
p53 Acetylation: Regulation and Consequences

Abstract
:1. Introduction to p53 and Acetylation
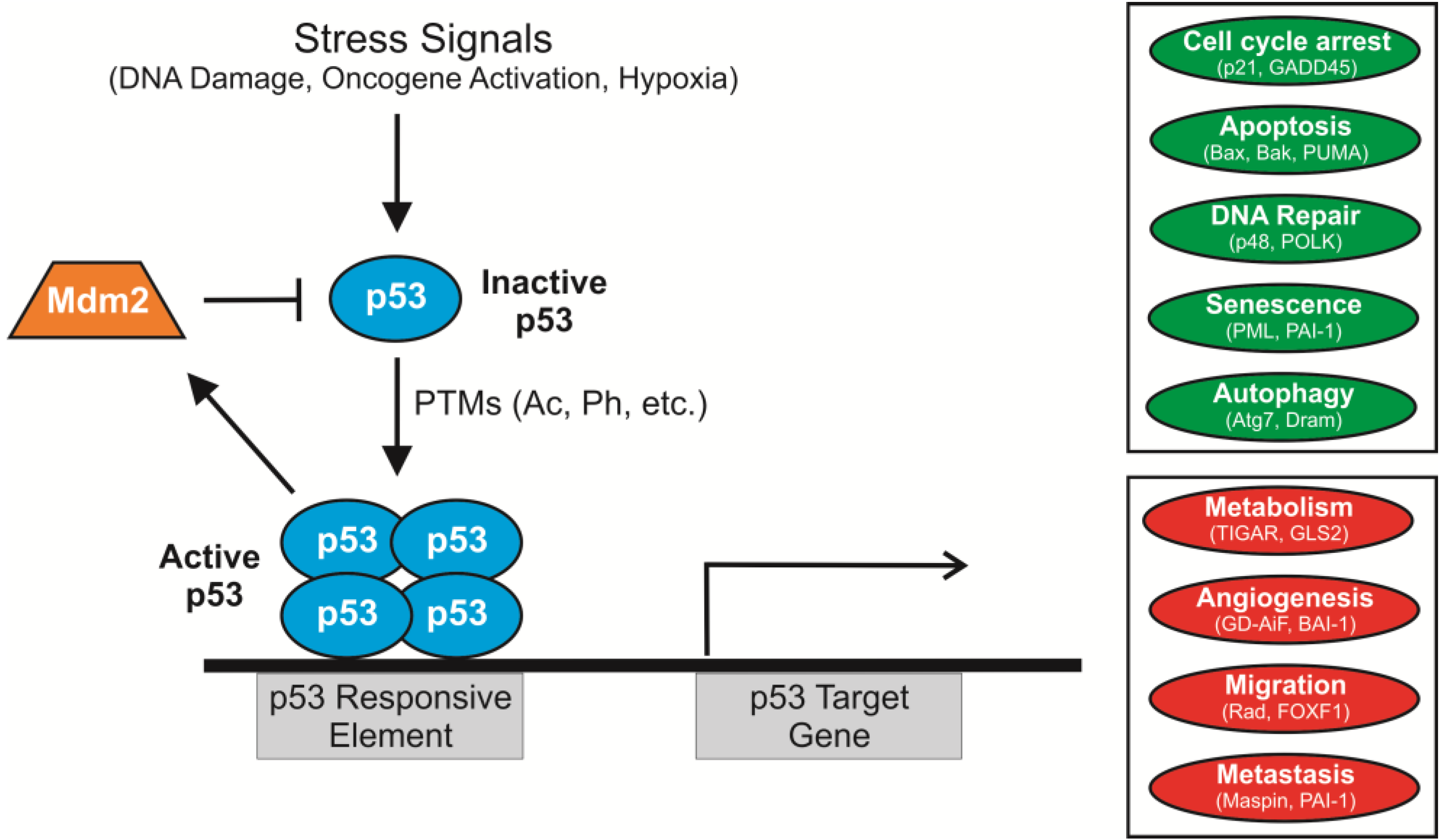
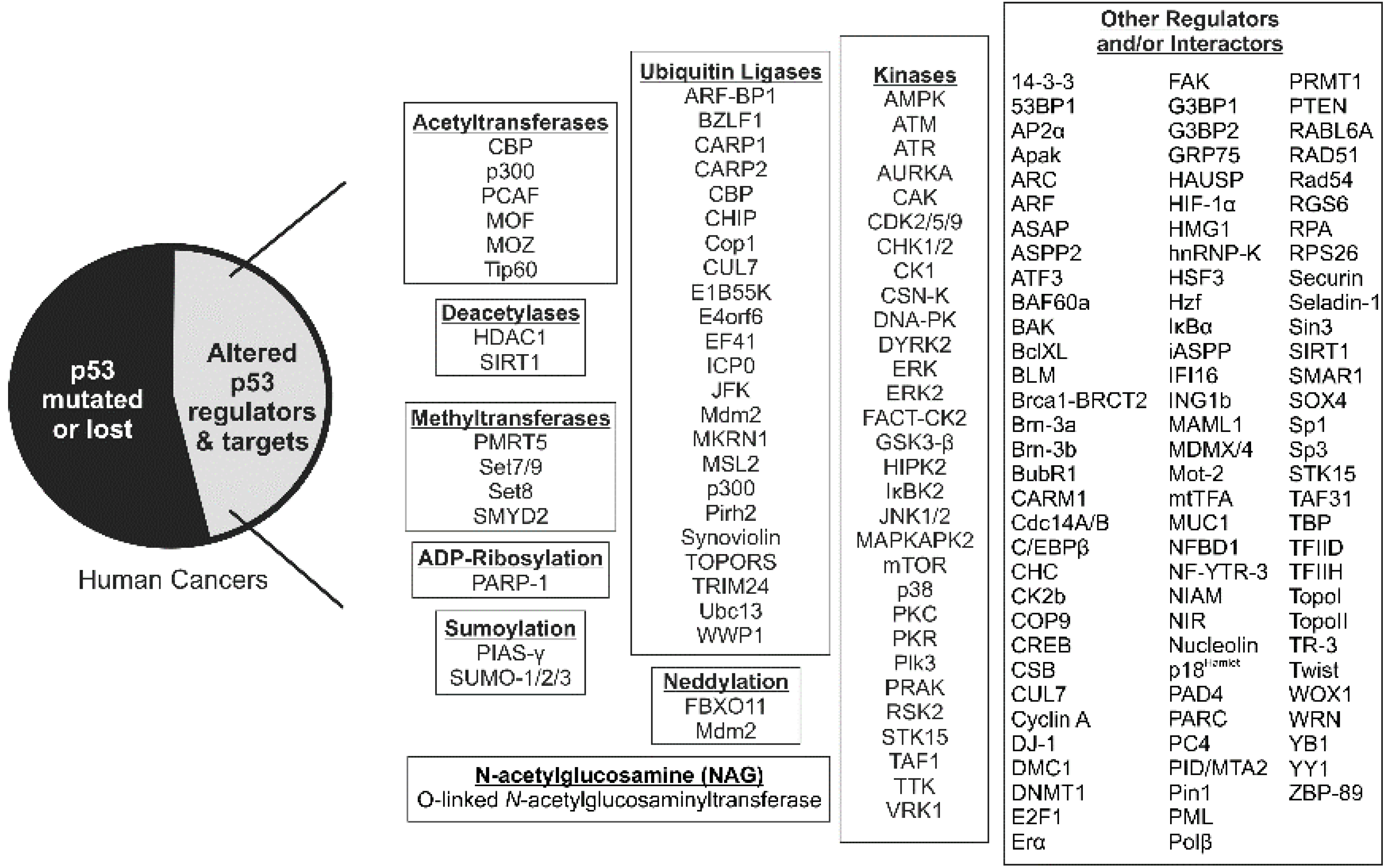
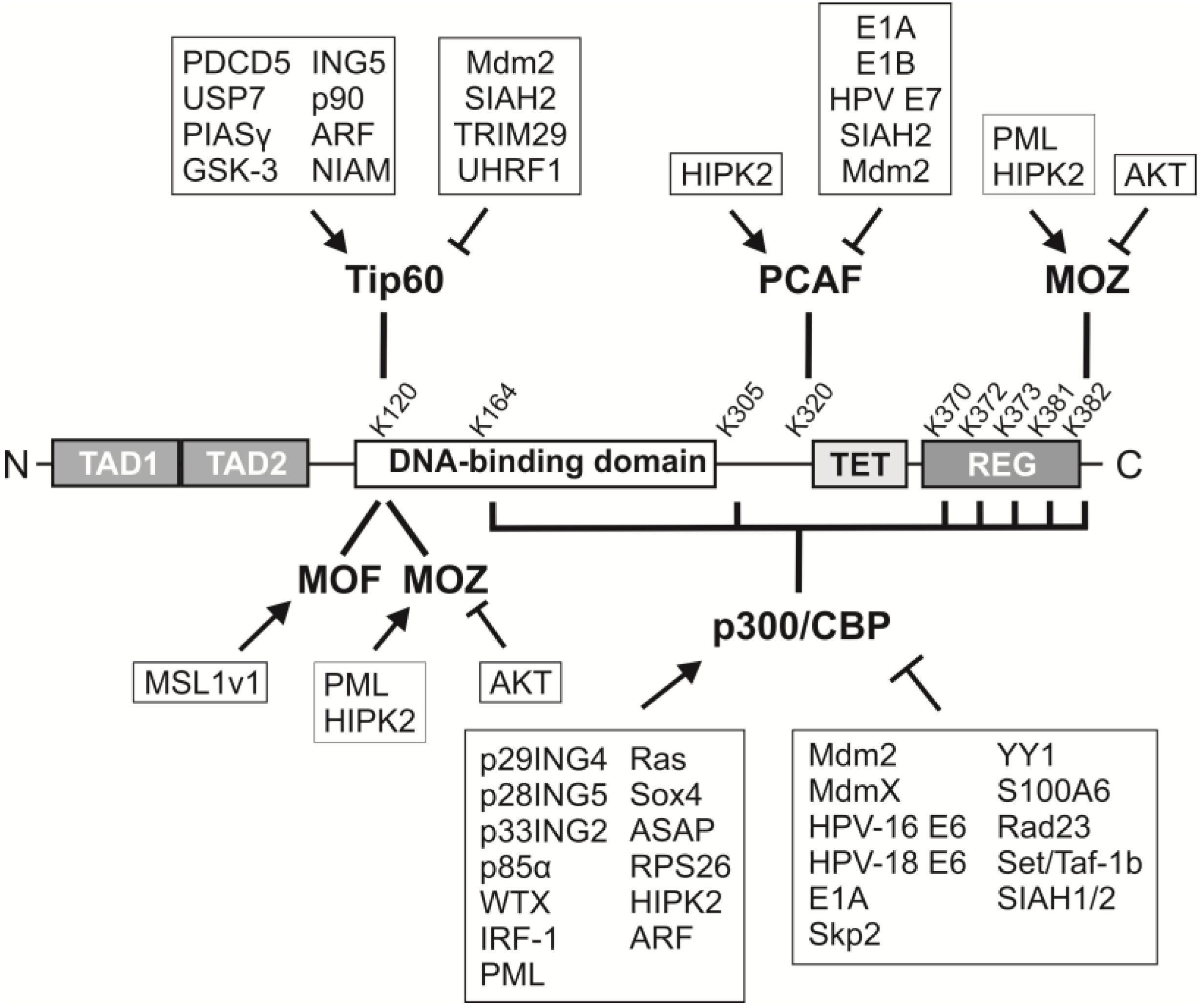
2. p53 Acetylation: Sites, Acetyltransferases and Consequences
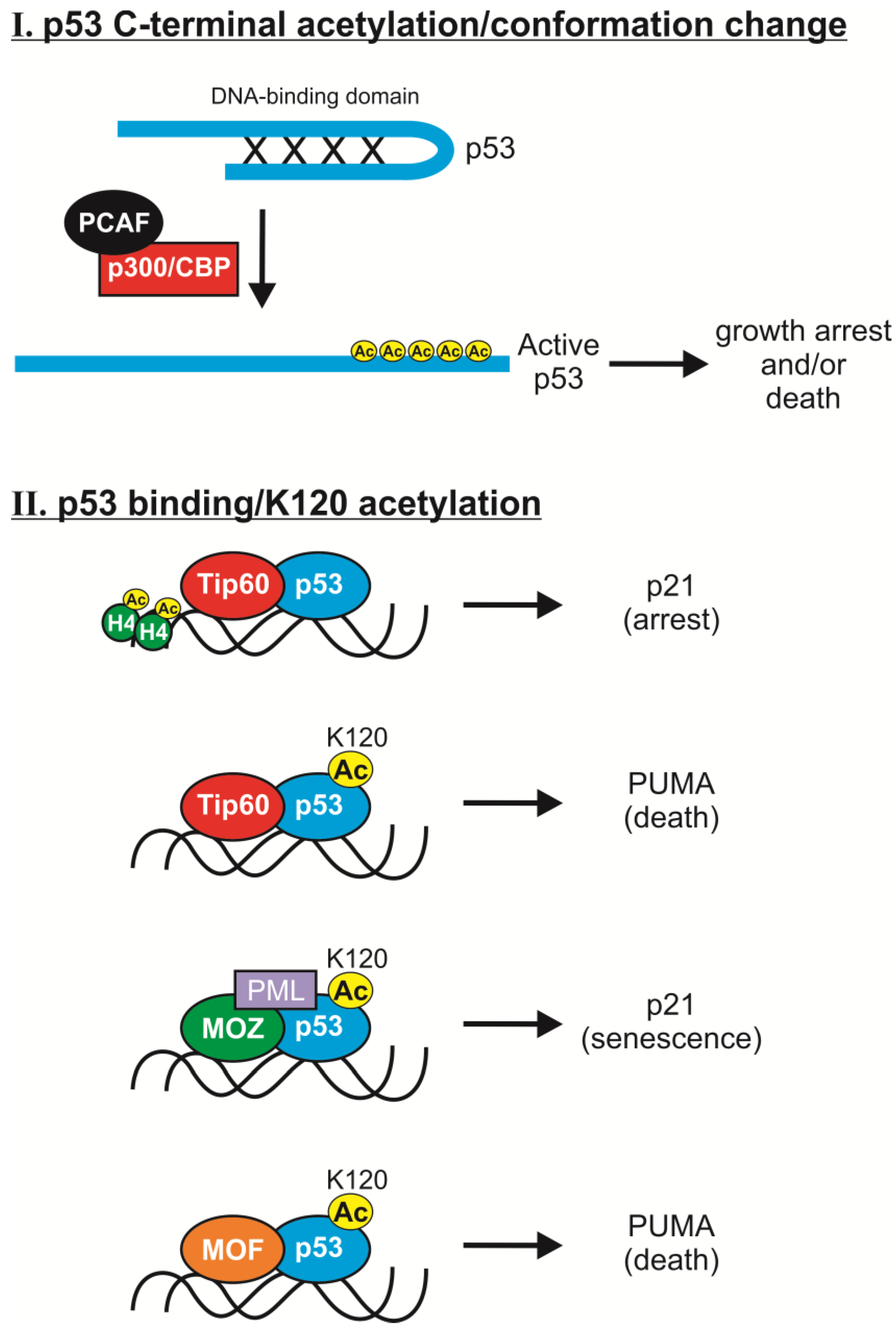
2.1. C-Terminal Acetylation
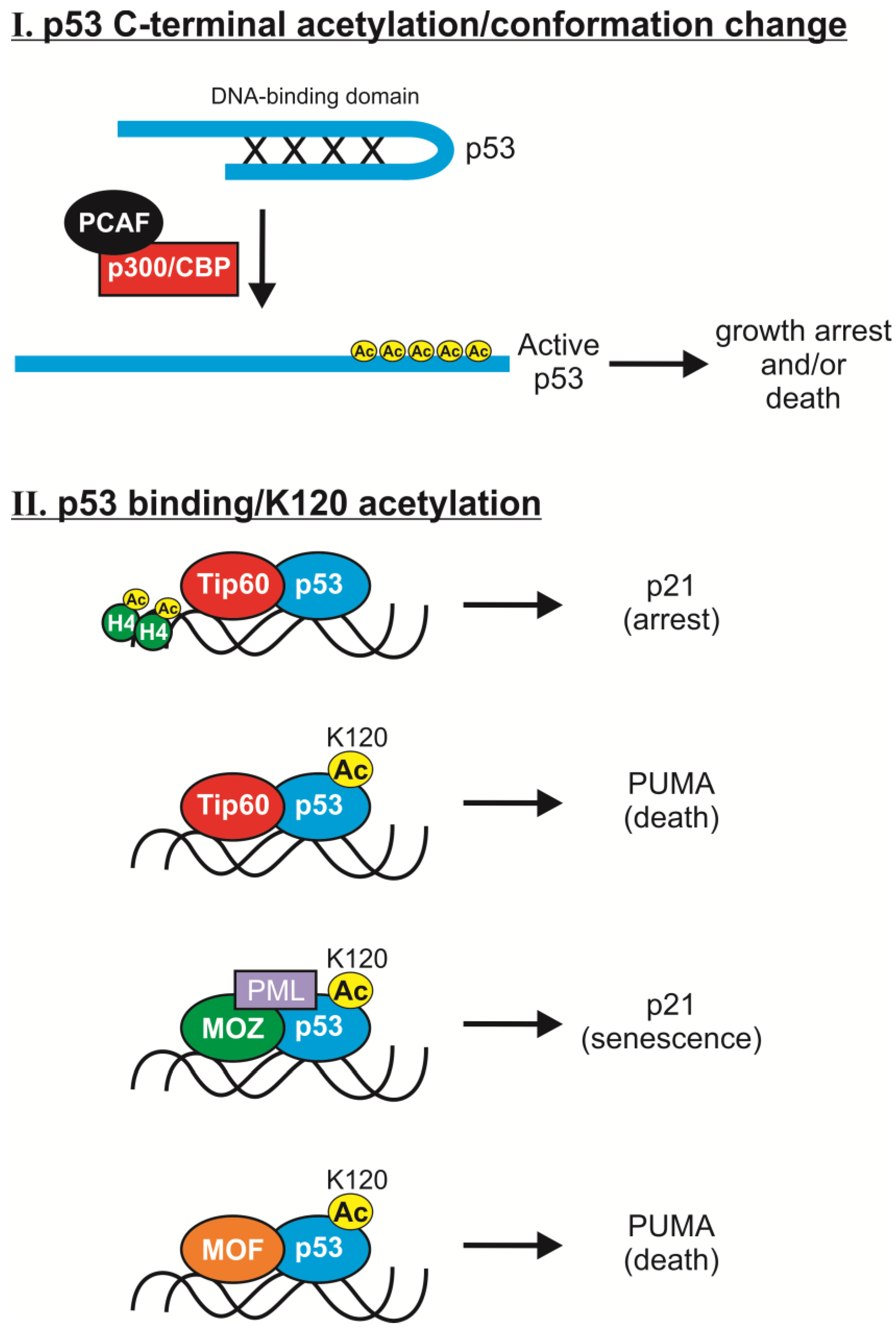
2.2. K120 Acetylation
3. p53 Deacetylation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sites of p53 Acetylation | p53 HATs Involved | Concurrent p53 Phosphorylation | Cell Stimulus | Key p53 Gene Targets | Molecular Phenotype | Biological Consequences |
|---|---|---|---|---|---|---|
| None | None | T377, S378 | Mitogens | Increased Mdm2,Pirh2 | p53 ubiquitylation & degradation | Cell survival & proliferation |
| C-terminal | P300/CBP, PCAF; binding by Tip60 w/o acetylation | N-terminal (including S15, T18, S20, S33, and S37) | DNA damage, other genotoxic stresses b | Increased p21, GADD45; Decreased Noxa, Pidd | Inhibition of Mdm2-p53 interaction; Inhibition of Cdk activity c | Cell cycle arrest (G1, G2 and/or S phase); DNA repair |
| K120, K320 and K382 | MOZ, PCAF, p300 | S15, S20 | DNA damage, oncogene activation (e.g., Ras) | Increased p21 | Localization of MOZ-p53 complexes in PML-NBs d | Cellular senescence |
| K120 and C-terminal | Tip60, MOF, p300/CBP, PCAF | S46 | DNA damage, other genotoxic stresses b | Increased Bax, Fas, Noxa and PUMA | p53 binding to low affinity, apoptotic gene promoters | Cellular apoptosis |
4. Crosstalk between p53 Ubiquitylation and Acetylation
5. Effects of p53 Phosphorylation on Acetylation
6. Acetylation vs. Other Post-Translational Modifications
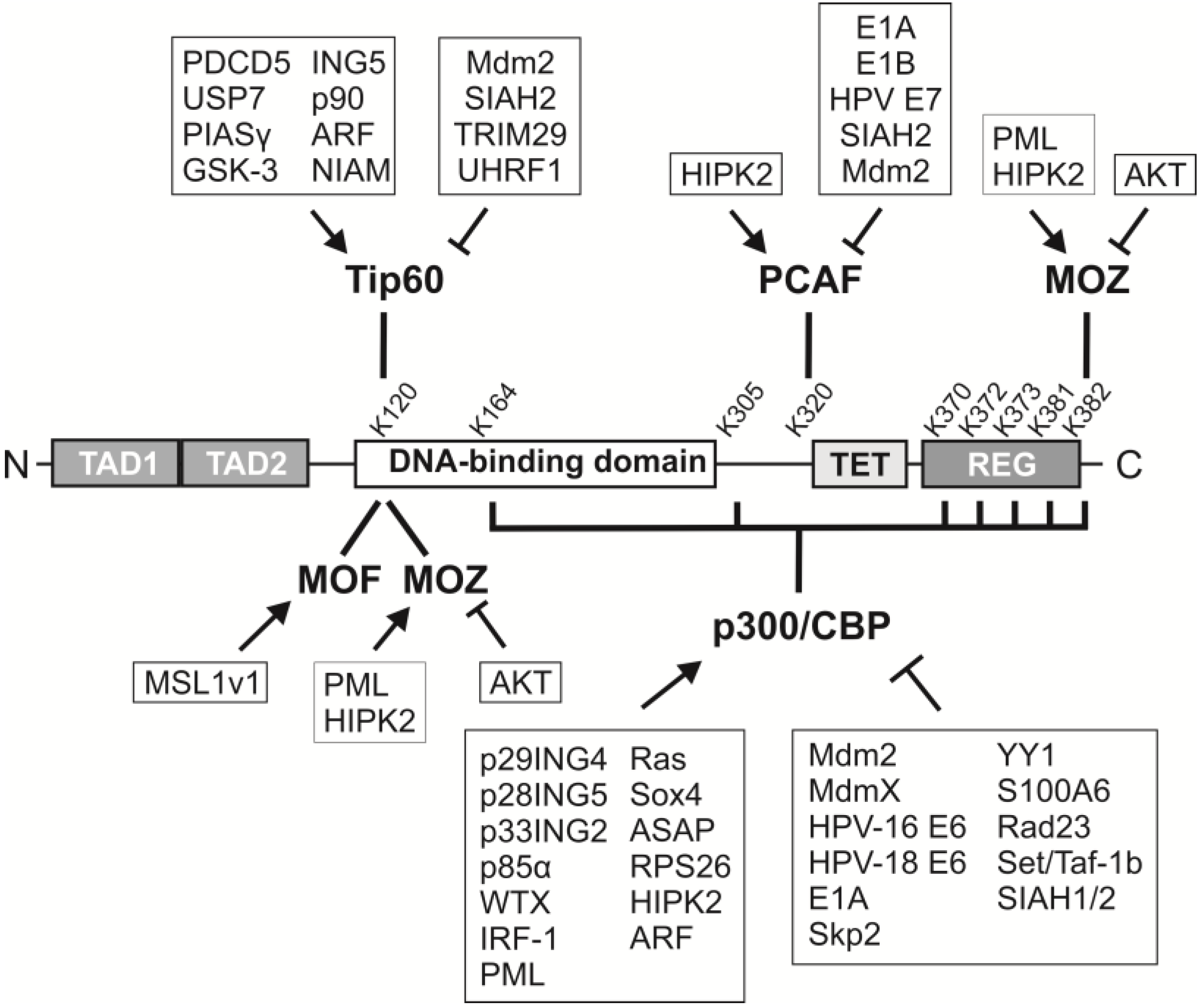
7. Regulators of Acetyltransferases
7.1. p300/CBP
7.2. PCAF
7.3. Tip60
7.4. MOF and MOZ
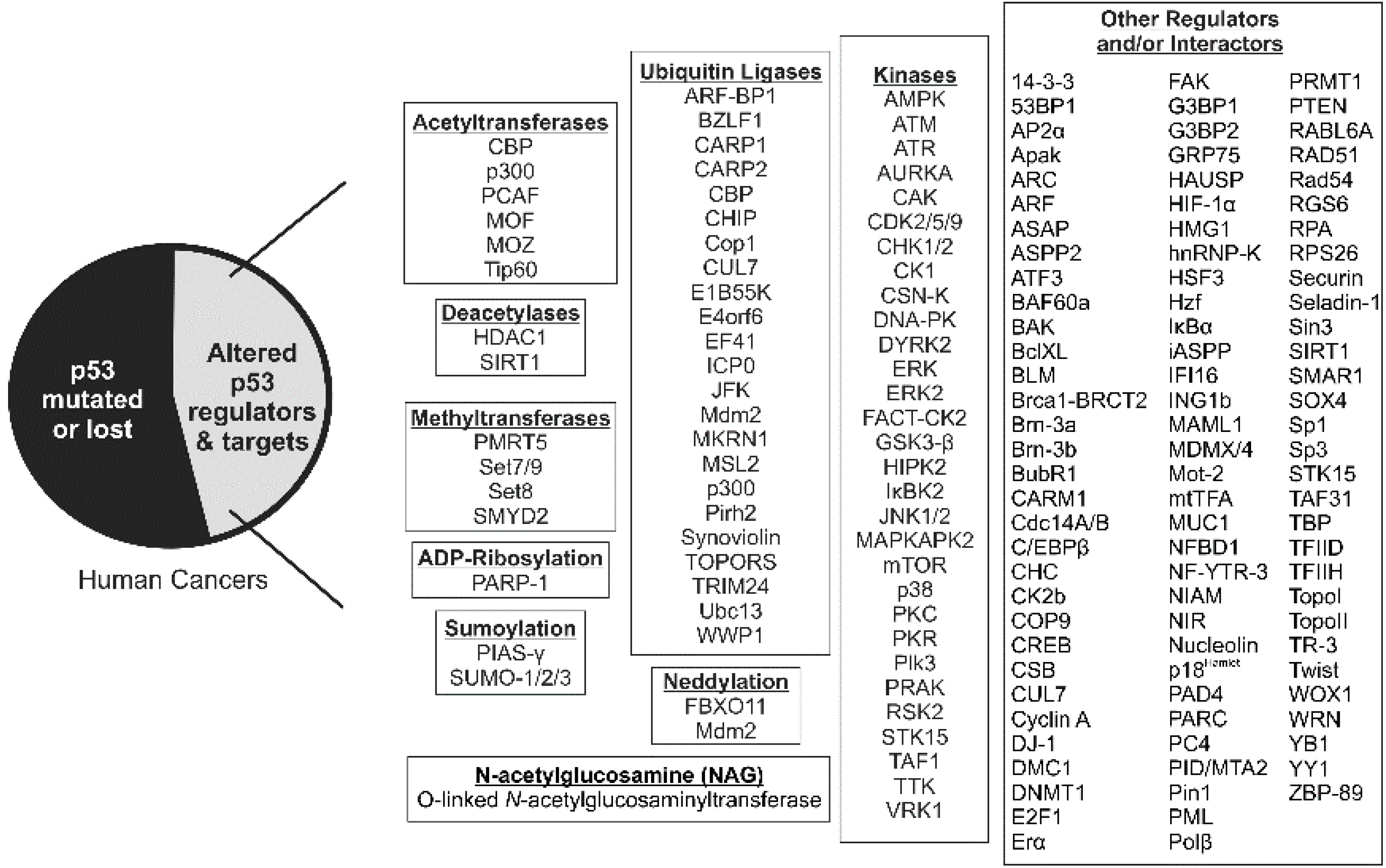
8. Role of p53 Acetyltransferases in Development and Cancer
9. p53 Acetylation and Tumor Suppression
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vousden, K.H.; Prives, C. Blinded by the light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Janus, F.; Albrechtsen, N.; Knippschild, U.; Wiesmuller, L.; Grosse, F.; Deppert, W. Different regulation of the p53 core domain activities 3'-to-5' exonuclease and sequence-specific DNA binding. Mol. Cell. Biol. 1999, 19, 2155–2168. [Google Scholar] [PubMed]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta-Bioenerg. 2009, 1787, 414–420. [Google Scholar] [CrossRef]
- Green, D.R.; Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009, 458, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Godefroy, N.; Lemaire, C.; Renaud, F.; Rincheval, V.; Perez, S.; Parvu-Ferecatu, I.; Mignotte, B.; Vayssiere, J.L. p53 Can promote mitochondria- and caspase-independent apoptosis. Cell Death Differ. 2004, 11, 785–787. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.J.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S. Regulation of p53 downstream genes. Sem. Cancer Biol. 1998, 8, 345–357. [Google Scholar] [CrossRef]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell. Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. p53, Guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Attardi, L.D. Engaging the p53 metabolic brake drives senescence. Cell Res. 2013, 23, 739–740. [Google Scholar] [CrossRef] [PubMed]
- Hock, A.K.; Vousden, K.H. Tumor suppression by p53: Fall of the triumvirate? Cell 2012, 149, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Wolff, S.; Speidel, D.; Deppert, W. Transcription-independent pro-apoptotic functions of p53. Curr. Opin. Cell Biol. 2005, 17, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Brady, C.A.; Johnson, T.M.; Lee, E.Y.; Park, E.J.; Scott, M.P.; Attardi, L.D. Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proc. Natl. Acad. Sci. USA 2011, 108, 17123–17128. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Broz, D.K.; Basak, S.; Park, E.J.; McLaughlin, M.E.; et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Remington, L.; Williams, B.O.; Schmitt, E.M.; Halachmi, S.; Bronson, R.T.; Weinberg, R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Garcia, P.B.; Attardi, L.D. Illuminating p53 function in cancer with genetically engineered mouse models. Sem. Cell Dev. Biol. 2014. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Sherr, C.J. Tumor suppression by Ink4a-Arf: Progress and puzzles. Curr. Opin. Genet. Dev. 2003, 13, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Achatz, M.I.W.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. Tp53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010. [Google Scholar] [CrossRef]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Nichols, K.E.; Malkin, D.; Garber, J.E.; Fraumeni, J.F.; Li, F.P. Germ-line p53 mutations predispose to a wide spectrum of early-onset cancers. Cancer Epidemiol. Biomark. Prev. 2001, 10, 83–87. [Google Scholar]
- Chen, F.; Wang, W.; El-Deiry, W.S. Current strategies to target p53 in cancer. Biochem. Pharmacol. 2010, 80, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P.; Cheok, C.F.; Lain, S. p53-Based cancer therapy. Cold Spring Harb. Perspect. Biol. 2010. [Google Scholar] [CrossRef]
- Cheok, C.F.; Verma, C.S.; Baselga, J.; Lane, D.P. Translating p53 into the clinic. Nat. Rev. Clin. Oncol. 2011, 8, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009. [Google Scholar] [CrossRef]
- Kruse, J.-P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef]
- Lee, J.T.; Gu, W. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 2010, 17, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Seong, M.W.; Jeon, Y.J.; Chung, C.H. Ubiquitin E3 ligases controlling p53 stability. Anim. Cells Syst. 2012, 16, 173–182. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Swygert, S.G.; Peterson, C.L. Chromatin dynamics: Interplay between remodeling enzymes and histone modifications. Biochim. Biophys. Acta 2014, 1839, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-Mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by Mdm2. EMBO J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Fontela, C.; Gonzalez, D.; Marcos-Villar, L.; Campagna, M.; Gallego, P.; Gonzalez-Santamaria, J.; Herranz, D.; Gu, W.; Serrano, M.; Aaronson, S.A.; et al. Acetylation is indispensable for p53 antiviral activity. Cell Cycle 2011, 10, 3701–3705. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.; Carbone, R.; Sebastiani, C.; Cioce, M.; Fagioli, M.; Saito, S.; Higashimoto, Y.; Appella, E.; Minucci, S.; Pandolfi, P.P.; et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 2000, 406, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.J.; Lin, T.X.; Uranishi, H.; Gu, W.; Xu, Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol. Cell. Biol. 2005, 25, 5389–5395. [Google Scholar] [CrossRef] [PubMed]
