mao, J.; Wang, L.; Zhu, L.; Li, H.; Wang, W.; Jin, X.; Zhu, J.; Chen, L. ClC-3 chloride channels are essential for cell proliferation and cell cycle progression in nasopharyngeal carcinoma cells. Acta Biochim. Biophys. Sin. (Shanghai) 2010, 42, 370–380. [Google Scholar] [CrossRef]
- Li, M.; Wang, B.; Lin, W. Cl-channel blockers inhibit cell proliferation and arrest the cell cycle of human ovarian cancer cells. Eur. J. Gynaecol. Oncol. 2008, 29, 267–271. [Google Scholar] [PubMed]
- Li, M.; Wu, D.B.; Wang, J. Effects of volume-activated chloride channels on the invasion and migration of human endometrial cancer cells. Eur. J. Gynaecol. Oncol. 2012, 34, 60–64. [Google Scholar]
- Garcia-Ferreiro, R.E.; Kerschensteiner, D.; Major, F.; Monje, F.; Stuhmer, W.; Pardo, L.A. Mechanism of block of hEag1 K+ channels by imipramine and astemizole. J. Gen. Physiol. 2004, 124, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Spitzner, M.; Ousingsawat, J.; Scheidt, K.; Kunzelmann, K.; Schreiber, R. Voltage-gated K+ channels support proliferation of colonic carcinoma cells. FASEB J. 2007, 21, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Asher, V.; Warren, A.; Shaw, R.; Sowter, H.; Bali, A.; Khan, R. The role of Eag and HERG channels in cell proliferation and apoptotic cell death in SK-OV-3 ovarian cancer cell line. Cancer Cell. Int. 2011, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zhong, D.; Lin, B.; Zhai, W.; Ding, Z.; Wu, J. p38 MAPK regulates the expression of ether a go-go potassium channel in human osteosarcoma cells. Radiol. Oncol. 2013, 47, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Jahchan, N.S.; Dudley, J.T.; Mazur, P.K.; Flores, N.; Yang, D.; Palmerton, A.; Zmoos, A.F.; Vaka, D.; Tran, K.Q.; Zhou, M.; et al. A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov. 2013, 3, 1364–1377. [Google Scholar] [CrossRef] [PubMed]
- Gavrilova-Ruch, O.; Schonherr, K.; Gessner, G.; Schonherr, R.; Klapperstuck, T.; Wohlrab, W.; Heinemann, S.H. Effects of imipramine on ion channels and proliferation of IGR1 melanoma cells. J. Membr. Biol. 2002, 188, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Ouadid-Ahidouch, H.; Le Bourhis, X.; Roudbaraki, M.; Toillon, R.A.; Delcourt, P.; Prevarskaya, N. Changes in the K+ current-density of MCF-7 cells during progression through the cell cycle: possible involvement of a h-ether.a-gogo K+ channel. Receptors Channels 2001, 7, 345–356. [Google Scholar] [PubMed]
- Garcia-Quiroz, J.; Garcia-Becerra, R.; Santos-Martinez, N.; Barrera, D.; Ordaz-Rosado, D.; Avila, E.; Halhali, A.; Villanueva, O.; Ibarra-Sanchez, M.J.; Esparza-Lopez, J.; et al. In vivo dual targeting of the oncogenic Ether-a-go-go-1 potassium channel by calcitriol and astemizole results in enhanced antineoplastic effects in breast tumors. BMC Cancer 2014, 14, 745. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Quiroz, J.; Camacho, J. Astemizole: An old anti-histamine as a new promising anti-cancer drug. Anticancer Agents Med. Chem. 2011, 11, 307–14. [Google Scholar] [CrossRef] [PubMed]
- Pillozzi, S.; Brizzi, M.F.; Balzi, M.; Crociani, O.; Cherubini, A.; Guasti, L.; Bartolozzi, B.; Becchetti, A.; Wanke, E.; Bernabei, P.A.; et al. HERG potassium channels are constitutively expressed in primary human acute myeloid leukemias and regulate cell proliferation of normal and leukemic hemopoietic progenitors. Leukemia 2002, 16, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Fiore, A.; Carraresi, L.; Morabito, A.; Polvani, S.; Fortunato, A.; Lastraioli, E.; Femia, A.P.; De Lorenzo, E.; Caderni, G.; Arcangeli, A. Characterization of hERG1 channel role in mouse colorectal carcinogenesis. Cancer Med. 2013, 2, 583–594. [Google Scholar] [PubMed]