- Krummel, K.A.; Lee, C.J.; Toledo, F.; Wahl, G.M. The C-terminal lysines fine-tune p53 stress responses in a mouse model but are not required for stability control or transactivation. Proc. Natl. Acad. Sci. USA 2005, 102, 10188–10193. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Marouco, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A. Lysine-specific modifications of p53: A matter of life and death? Oncotarget 2013, 4, 1556–1571. [Google Scholar] [PubMed]
- Benkirane, M.; Sardet, C.; Coux, O. Lessons from interconnected ubiquitylation and acetylation of p53: Think metastable networks. Biochem. Soc. Trans. 2010, 38, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Avantaggiati, M.L.; Ogryzko, V.; Gardner, K.; Giordano, A.; Levine, A.S.; Kelly, K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell 1997, 89, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Lill, N.L.; Grossman, S.R.; Ginsberg, D.; DeCaprio, J.; Livingston, D.M. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997, 387, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.Y.; Li, M.Y.; Tang, Y.; Laszkowska, M.; Roeder, R.G.; Gu, W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl. Acad. Sci. USA 2004, 101, 2259–2264. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, K.; Herrera, J.E.; Saito, S.; Miki, T.; Bustin, M.; Vassilev, A.; Anderson, C.W.; Appella, E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 1998, 12, 2831–2841. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P.; Madhumalar, A.; Lee, A.P.; Tay, B.-H.; Verma, C.; Brenner, S.; Venkatesh, B. Conservation of all three p53 family members and Mdm2 and Mdm4 in the cartilaginous fish. Cell Cycle 2011, 10, 4272–4279. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Tsay, Y.G.; Tan, B.C.M.; Lo, W.Y.; Lee, S.C. Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J. Biol. Chem. 2003, 278, 25568–25576. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Scolnick, D.M.; Trievel, R.C.; Zhang, H.B.; Marmorstein, R.; Halazonetis, T.D.; Berger, S.L. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 1999, 19, 1202–1209. [Google Scholar] [PubMed]
- Prives, C.; Manley, J.L. Why is p53 acetylated? Cell 2001, 107, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Snowden, A.W.; Perkins, N.D. Cell cycle regulation of the transcriptional coactivators p300 and CREB binding protein. Biochem. Pharmacol. 1998, 55, 1947–1954. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.M.; la Thangue, N.B. p300/CBP Proteins: Hats for transcriptional bridges and scaffolds. J. Cell Sci. 2001, 114, 2363–2373. [Google Scholar] [PubMed]
- Arany, Z.; Sellers, W.R.; Livingston, D.M.; Eckner, R. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell 1994, 77, 799–800. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. The CBP co-activator is a histone acetyltransferase. Nature 1996, 384, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef]
- Buschmann, T.; Adler, V.; Matusevich, E.; Fuchs, S.Y.; Ronai, Z. p53 phosphorylation and association with Murine double minute 2, c-Jun NH2-terminal kinase, p14(ARF), and p300/CBP during the cell cycle and after exposure to ultraviolet irradiation. Cancer Res. 2000, 60, 896–900. [Google Scholar] [PubMed]
- Wadgaonkar, R.; Phelps, K.M.; Haque, Z.; Williams, A.J.; Silverman, E.S.; Collins, T. CREB-binding protein is a nuclear integrator of nuclear factor-kappa B and p53 signaling. J. Biol. Chem. 1999, 274, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Scolnick, D.M.; Chehab, N.H.; Stavridi, E.S.; Lien, M.C.; Caruso, L.; Moran, E.; Berger, S.L.; Halazonetis, T.D. CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res. 1997, 57, 3693–3696. [Google Scholar] [PubMed]
- Grossman, S.R.; Perez, M.; Kung, A.L.; Joseph, M.; Mansur, C.; Ziao, Z.X.; Kumar, S.; Howley, P.M.; Livingston, D.M. p300/Mdm2 complexes participate in Mdm2-mediated p53 degradation. Mol. Cell 1998, 2, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Shi, X.-L.; Roeder, R.G. Synergistic activation of transcription by CBP and p53. Nature 1997, 387, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.G.; Chin, S.F.; Ozdag, H.; Daigo, Y.; Hu, D.E.; Cariati, M.; Brindle, K.; Aparicio, S.; Caldas, C. p300 Regulates p53-dependent apoptosis after DNA damage in colorectal cancer cells by modulation of PUMA/p21 levels. Proc. Natl. Acad. Sci. USA 2004, 101, 7386–7391. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.M.; Huang, Y.Y.; Ishiko, T.; Nakada, S.; Utsugisawa, T.; Shioya, H.; Utsugisawa, Y.; Yokoyama, K.; Weichselbaum, R.; Shi, Y.; et al. Role for p300 in stabilization of p53 in the response to DNA damage. J. Biol. Chem. 1999, 274, 1883–1886. [Google Scholar] [CrossRef] [PubMed]
- Romanov, V.S.; Abramova, M.V.; Svetlikova, S.B.; Bykova, T.V.; Zubova, S.G.; Aksenov, N.D.; Fornace, A.J., Jr.; Pospelova, T.V.; Pospelov, V.A. p21(waf1) Is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle 2010, 9, 3945–3955. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G(1)-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.H.; Thomas, M.C.; Zambetti, G.P.; Brindle, P.K. Double null cells reveal that CBP and p300 are dispensable for p53 targets p21 and Mdm2 but variably required for target genes of other signaling pathways. Cell Cycle 2011, 10, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Brochier, C.; Dennis, G.; Rivieccio, M.A.; McLaughlin, K.; Coppola, G.; Ratan, R.R.; Langley, B. Specific acetylation of p53 by HDAC inhibition prevents DNA damage-induced apoptosis in neurons. J. Neurosci. 2013, 33, 8621–8632. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Ogryzko, V.V.; Nishikawa, J.; Howard, B.H.; Nakatani, Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 1996, 382, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Knights, C.D.; Catania, J.; di Giovanni, S.; Muratoglu, S.; Perez, R.; Swartzbeck, A.; Quong, A.A.; Zhang, X.J.; Beerman, T.; Pestell, R.G.; et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J. Cell Biol. 2006, 173, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.; Wu, Z.; Mazur, S.J.; Borges, H.; Rossi, M.; Lin, T.; Wang, J.Y.J.; Anderson, C.W.; Appella, E.; Xu, Y. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol. Cell. Biol. 2006, 26, 6859–6869. [Google Scholar] [CrossRef] [PubMed]
- Love, I.M.; Sekaric, P.; Shi, D.; Grossman, S.R.; Androphy, E.J. The histone acetyltransferase PCAF regulates p21 transcription through stress-induced acetylation of histone H3. Cell Cycle 2012, 11, 2458–2466. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Luo, J.; Zhang, W.; Gu, W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Marmorstein, R.; Lane, W.S.; McMahon, S.B. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 2006, 24, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Mellert, H.; Sykes, S.M.; Murphy, M.E.; McMahon, S.B. The ARF/oncogene pathway activates p53 acetylation within the DNA binding domain. Cell Cycle 2007, 6, 1304–1306. [Google Scholar] [CrossRef] [PubMed]
- Deissler, H.; Kafka, A.; Schuster, E.; Sauer, G.; Kreienberg, R.; Zeillinger, R. Spectrum of p53 mutations in biopsies from breast cancer patients selected for preoperative chemotherapy analysed by the functional yeast assay to predict therapeutic response. Oncol. Rep. 2004, 11, 1281–1286. [Google Scholar] [PubMed]
- Hashimoto, T.; Tokuchi, Y.; Hayashi, M.; Kobayashi, Y.; Nishida, K.; Hayashi, S.; Ishikawa, Y.; Tsuchiya, S.; Nakagawa, K.; Hayashi, J.; et al. p53 Null mutations undetected by immunohistochemical staining predict a poor outcome with early-stage non-small cell lung carcinomas. Cancer Res. 1999, 59, 5572–5577. [Google Scholar] [PubMed]
- Hayes, V.M.; Dirven, C.M.F.; Dam, A.; Verlind, E.; Molenaar, W.M.; Mooij, J.J.A.; Hofstra, R.M.W.; Buys, C. High frequency of TP53 mutations in juvenile pilocytic astrocytomas indicates role of TP53 in the development of these tumors. Brain Pathol. 1999, 9, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Leitao, M.M.; Soslow, R.A.; Baergen, R.N.; Olvera, N.; Arroyo, C.; Boyd, J. Mutation and expression of the TP53 gene in early stage epithelial ovarian carcinoma. Gynecol. Oncol. 2004, 93, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Meyers, F.J.; Chi, S.G.; Fishman, J.R.; White, R.W.D.; Gumerlock, P.H. p53 Mutations in benign prostatic hyperplasia. J. Natl. Cancer Inst. 1993, 85, 1856–1858. [Google Scholar] [CrossRef] [PubMed]
- Rokudai, S.; Laptenko, O.; Arnal, S.M.; Taya, Y.; Kitabayashi, I.; Prives, C. MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc. Natl. Acad. Sci. USA 2013, 110, 3895–3900. [Google Scholar] [CrossRef] [PubMed]
- Squatrito, M.; Gorrini, C.; Amati, B. Tip60 in DNA damage response and growth control: Many tricks in one hat. Trends Cell Biol. 2006, 16, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.; Hijmans, E.M.; Mullenders, J.; Brummelkamp, T.R.; Velds, A.; Heimerikx, M.; Kerkhoven, R.M.; Madiredjo, M.; Nijkamp, W.; Weigelt, B.; et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 2004, 428, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Legube, G.; Linares, L.K.; Tyteca, S.; Caron, C.; Scheffner, M.; Chevillard-Briet, M.; Trouche, D. Role of the histone acetyl transferase Tip60 in the p53 pathway. J. Biol. Chem. 2004, 279, 44825–44833. [Google Scholar] [CrossRef] [PubMed]
- Dohmesen, C.; Koeppel, M.; Dobbelstein, M. Specific inhibition of Mdm2-mediated neddylation by Tip60. Cell Cycle 2008, 7, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Legube, G.; Linares, L.K.; Lemercier, C.; Scheffner, M.; Khochbin, S.; Trouche, D. Tip60 is targeted to proteasome-mediated degradation by Mdm2 and accumulates after UV irradiation. EMBO J. 2002, 21, 1704–1712. [Google Scholar] [CrossRef] [PubMed]
- Xirodimas, D.P.; Saville, M.K.; Bourdon, J.C.; Hay, R.T.; Lane, D.P. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 2004, 118, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wang, J.; Wang, J.; Wang, R.; Liu, Z.; Yu, Y.; Lu, H. ING5 is a Tip60 cofactor that acetylates p53 in response to DNA damage. Cancer Res. 2013, 73, 3749–3760. [Google Scholar] [CrossRef] [PubMed]
- Arbely, E.; Natan, E.; Brandt, T.; Allen, M.D.; Veprintsev, D.B.; Robinson, C.V.; Chin, J.W.; Joerger, A.C.; Fersht, A.R. Acetylation of lysine 120 of p53 endows DNA-binding specificity at effective physiological salt concentration. Proc. Natl. Acad. Sci. USA 2011, 108, 8251–8256. [Google Scholar] [CrossRef]
- Li, X.; Wu, L.; Corsa, C.A.S.; Kunkel, S.; Dou, Y. Two mammalian mof complexes regulate transcription activation by distinct mechanisms. Mol. Cell 2009, 36, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker, A.; HilfikerKleiner, D.; Pannuti, A.; Lucchesi, J.C. MOF, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. EMBO J. 1997, 16, 2054–2060. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.; Becker, P.B. Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol. Cell 2000, 5, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Rea, S.; Richter, K.; Vilar, A.; Lichter, P.; Imhof, A.; Akhtar, A. HMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol. Cell. Biol. 2005, 25, 6798–6810. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Pannuti, A.; Gu, W.G.; Steurnagel, A.; Cook, R.G.; Allis, C.D.; Lucchesi, J.C. The Drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol. Cell. Biol. 2000, 20, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.R.; Cayrou, C.; Huang, R.; Lane, W.S.; Cote, J.; Lucchesi, J.C. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Mol. Cell. Biol. 2005, 25, 9175–9188. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.L.; Milne, T.A.; Tackett, A.J.; Smith, E.R.; Fukuda, A.; Wysocka, J.; Allis, C.D.; Chait, B.T.; Hess, J.L.; Roeder, R.G. Physical association and coordinate function of the H3K4 methyltransferase MLL1 and the H4K16 acetyltransferase MOF. Cell 2005, 121, 873–885. [Google Scholar] [CrossRef] [PubMed]
- Katsumoto, T.; Yoshida, N.; Kitabayashi, I. Roles of the histone acetyltransferase monocytic leukemia zinc finger protein in normal and malignant hematopoiesis. Cancer Sci. 2008, 99, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Rokudai, S.; Aikawa, Y.; Tagata, Y.; Tsuchida, N.; Taya, Y.; Kitabayashi, I. Monocytic leukemia zinc finger (MOZ) interacts with p53 to induce p21 expression and cell-cycle arrest. J. Biol. Chem. 2009, 284, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.Y.; Su, F.; Chen, D.L.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.Y.; Nikolaev, A.Y.; Imai, S.; Chen, D.L.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by SIR2 alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Eagon, E.N.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) Functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Kawaguchi, Y.; Lai, C.H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.P. Mdm2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002, 21, 6236–6245. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Luo, J.Y.; Brooks, C.L.; Gu, W. Acetylation of p53 inhibits its ubiquitination by Mdm2. J. Biol. Chem. 2002, 277, 50607–50611. [Google Scholar] [CrossRef] [PubMed]