- Kallergis, E.M.; Goudis, C.A.; Simantirakis, E.N.; Kochiadakis, G.E.; Vardas, P.E. Mechanisms, risk factors, and management of acquired long QT syndrome: A comprehensive review. Sci. World J. 2012. [Google Scholar] [CrossRef]
- Ganapathi, S.B.; Kester, M.; Elmslie, K.S. State-dependent block of HERG potassium channels by R-roscovitine: Implications for cancer therapy. Am. J. Physiol. Cell. Physiol. 2009, 296, C701–C710. [Google Scholar] [CrossRef] [PubMed]
- Nair, B.C.; Vallabhaneni, S.; Tekmal, R.R.; Vadlamudi, R.K. Roscovitine confers tumor suppressive effect on therapy-resistant breast tumor cells. Breast Cancer Res. 2011, 13, R80. [Google Scholar] [CrossRef] [PubMed]
- Benson, C.; White, J.; de Bono, J.; O’Donnell, A.; Raynaud, F.; Cruickshank, C.; McGrath, H.; Walton, M.; Workman, P.; Kaye, S.; et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br. J. Cancer 2007, 96, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Chouabe, C.; Drici, M.D.; Romey, G.; Barhanin, J.; Lazdunski, M. HERG and KvLQT1/IsK, the cardiac K+ channels involved in long QT syndromes, are targets for calcium channel blockers. Mol. Pharmacol. 1998, 54, 695–703. [Google Scholar] [PubMed]
- Millward, M.J.; Cantwell, B.M.; Munro, N.C.; Robinson, A.; Corris, P.A.; Harris, A.L. Oral verapamil with chemotherapy for advanced non-small cell lung cancer: A randomised study. Br. J. Cancer 1993, 67, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Mouhat, S.; Andreotti, N.; Jouirou, B.; Sabatier, J.M. Animal toxins acting on voltage-gated potassium channels. Curr. Pharm Des. 2008, 14, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Loret, E.; Bruhn, T.; Beress, L.; Lazdunski, M. APETx1, a new toxin from the sea anemone Anthopleura elegantissima, blocks voltage-gated human ether-a-go-go-related gene potassium channels. Mol. Pharmacol. 2003, 64, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Restano-Cassulini, R.; Korolkova, Y.V.; Diochot, S.; Gurrola, G.; Guasti, L.; Possani, L.D.; Lazdunski, M.; Grishin, E.V.; Arcangeli, A.; Wanke, E. Species diversity and peptide toxins blocking selectivity of ether-a-go-go-related gene subfamily K+ channels in the central nervous system. Mol. Pharmacol. 2006, 69, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Pennington, M.W.; Norton, R.S. Analogs of the sea anemone potassium channel blocker ShK for the treatment of autoimmune diseases. Inflamm. Allergy Drug Targets 2011, 10, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Kozminski, D.J.; Wold, L.A.; Modak, R.; Calhoun, J.D.; Isom, L.L.; Brackenbury, W.J. Therapeutic potential for phenytoin: targeting Na(v)1.5 sodium channels to reduce migration and invasion in metastatic breast cancer. Breast Cancer Res. Treat. 2012, 134, 603–615. [Google Scholar] [CrossRef]
- Abdul, M.; Hoosein, N. Inhibition by anticonvulsants of prostate-specific antigen and interleukin-6 secretion by human prostate cancer cells. Anticancer Res. 2001, 21, 2045–2048. [Google Scholar]
- Nelson, M.; Yang, M.; Dowle, A.A.; Thomas, J.R.; Brackenbury, W.J. The sodium channel-blocking antiepileptic drug phenytoin inhibits breast tumour growth and metastasis. Mol. Cancer 2015, 14, 13. [Google Scholar] [CrossRef] [PubMed]
- Fraser, S.P.; Ozerlat-Gunduz, I.; Brackenbury, W.J.; Fitzgerald, E.M.; Campbell, T.M.; Coombes, R.C.; Djamgoz, M.B. Philos. Regulation of voltage-gated sodium channel expression in cancer: Hormones, growth factors and auto-regulation. Trans. R Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef]