- Juan, L.J.; Shia, W.J.; Chen, M.H.; Yang, W.M.; Seto, E.; Lin, Y.S.; Wu, C.W. Histone deacetylases specifically down-regulate p53-dependent gene activation. J. Biol. Chem. 2000, 275, 20436–20443. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Liu, H.-D.; Huang, Q.; Liang, H.-X.; Ding, Z.-H.; Liao, Z.-J.; Huang, G. HDAC6 promotes hepatocellular carcinoma progression by inhibiting p53 transcriptional activity. FEBS Lett. 2013, 587, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Langley, E.; Pearson, M.; Faretta, M.; Bauer, U.M.; Frye, R.A.; Minucci, S.; Pelicci, P.G.; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002, 21, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Kruse, J.-P.; Tang, Y.; Jung, S.Y.; Qin, J.; Gu, W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature 2008, 451, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Chen, J.; Lou, Z. DBC1 is a negative regulator of SIRT1. Nature 2008, 451, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-J.; Kho, J.-H.; Kang, M.-R.; Um, S.-J. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol. Cell 2007, 28, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Lain, S.; Hollick, J.J.; Campbell, J.; Staples, O.D.; Higgins, M.; Aoubala, M.; McCarthy, A.; Appleyard, V.; Murray, K.E.; Baker, L.; et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008, 13, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-H.; Sengupta, K.; Li, C.; Kim, H.-S.; Cao, L.; Xiao, C.; Kim, S.; Xu, X.; Zheng, Y.; Chilton, B.; et al. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 2008, 14, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-L.; Mostoslavsky, R.; Saito, S.I.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in SIR2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef] [PubMed]
- Chua, K.F.; Mostoslavsky, R.; Lombard, D.B.; Pang, W.W.; Saito, S.; Franco, S.; Kaushal, D.; Cheng, H.L.; Fischer, M.R.; Stokes, N.; et al. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell MeTable 2005, 2, 67–76. [Google Scholar] [CrossRef]
- Fan, W.; Luo, J. SIRT1 regulates uv-induced DNA repair through deacetylating XPA. Mol. Cell 2010, 39, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Zhang, X.; Sengupta, N.; Lane, W.S.; Seto, E. SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol. Cell 2007, 27, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Casta, A.; Wang, R.; Lozada, E.; Fan, W.; Kane, S.; Ge, Q.; Gu, W.; Orren, D.; Luo, J. Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. J. Biol. Chem. 2008, 283, 7590–7598. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-Mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [PubMed]
- Picksley, S.M.; Lane, D.P. The p53-mdm2 autoregulatory feedback loop: A paradigm for the regulation of growth-control by p53. Bioessays 1993, 15, 689–690. [Google Scholar] [CrossRef] [PubMed]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein Mdm2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; Jochemsen, A.G. MdmX as an essential regulator of p53 activity. Biochem. Biophys. Res. Comm. 2005, 331, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar] [CrossRef] [PubMed]
- Montes de Oca Luna, R.; Wagner, D.S.; Lozano, G. Rescue of early embryonic lethality in Mdm2-deficient mice by deletion of p53. Nature 1995, 378, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Parant, J.; Chavez-Reyes, A.; Little, N.A.; Yan, W.; Reinke, V.; Jochemsen, A.G.; Lozano, G. Rescue of embryonic lethality in Mdm4-null mice by loss of TRP53 suggests a nonoverlapping pathway with Mdm2 to regulate p53. Nat. Genet. 2001, 29, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Barboza, J.A.; Iwakuma, T.; Terzian, T.; El-Naggar, A.K.; Lozano, G. Mdm2 and Mdm4 loss regulates distinct p53 activities. Mol. Cancer Res. 2008, 6, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Barton, M.C. Making sense of ubiquitin ligases that regulate p53. Cancer Biol. Ther. 2010, 10, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Li, M.Y.; Brooks, C.L.; Wu-Baer, F.; Chen, D.L.; Baer, R.; Gu, W. Mono-versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Lohrum, M.A.E.; Woods, D.B.; Ludwig, R.L.; Balint, E.; Vousden, K.H. C-Terminal ubiquitination of p53 contributes to nuclear export. Mol. Cell. Biol. 2001, 21, 8521–8532. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Li, M.; Gu, W. Mechanistic studies of Mdm2-mediated ubiquitination in p53 regulation. J. Biol. Chem. 2007, 282, 22804–22815. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Erster, S.; Zaika, A.; Petrenko, O.; Chittenden, T.; Pancoska, P.; Moll, U.M. p53 Has a direct apoptogenic role at the mitochondria. Mol. Cell 2003, 11, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.; Schuler, M.; Green, D.R. Direct activation of bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Kobet, E.; Zeng, X.Y.; Zhu, Y.; Keller, D.; Lu, H. Mdm2 inhibits p300-mediated p53 acetylation and activation by forming a ternary complex with the two proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 12547–12552. [Google Scholar] [CrossRef] [PubMed]
- Wadgaonkar, R.; Collins, T. Murine double minute (Mdm2) blocks p53-coactivator interaction, a new mechanism for inhibition of p53-dependent gene expression. J. Biol. Chem. 1999, 274, 13760–13767. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, J.C.; Lee, C.W.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and Hdm2. Proc. Natl. Acad. Sci. USA 2009, 106, 6591–6596. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.T.; Zeng, S.X.; Dai, M.S.; Yang, X.J.; Lu, H. Mdm2 inhibits PCAF (p300/CREB-binding protein-associated factor)-mediated p53 acetylation. J. Biol. Chem. 2002, 277, 30838–30843. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Roth, J.A.; Mukhopadhyay, T. Multiple lysine mutations in the C-terminal domain of p53 interfere with Mdm2-dependent protein degradation and ubiquitination. Mol. Cell. Biol. 2000, 20, 9391–9398. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.S.; Desterro, J.M.P.; Lain, S.; Lane, D.P.; Hay, R.T. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol. Cell. Biol. 2000, 20, 8458–8467. [Google Scholar] [CrossRef] [PubMed]