- Sikes, R.A.; Walls, A.M.; Brennen, W.N.; Anderson, J.D.; Choudhury-Mukherjee, I.; Schenck, H.A.; Brown, M.L. Therapeutic approaches targeting prostate cancer progression using novel voltage-gated ion channel blockers. Clin. Prostate Cancer 2003, 2, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Jacob, T.J.; Valverde, M.A.; Hardy, S.P.; Mintenig, G.M.; Sepulveda, F.V.; Gill, D.R.; Hyde, S.C.; Trezise, A.E.; Higgins, C.F. Tamoxifen blocks chloride channels. A possible mechanism for cataract formation. J. Clin. Invest. 1994, 94, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhao, J.; Qiao, W.; Chen, K. Recent advances in diagnosis and treatment of gliomas using chlorotoxin-based bioconjugates. Am. J. Nucl. Med. Mol. Imaging 2014, 4, 385–405. [Google Scholar] [PubMed]
- Deshane, J.; Garner, C.C.; Sontheimer, H. Chlorotoxin inhibits glioma cell invasion via matrix metalloproteinase-2. J. Biol. Chem. 2003, 278, 4135–4144. [Google Scholar] [CrossRef] [PubMed]
- The MICAD Research Team. Molecular Imaging and Contrast Agent Database (MICAD) [Internet]. In 131I-Chlorotoxin; National Center for Biotechnology Information (U.S.): Bethesda, MD, USA, 17 July 2007; updated 08 August 2007. [Google Scholar]
- Dardevet, L.; Rani, D.; Aziz, T.A.; Bazin, I.; Sabatier, J.M.; Fadl, M.; Brambilla, E.; de Waard, M. Chlorotoxin: A helpful natural scorpion peptide to diagnose glioma and fight tumor invasion. Toxins (Basel) 2015, 7, 1079–1101. [Google Scholar] [CrossRef]
- Zhao, J.; Wei, X.L.; Jia, Y.S.; Zheng, J.Q. Silencing of herg gene by shRNA inhibits SH-SY5Y cell growth in vitro and in vivo. Eur. J. Pharmacol. 2008, 579, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, H. An unexpected role for ion channels in brain tumor metastasis. Exp. Biol. Med. (Maywood) 2008, 233, 779–791. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, H.; Zuo, W.; Yang, L.; Zhang, H.; Ye, W.; Mao, J.; Chen, L.; Wang, L. Differential expression and roles of volume-activated chloride channels in control of growth of normal and cancerous nasopharyngeal epithelial cells. Biochem. Pharmacol. 2012, 83, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Chittajallu, R.; Chen, Y.; Wang, H.; Yuan, X.; Ghiani, C.A.; Heckman, T.; McBain, C.J.; Gallo, V. Regulation of Kv1 subunit expression in oligodendrocyte progenitor cells and their role in G1/S phase progression of the cell cycle. Proc. Natl. Acad. Sci. USA 2002, 99, 2350–2355. [Google Scholar] [CrossRef] [PubMed]
- Habela, C.W.; Olsen, M.L.; Sontheimer, H. ClC3 is a critical regulator of the cell cycle in normal and malignant glial cells. J. Neurosci. 2008, 28, 9205–9217. [Google Scholar] [CrossRef] [PubMed]
- Lansu, K.; Gentile, S. Potassium channel activation inhibits proliferation of breast cancer cells by activating a senescence program. Cell. Death Dis. 2013, 4, e652. [Google Scholar] [CrossRef] [PubMed]
- Perez-Neut, M.; Rao, V.R.; Gentile, S. hERG1/Kv11.1 activation stimulates transcription of p21waf/cip in breast cancer cells via a calcineurin-dependent mechanism. Oncotarget. 2015, in press. [Google Scholar]
- Liu, J.; Zhang, D.; Li, Y.; Chen, W.; Ruan, Z.; Deng, L.; Wang, L.; Tian, H.; Yiu, A.; Fan, C. Discovery of bufadienolides as a novel class of ClC-3 chloride channel activators with antitumor activities. J. Med. Chem. 2013, 56, 5734–5743. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rao, V.R.; Perez-Neut, M.; Kaja, S.; Gentile, S. Voltage-Gated Ion Channels in Cancer Cell Proliferation. Cancers 2015, 7, 849-875. https://doi.org/10.3390/cancers7020813
Rao VR, Perez-Neut M, Kaja S, Gentile S. Voltage-Gated Ion Channels in Cancer Cell Proliferation. Cancers. 2015; 7(2):849-875. https://doi.org/10.3390/cancers7020813
Chicago/Turabian StyleRao, Vidhya R., Mathew Perez-Neut, Simon Kaja, and Saverio Gentile. 2015. "Voltage-Gated Ion Channels in Cancer Cell Proliferation" Cancers 7, no. 2: 849-875. https://doi.org/10.3390/cancers7020813
APA StyleRao, V. R., Perez-Neut, M., Kaja, S., & Gentile, S. (2015). Voltage-Gated Ion Channels in Cancer Cell Proliferation. Cancers, 7(2), 849-875. https://doi.org/10.3390/cancers7020813