- Le Cam, L.; Linares, L.K.; Paul, C.; Julien, E.; Lacroix, M.; Hatchi, E.; Triboulet, R.; Bossis, G.; Shmueli, A.; Rodriguez, M.S.; et al. E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell 2006, 127, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Pop, M.S.; Kulikov, R.; Love, I.M.; Kung, A.; Grossman, S.R. CBP and p300 are cytoplasmic E4 polyubiquitin ligases for p53. Proc. Natl. Acad. Sci. USA 2009, 106, 16275–16280. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.R.; Deato, M.E.; Brignone, C.; Chan, H.M.; Kung, A.L.; Tagami, H.; Nakatani, Y.; Livingston, D.M. Polyubiquitination of p53 by a ubiquitin ligase activity of p300. Science 2003, 300, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Linares, L.K.; Kiernan, R.; Triboulet, R.; Chable-Bessia, C.; Latreille, D.; Cuvier, O.; Lacroix, M.; Le Cam, L.; Coux, O.; Benkirane, M. Intrinsic ubiquitination activity of PCAF controls the stability of the oncoprotein Mdm2. Nat. Cell Biol. 2007, 9, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Banin, S.; Moyal, L.; Shieh, S.Y.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef] [PubMed]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Leesmiller, S.P.; Sakaguchi, K.; Ullrich, S.J.; Appella, E.; Anderson, C.W. Human DNA-activated protein-kinase phosphorylates serine-15 and serine-37 in the amino-terminal transactivation domain of human p53. Mol. Cell. Biol. 1992, 12, 5041–5049. [Google Scholar] [PubMed]
- Shieh, S.Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by Mdm2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Canman, C.E.; Taya, Y.; Sakaguchi, K.; Appella, E.; Kastan, M.B. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997, 11, 3471–3481. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, R.S.; Brumbaugh, K.M.; Williams, J.M.; Sarkaria, J.N.; Cliby, W.A.; Shieh, S.Y.; Taya, Y.; Prives, C.; Abraham, R.T. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999, 13, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.K.; Keating, K.E.; Kozlov, S.; Scott, S.; Gatei, M.; Hobson, K.; Taya, Y.; Gabrielli, B.; Chan, D.; Lees-Miller, S.P.; et al. ATM associates with and phosphorylates p53: Mapping the region of interaction. Nat. Genet. 1998, 20, 398–400. [Google Scholar] [CrossRef] [PubMed]
- Chehab, N.H.; Malikzay, A.; Appel, M.; Halazonetis, T.D. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev. 2000, 14, 278–288. [Google Scholar] [PubMed]
- Shieh, S.Y.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000, 14, 289–300. [Google Scholar] [PubMed]
- Dumaz, N.; Milne, D.M.; Jardine, L.J.; Meek, D.W. Critical roles for the serine 20, but not the serine 15, phosphorylation site and for the polyproline domain in regulating p53 turnover. Biochem. J. 2001, 359, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Unger, T.; Juven-Gershon, T.; Moallem, E.; Berger, M.; Vogt Sionov, R.; Lozano, G.; Oren, M.; Haupt, Y. Critical role for Ser20 of human p53 in the negative regulation of p53 by Mdm2. EMBO J. 1999, 18, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- D’Orazi, G.; Cecchinelli, B.; Bruno, T.; Manni, I.; Higashimoto, Y.; Saito, S.; Gostissa, M.; Coen, S.; Marchetti, A.; del Sal, G.; et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 2002, 4, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, T.G.; Moller, A.; Sirma, H.; Zentgraf, H.; Taya, Y.; Droge, W.; Will, H.; Schmitz, M.L. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 2002, 4, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ko, L.J.; Shieh, S.Y.; Chen, X.; Jayaraman, L.; Tamai, K.; Taya, Y.; Prives, C.; Pan, Z.Q. p53 Is phosphorylated by CDK7-cyclin H in a p36MAT1-dependent manner. Mol. Cell. Biol. 1997, 17, 7220–7229. [Google Scholar] [PubMed]
- Teufel, D.P.; Bycroft, M.; Fersht, A.R. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009, 28, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Ferreon, J.C.; Ferreon, A.C.M.; Arai, M.; Wright, P.E. Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc. Natl. Acad. Sci. USA 2010, 107, 19290–19295. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, L.M.M.; Yamaguchi, H.; Hayashi, R.; Cherry, S.; Tropea, J.E.; Miller, M.; Wlodawer, A.; Appella, E.; Mazur, S.J. Two distinct motifs within the p53 transactivation domain bind to the Taz2 domain of p300 and are differentially affected by phosphorylation. Biochemistry 2009, 48, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Sakaguchi, K.; Shimohigashi, Y.; Samaddar, S.; Banerjee, R.; Basu, G.; Swaminathan, V.; Kundu, T.K.; Roy, S. Effect of phosphorylation on the structure and fold of transactivation domain of p53. J. Biol. Chem. 2002, 277, 15579–15585. [Google Scholar] [CrossRef] [PubMed]
- Lambert, P.F.; Kashanchi, F.; Radonovich, M.F.; Shiekhattar, R.; Brady, J.N. Phosphorylation of p53 serine 15 increases interaction with CBP. J. Biol. Chem. 1998, 273, 33048–33053. [Google Scholar] [CrossRef] [PubMed]
- Dumaz, N.; Meek, D.W. Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with Hdm2. EMBO J. 1999, 18, 7002–7010. [Google Scholar] [CrossRef] [PubMed]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Mapping the interactions of the p53 transactivation domain with the KIX domain of CBP. Biochemistry 2009, 48, 2115–2124. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Goodarzi, A.A.; Higashimoto, Y.; Noda, Y.; Lees-Miller, S.P.; Appella, E.; Anderson, C.W. ATM mediates phosphorylation at multiple p53 sites, including Ser(46), in response to ionizing radiation. J. Biol. Chem. 2002, 277, 12491–12494. [Google Scholar] [CrossRef] [PubMed]
- Puca, R.; Nardinocchi, L.; Sacchi, A.; Rechavi, G.; Givol, D.; D’Orazi, G. HIPK2 modulates p53 activity towards pro-apoptotic transcription. Mol. Cancer 2009. [Google Scholar] [CrossRef]
- Chao, C.; Saito, S.; Anderson, C.W.; Appella, E.; Xu, Y. Phosphorylation of murine p53 at Ser-18 regulates the p53 responses to DNA damage. Proc. Natl. Acad. Sci. USA 2000, 97, 11936–11941. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.H.; Chung, P.H.; Sun, T.P.; Shieh, S.Y. p53 C-terminal phosphorylation by CHM and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Mol. Biol. Cell 2005, 16, 1684–1695. [Google Scholar] [CrossRef] [PubMed]
- Kurash, J.K.; Lei, H.; Shen, Q.; Marston, W.L.; Granda, B.W.; Fan, H.; Wall, D.; Li, E.; Gaudet, F. Methylation of p53 by Set7/9 mediates p53 acetylation and activity in vivo. Mol. Cell 2008, 29, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Perez-Burgos, L.; Placek, B.J.; Sengupta, R.; Richter, M.; Dorsey, J.A.; Kubicek, S.; Opravil, S.; Jenuwein, T.; Berger, S.L. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006, 444, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, G.S.; Ivanova, T.; Kurash, J.; Ivanov, A.; Chuikov, S.; Gizatullin, F.; Herrera-Medina, E.M.; Rauscher, F., III; Reinberg, D.; Barlev, N.A. Methylation-acetylation interplay activates p53 in response to DNA damage. Mol. Cell. Biol. 2007, 27, 6756–6769. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Kachirskaia, L.; Yamaguchi, H.; West, L.E.; Wen, H.; Wang, E.W.; Dutta, S.; Appella, E.; Gozani, O. Modulation of p53 function by Set8-mediated methylation at lysine 382. Mol. Cell 2007, 27, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Chuikov, S.; Kurash, J.K.; Wilson, J.R.; Xiao, B.; Justin, N.; Ivanov, G.S.; McKinney, K.; Tempst, P.; Prives, C.; Gamblin, S.J.; et al. Regulation of p53 activity through lysine methylation. Nature 2004, 432, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Campaner, S.; Spreafico, F.; Burgold, T.; Doni, M.; Rosato, U.; Amati, B.; Testa, G. The methyltransferase Set7/9 (Setd7) is dispensable for the p53-mediated DNA damage response in vivo. Mol. Cell 2011, 43, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Lehnertz, B.; Rogalski, J.C.; Schulze, F.M.; Yi, L.; Lin, S.; Kast, J.; Rossi, F.M.V. p53-Dependent transcription and tumor suppression are not affected in Set7/9-deficient mice. Mol. Cell 2011, 43, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Gostissa, M.; Hengstermann, A.; Fogal, V.; Sandy, P.; Schwarz, S.E.; Scheffner, M.; del Sal, G. Activation of p53 by conjugation to the ubiquitin-like protein SUMO-1. EMBO J. 1999, 18, 6462–6471. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.S.; Desterro, J.M.P.; Lain, S.; Midgley, C.A.; Lane, D.P.; Hay, R.T. SUMO-1 modification activates the transcriptional response of p53. EMBO J. 1999, 18, 6455–6461. [Google Scholar] [CrossRef] [PubMed]
- Fogal, V.; Gostissa, M.; Sandy, P.; Zacchi, P.; Sternsdorf, T.; Jensen, K.; Pandolfi, P.P.; Will, H.; Schneider, C.; del Sal, G. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 2000, 19, 6185–6195. [Google Scholar] [CrossRef] [PubMed]
- Kwek, S.S.S.; Derry, J.; Tyner, A.L.; Shen, Z.Y.; Gudkov, A.V. Functional analysis and intracellular localization of p53 modified by SUMO-1. Oncogene 2001, 20, 2587–2599. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.; Bischof, O.; Dejean, A.; Vousden, K.H. C-Terminal modifications regulate Mdm2 dissociation and nuclear export of p53. Nat. Cell Biol. 2007, 9, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Naidu, S.R.; Lakhter, A.J.; Androphy, E.J. PIASy-mediated Tip60 sumoylation regulates p53-induced autophagy. Cell Cycle 2012, 11, 2717–2728. [Google Scholar] [CrossRef] [PubMed]
- Hay, R.T. SUMO: A history of modification. Mol. Cell 2005, 18, 1–12. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Chiang, C.-M. Crosstalk between sumoylation and acetylation regulates p53-dependent chromatin transcription and DNA binding. EMBO J. 2009, 28, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.M.; Nikolaev, A.; Zhao, W.; Zhang, W.; Gu, W. FBXO11 promotes the neddylation of p53 and inhibits its transcriptional activity. J. Biol. Chem. 2007, 282, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Shiseki, M.; Nagashima, M.; Pedeux, R.M.; Kitahama-Shiseki, M.; Miura, K.; Okamura, S.; Onogi, H.; Higashimoto, Y.; Appella, E.; Yokota, J.; et al. p29ING4 And p28ING5 bind to p53 and p300, and enhance p53 activity. Cancer Res. 2003, 63, 2373–2378. [Google Scholar] [PubMed]
- Pedeux, R.; Sengupta, S.; Shen, J.C.; Demidov, O.N.; Saito, S.; Onogi, H.; Kumamoto, K.; Wincovitch, S.; Garfield, S.H.; McMenamin, M.; et al. ING2 regulates the onset of replicative senescence by induction of p300-dependent p53 acetylation. Mol. Cell. Biol. 2005, 25, 6639–6648. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, M.; Shiseki, M.; Miura, K.; Hagiwara, K.; Linke, S.P.; Pedeux, R.; Wang, X.W.; Yokota, J.; Riabowol, K.; Harris, C.C. DNA damage-inducible gene p33ING2 negatively regulates cell proliferation through acetylation of p53. Proc. Natl. Acad. Sci. USA 2001, 98, 9671–9676. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Gao, M.; Dong, W.; Hu, M.; Li, J.; Shi, X.; Hao, Y.; Li, Y.; Huang, C. p85 Alpha mediates p53 K370 acetylation by p300 and regulates its promoter-specific transactivity in the cellular UVB response. Oncogene 2011, 30, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Dornan, D.; Eckert, M.; Wallace, M.; Shimizu, H.; Ramsay, E.; Hupp, T.R.; Ball, K.L. Interferon regulatory factor 1 binding to p300 stimulates DNA-dependent acetylation of p53. Mol. Cell. Biol. 2004, 24, 10083–10098. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Rivera, M.N.; Coffman, E.J.; Haber, D.A. The WTX tumor suppressor enhances p53 acetylation by CBP/p300. Mol. Cell 2012, 45, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Zhao, J.; Zhang, W.-N.; Li, H.-Y.; Mu, R.; Zhou, T.; Zhang, H.-Y.; Gong, W.-L.; Yu, M.; Man, J.-H.; et al. Induction of SOX4 by DNA damage is critical for p53 stabilization and function. Proc. Natl. Acad. Sci. USA 2009, 106, 3788–3793. [Google Scholar] [CrossRef] [PubMed]
- Basbous, J.; Knani, D.; Bonneaud, N.; Giorgi, D.; Brondello, J.-M.; Rouquier, S. Induction of ASAP (MAP9) contributes to p53 stabilization in response to DNA damage. Cell Cycle 2012, 11, 2380–2390. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Li, L.; Lou, H.; Sun, H.; Ngai, S.-M.; Shao, G.; Tang, J. The ribosomal protein S26 regulates p53 activity in response to DNA damage. Oncogene 2014, 33, 2225–2235. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, Y.; Nguyen, L.A.; Isono, K.; Takakura, N.; Tagata, Y.; Schmitz, M.L.; Koseki, H.; Kitabayashi, I. Roles of HIPK1 and HIPK2 in AML1-and p300-dependent transcription, hematopoiesis and blood vessel formation. EMBO J. 2006, 25, 3955–3965. [Google Scholar] [CrossRef] [PubMed]
- Sabbatini, P.; McCormick, F. MdmX inhibits the p300/CBP-mediated acetylation of p53. DNA Cell Biol. 2002, 21, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Grishina, I.; Debus, K.; Garcia-Limones, C.; Schneider, C.; Shresta, A.; Garcia, C.; Calzado, M.A.; Schmitz, M.L. SIAH-mediated ubiquitination and degradation of acetyl-transferases regulate the p53 response and protein acetylation. Biochim. Biophys. Acta 2012, 1823, 2287–2296. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus type-16 and type-18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Huang, S.M.; Baglia, L.A.; McCance, D.J. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. EMBO J. 1999, 18, 5061–5072. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [PubMed]
- Chakravarti, D.; Ogryzko, V.; Kao, H.Y.; Nash, A.; Chen, H.W.; Nakatani, Y.; Evans, R.M. A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell 1999, 96, 393–403. [Google Scholar] [CrossRef]
- Kitagawa, M.; Lee, S.H.; McCormick, F. Skp2 suppresses p53-dependent apoptosis by inhibiting p300. Mol. Cell 2008, 29, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Gronroos, E.; Terentiev, A.A.; Punga, T.; Ericsson, J. YY1 inhibits the activation of the p53 tumor suppressor in response to genotoxic stress. Proc. Natl. Acad. Sci. USA 2004, 101, 12165–12170. [Google Scholar] [CrossRef] [PubMed]
- Graczyk, A.; Slomnicki, L.P.; Lesniak, W. S100A6 competes with the TAZ2 domain of p300 for binding to p53 and attenuates p53 acetylation. J. Mol. Biol. 2013, 425, 3488–3494. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.Z.; Wani, G.; Wani, M.A.; Wani, A.A. Human homologue of yeast rad23 protein A interacts with p300/cyclic AMP-responsive element binding (CREB)-binding protein to down-regulate transcriptional activity of p53. Cancer Res. 2001, 61, 64–70. [Google Scholar] [PubMed]
- Kim, J.-Y.; Lee, K.-S.; Seol, J.-E.; Yu, K.; Chakravarti, D.; Seo, S.-B. Inhibition of p53 acetylation by INHAT subunit SET/TAF-Iβ represses p53 activity. Nuc. Acids Res. 2012, 40, 75–87. [Google Scholar] [CrossRef]
- Liu, Y.; Colosimo, A.L.; Yang, X.J.; Liao, D.Q. Adenovirus E1b 55-kilodalton oncoprotein inhibits p53 acetylation by PCAF. Mol. Cell. Biol. 2000, 20, 5540–5553. [Google Scholar] [CrossRef] [PubMed]
- Avvakumov, N.; Torchia, J.; Mymryk, J.S. Interaction of the HPV E7 proteins with the PCAF acetyltransferase. Oncogene 2003, 22, 3833–3841. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.T.; Zeng, S.X.; Lee, H.; Lu, H. Mdm2 mediates p300/CREB-binding protein-associated factor ubiquitination and degradation. J. Biol. Chem. 2004, 279, 20035–20043. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, V.; Soddu, S.; Sacchi, A.; D’Orazi, G. HIPK2 contributes to PCAF-mediated p53 acetylation and selective transactivation of p21(WAF1) after nonapoptotic DNA damage. Oncogene 2005, 24, 5431–5442. [Google Scholar] [CrossRef] [PubMed]
- Sho, T.; Tsukiyama, T.; Sato, T.; Kondo, T.; Cheng, J.; Saku, T.; Asaka, M.; Hatakeyama, S. TRIM29 negatively regulates p53 via inhibition of Tip60. Biochim. Biophys. Acta 2011, 1813, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Shi, D.; Gu, W. Negative regulation of the acetyltransferase Tip60-p53 interplay by UHRF1 (Ubiquitin-like with PHD and RING finger domains 1). J. Biol. Chem. 2013, 288, 19581–19592. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Chen, Y.; Song, Q.; Xu, D.; Wang, Y.; Ma, D. PDCD5 interacts with Tip60 and functions as a cooperator in acetyltransferase activity and DNA damage-induced apoptosis. Neoplasia 2009, 11, 345–354. [Google Scholar] [PubMed]
- Dar, A.; Shibata, E.; Dutta, A. Deubiquitination of Tip60 by USP7 determines the activity of the p53-dependent apoptotic pathway. Mol. Cell. Biol. 2013, 33, 3309–3320. [Google Scholar] [CrossRef]
- Charvet, C.; Wissler, M.; Brauns-Schubert, P.; Wang, S.-J.; Tang, Y.; Sigloch, F.C.; Mellert, H.; Brandenburg, M.; Lindner, S.E.; Breit, B.; et al. Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53. Mol. Cell 2011, 42, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Tang, Y.; Jung, S.Y.; Qin, J.; Aaronson, S.A.; Gu, W. Differential effects on p53-mediated cell cycle arrest vs. apoptosis by p90. Proc. Natl. Acad. Sci. USA 2011, 108, 18937–18942. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Yang, J.; Maity, B.; Mayuzumi, D.; Fisher, R.A. Regulator of G protein signaling 6 mediates doxorubicin-induced ATM and p53 activation by a reactive oxygen species-dependent mechanism. Cancer Res. 2011, 71, 6310–6319. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Stewart, A.; Maity, B.; Hagen, J.; Fagan, R.L.; Yang, J.; Quelle, D.E.; Brenner, C.; Fisher, R.A. RGS6 suppresses Ras-induced cellular transformation by facilitating Tip60-mediated DNMT1 degradation and promoting apoptosis. Oncogene 2013. [Google Scholar] [CrossRef]
- Sherr, C.J. Divorcing ARF and p53: An unsettled case. Nat. Rev. Cancer 2006, 6, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Eymin, B.; Claverie, P.; Salon, C.; Leduc, C.; Col, E.; Brambilla, E.; Khochbin, S.; Gazzeri, S. P14(ARF) activates a Tip60-dependent and p53-independent ATM/ATR/Chk pathway in response to genotoxic stress. Mol. Cell. Biol. 2006, 26, 4339–4350. [Google Scholar] [CrossRef] [PubMed]
- Tompkins, V.S.; Hagen, J.; Frazier, A.A.; Lushnikova, T.; Fitzgerald, M.P.; di Tommaso, A.; Ladeveze, V.; Domann, F.E.; Eischen, C.M.; Quelle, D.E. A novel nuclear interactor of ARF and Mdm2 (NIAM) that maintains chromosomal stability. J. Biol. Chem. 2007, 282, 1322–1333. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.M.; Hagen, J.; Tompkins, V.; Thies, K.; Quelle, F.W.; Quelle, D.E. Nuclear interactor of ARF and Mdm2 regulates multiple pathways to activate p53. Cell Cycle 2014, 13, 1288–1298. [Google Scholar] [CrossRef] [PubMed]
- Kitabayashi, I.; Yokoyama, A.; Shimizu, K.; Ohki, M. Interaction and functional cooperation of the leukemia-associated factors AML1 and p300 in myeloid cell differentiation. EMBO J. 1998, 17, 2994–3004. [Google Scholar] [CrossRef] [PubMed]
- Kung, A.L.; Rebel, V.I.; Bronson, R.T.; Ch’ng, L.E.; Sieff, C.A.; Livingston, D.M.; Yao, T.P. Gene dose-dependent control of hematopoiesis and hematologic tumor suppression by CBP. Genes Dev. 2000, 14, 272–277. [Google Scholar] [PubMed]
- Gayther, S.A.; Batley, S.J.; Linger, L.; Bannister, A.; Thorpe, K.; Chin, S.F.; Daigo, Y.; Russell, P.; Wilson, A.; Sowter, H.M.; et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000, 24, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.G.; Ozdag, H.; Caldas, C. P300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [PubMed]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577. [Google Scholar]
- Borrow, J.; Stanton, V.P.; Andresen, J.M.; Becher, R.; Behm, F.G.; Chaganti, R.S.K.; Civin, C.I.; Disteche, C.; Dube, I.; Frischauf, A.M.; et al. The translocation t(8;l6)(p11, p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB binding protein. Nat. Genet. 1996, 14, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, M.; Konishi, M.; KikuchiYanoshita, R.; Tanaka, K.; Shitara, N.; Chong, J.M.; Iwama, T.; Miyaki, M. p300 gene alterations in colorectal and gastric carcinomas. Oncogene 1996, 12, 1565–1569. [Google Scholar] [PubMed]
- Perez, R.E.; Knights, C.D.; Sahu, G.; Catania, J.; Kolukula, V.K.; Stoler, D.; Graessmann, A.; Ogryzko, V.; Pishvaian, M.; Albanese, C.; et al. Restoration of DNA-binding and growth-suppressive activity of mutant forms of p53 via a PCAF-mediated acetylation pathway. J. Cell. Physiol. 2010, 225, 394–405. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.-Z.; Wang, J.-J.; Li, D.-W.; Yu, G.-Z.; Wang, X.; Pan, J.; Chen, Y.; He, M.-X. The p300/CBP associated factor is frequently downregulated in intestinal-type gastric carcinoma and constitutes a biomarker for clinical outcome. Cancer Biol. Ther. 2010, 9, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Squatrito, M.; Luise, C.; Syed, N.; Perna, D.; Wark, L.; Martinato, F.; Sardella, D.; Verrecchia, A.; Bennett, S.; et al. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 2007, 448, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Lleonart, M.E.; Vidal, F.; Gallardo, D.; Diaz-Fuertes, M.; Rojo, F.; Cuatrecasas, M.; Lopez-Vicente, L.; Kondoh, H.; Blanco, C.; Carnero, A.; et al. New p53 related genes in human tumors: Significant downregulation in colon and lung carcinomas. Oncol. Reports 2006, 16, 603–608. [Google Scholar]
- Sakuraba, K.; Yokomizo, K.; Shirahata, A.; Goto, T.; Saito, M.; Ishibashi, K.; Kigawa, G.; Nemoto, H.; Hibi, K. Tip60 as a potential marker for the malignancy of gastric cancer. Anticancer Res. 2011, 31, 77–79. [Google Scholar] [PubMed]
- Sakuraba, K.; Yasuda, T.; Sakata, M.; Kitamura, Y.-H.; Shirahata, A.; Goto, T.; Mizukami, H.; Saito, M.; Ishibashi, K.; Kigawa, G.; et al. Down-regulation of Tip60 gene as a potential marker for the malignancy of colorectal cancer. Anticancer Res. 2009, 29, 3953–3955. [Google Scholar] [PubMed]
- Zhao, H.; Jin, S.; Gewirtz, A.M. The histone acetyltransferase Tip60 interacts with c-Myb and inactivates its transcriptional activity in human leukemia. J. Biol. Chem. 2012, 287, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Cheng, Y.; Tang, Y.; Martinka, M.; Li, G. Role of Tip60 in human melanoma cell migration, metastasis, and patient survival. J. Invest. Derm. 2012, 132, 2632–2641. [Google Scholar] [CrossRef] [PubMed]
- Huntly, B.J.P.; Shigematsu, H.; Deguchi, K.; Lee, B.H.; Mizuno, S.; Duclos, N.; Rowan, R.; Amaral, S.; Curley, D.; Williams, I.R.; et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 2004, 6, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, K.; Ayton, P.M.; Carapeti, M.; Kutok, J.L.; Snyder, C.S.; Williams, I.R.; Cross, N.C.P.; Glass, C.K.; Cleary, M.L.; Gilliland, D.G. MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell 2003, 3, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Ida, K.; Kitabayashi, I.; Taki, T.; Taniwaki, M.; Noro, K.; Yamamoto, M.; Ohki, M.; Hayashi, Y. Adenoviral E1A-associated protein p300 is involved in acute myeloid leukemia with t(11;22)(q23;q13). Blood 1997, 90, 4699–4704. [Google Scholar] [PubMed]
- Sobulo, O.M.; Borrow, J.; Tomek, R.; Reshmi, S.; Harden, A.; Schlegelberger, B.; Housman, D.; Doggett, N.A.; Rowley, J.D.; ZeleznikLe, N.J. MLL is fused to CBP, a histone acetyltransferase, in therapy-related acute myeloid leukemia with a t(11;16)(q23;p13.3). Proc. Natl. Acad. Sci. USA 1997, 94, 8732–8737. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Sharma, G.G.; Young, C.S.H.; Agarwal, M.; Smith, E.R.; Paull, T.T.; Lucchesi, J.C.; Khanna, K.K.; Ludwig, T.; Pandita, T.K. Involvement of human MOF in ATM function. Mol. Cell. Biol. 2005, 25, 5292–5305. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Guerin-Peyrou, T.G.; Sharma, G.G.; Park, C.; Agarwal, M.; Ganju, R.K.; Pandita, S.; Choi, K.; Sukumar, S.; Pandita, R.K.; et al. The mammalian ortholog of Drosophila MOF that acetylates histone H4 lysine 16 is essential for embryogenesis and oncogenesis. Mol. Cell. Biol. 2008, 28, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Shogren-Knaak, M.; Ishii, H.; Sun, J.M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4-k16 acetylation controls chromatin structure and protein interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Sapountzi, V.; Logan, I.R.; Robson, C.N. Cellular functions of Tip60. Int. J. Biochem. Cell Biol. 2006, 38, 1496–1509. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.A.; Kuo, Y.-M.; Andrews, A.J. Differences in specificity and selectivity between CBP and p300 acetylation of histone H3 and H3/H4. Biochemistry 2013, 52, 5746–5759. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Fisher, J.B.; Koprowski, S.; McAllister, D.; Kim, M.-S.; Lough, J. Homozygous disruption of the Tip60 gene causes early embryonic lethality. Develop. Dynamics 2009, 238, 2912–2921. [Google Scholar] [CrossRef]
- Katsumoto, T.; Aikawa, Y.; Iwama, A.; Ueda, S.; Ichikawa, H.; Ochiya, T.; Kitabayashi, I. MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev. 2006, 20, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.P.; Oh, S.P.; Fuchs, M.; Zhou, N.D.; Ch’ng, L.E.; Newsome, D.; Bronson, R.T.; Li, E.; Livingston, D.M.; Eckner, R. Gene dosage-dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 1998, 93, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Sah, V.P.; Attardi, L.D.; Mulligan, G.J.; Williams, B.O.; Bronson, R.T.; Jacks, T. A subset of p53-deficient embryos exhibit exencephaly. Nat. Genet. 1995, 10, 175–180. [Google Scholar] [CrossRef] [PubMed]
- McKeller, R.N.; Fowler, J.L.; Cunningham, J.J.; Warner, N.; Smeyne, R.J.; Zindy, F.; Skapek, S.X. The ARF tumor suppressor gene promotes hyaloid vascular regression during mouse eye development. Proc. Natl. Acad. Sci. USA 2002, 99, 3848–3853. [Google Scholar] [CrossRef] [PubMed]
- Reichel, M.B.; Ali, R.R.; D’Esposito, F.; Clarke, A.R.; Luthert, P.J.; Bhattacharya, S.S.; Hunt, D.M. High frequency of persistent hyperplastic primary vitreous and cataracts in p53-deficient mice. Cell Death Differ. 1998, 5, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Hawes, N.L.; Chang, B.; Avery, C.S.; Smith, R.S.; Nishina, P.M. Severe ocular abnormalities in C57BL/6 but not in 129/SV p53-deficient mice. Invest. Ophthalmol. Vis. Sci. 1999, 40, 1874–1878. [Google Scholar] [PubMed]
- Gannon, H.S.; Jones, S.N. Using mouse models to explore Mdm-p53 signaling in development, cell growth, and tumorigenesis. Genes Cancer 2012, 3, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed]
- Christophorou, M.A.; Ringshausen, I.; Finch, A.J.; Swigart, L.B.; Evan, G.I. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 2006, 443, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Mancuso, A.; Wellen, K.E.; Yang, X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013, 493, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reed, S.M.; Quelle, D.E. p53 Acetylation: Regulation and Consequences. Cancers 2015, 7, 30-69. https://doi.org/10.3390/cancers7010030
Reed SM, Quelle DE. p53 Acetylation: Regulation and Consequences. Cancers. 2015; 7(1):30-69. https://doi.org/10.3390/cancers7010030
Chicago/Turabian StyleReed, Sara M., and Dawn E. Quelle. 2015. "p53 Acetylation: Regulation and Consequences" Cancers 7, no. 1: 30-69. https://doi.org/10.3390/cancers7010030
APA StyleReed, S. M., & Quelle, D. E. (2015). p53 Acetylation: Regulation and Consequences. Cancers, 7(1), 30-69. https://doi.org/10.3390/cancers7010030