The Role of STAT3 in Thyroid Cancer

Abstract
:1. Introduction

{kind=link}
| Tumor type | Cells of origin | Prevalence (% of total thyroid cancers) | Frequently detected genetic/epigenetic alterations | Frequently detected aberrant pathway signaling |
|---|---|---|---|---|
| Papillary thyroid carcinoma (PTC) | Follicular thyrocytes | 80%–85% | BRAFV600E (45%) | MAPK pathway PI3K/AKT pathway IDH1-associated metabolic pathways |
| RET/PTC translocation (20%) | ||||
| IDH1 mutation (10%) | ||||
| EGFR mutation (5%) | ||||
| RASAL1 mutation or hypermethylation (3%) | ||||
| PTEN mutation (1%–2%) | ||||
| PIK3CA mutations (1%–2%) | ||||
| Follicular thyroid carcinoma (FTC) | Follicular thyrocytes | 10%–15% | PAX8/PPARγ rearrangement (40%–60%) | MAPK pathway PI3K/AKT pathway IDH1-associated metabolic pathways |
| HRAS, KRAS, or NRAS mutation (30%–45%) | ||||
| RASAL1 mutation or hypermethylation (32%) | ||||
| PTEN deletion (30%) | ||||
| PTEN mutation (10%–15%) | ||||
| PIK3CA mutation (5%–15%) | ||||
| IDH1 mutation (5%–15%) | ||||
| Poorly differentiated thyroid carcinoma (PDTC) | Follicular thyrocytes | 5%–10% | HRAS, KRAS, or NRAS mutation (20%–40%) | MAPK pathway PI3K/AKT pathway WNT/β-catenin pathway p53-regulated pathways |
| CTNNB1 mutation (25%) | ||||
| TP53 mutation (25%) | ||||
| Anaplastic thyroid carcinoma (ATC) | Follicular thyrocytes | 2%–3% | BRAFV600E mutation (25%–50%) | MAPK pathway PI3K/AKT pathway WNT/β-catenin pathway p53-regulated pathways IDH1- associated metabolic pathways |
| HRAS, KRAS, or NRAS (20%–30%) | ||||
| PTEN mutation (10%–20%) | ||||
| PIK3CA mutation (15%–25%) | ||||
| CTNNB1 mutation (60%–65%) | ||||
| TP53 mutation (70%–80%) | ||||
| IDH1 mutation (5%–15%) | ||||
| ALK mutation (10%) | ||||
| RASAL1 mutations or hypermethylation (33%) | ||||
| Medullary thyroid cancer (MTC) | Parafollicular C-cells | 2%–6% | RET mutation (99% of the familial, 30%–50% of the sporadic cases) | MAPK pathway |
| RAS mutation (10% of the sporadic cases) | PI3K/AKT pathway |
2. The JAK/STAT Pathway
2.1. STAT3
2.2. Activation/Inactivation of STAT3
2.3. Non-Canonical Activity of STAT3
3. JAK/STAT Signaling in Different Cancer Types
4. STAT3 in Thyroid
4.1. STAT3 in Non-Malignant Thyroid Epithelium
4.2. STAT3 in Thyroid Cancer
4.2.1. Expression of STAT3 in Thyroid Cancer
4.2.2. Mechanism of STAT3 Activation in Thyroid Cancer
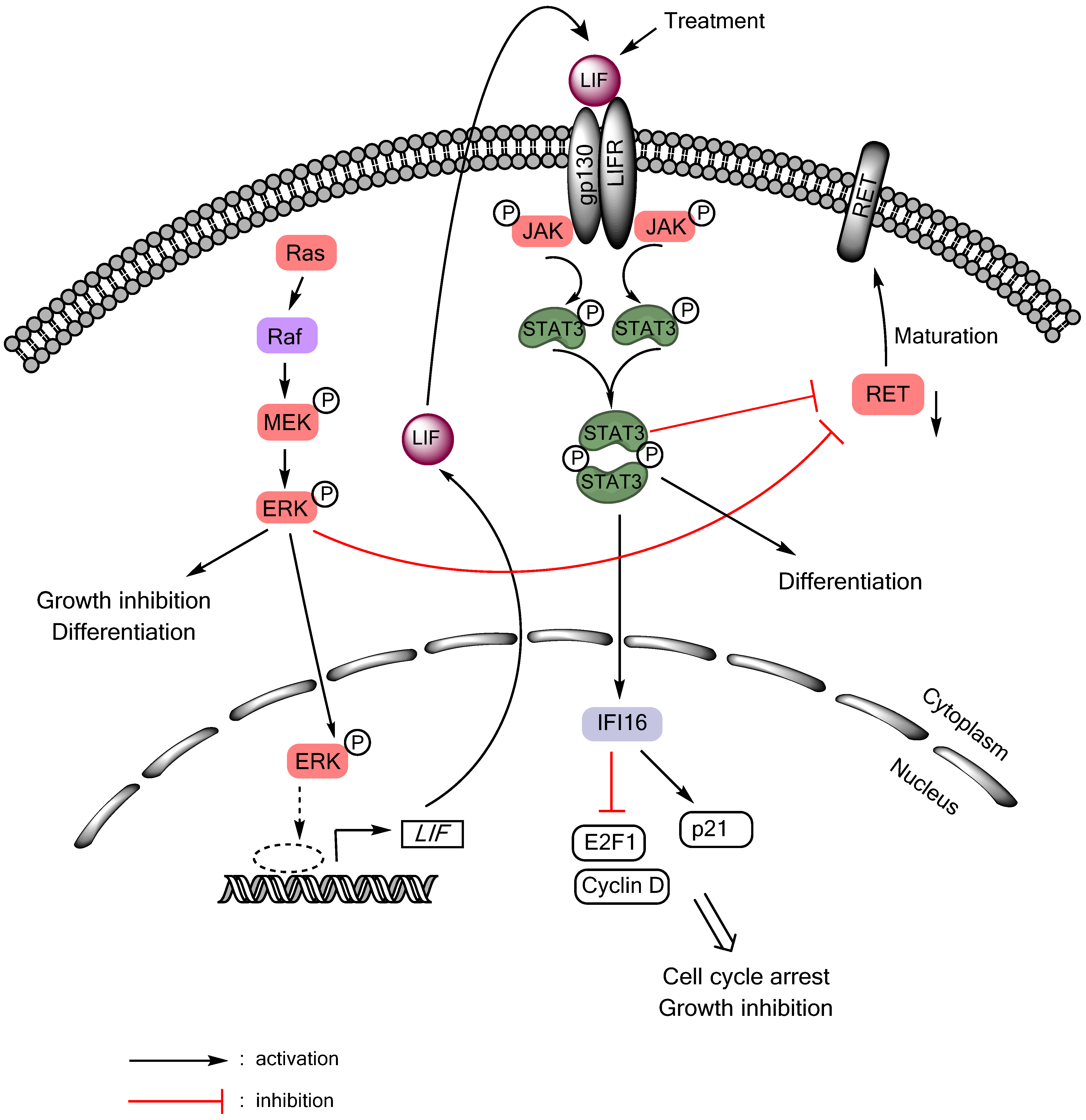
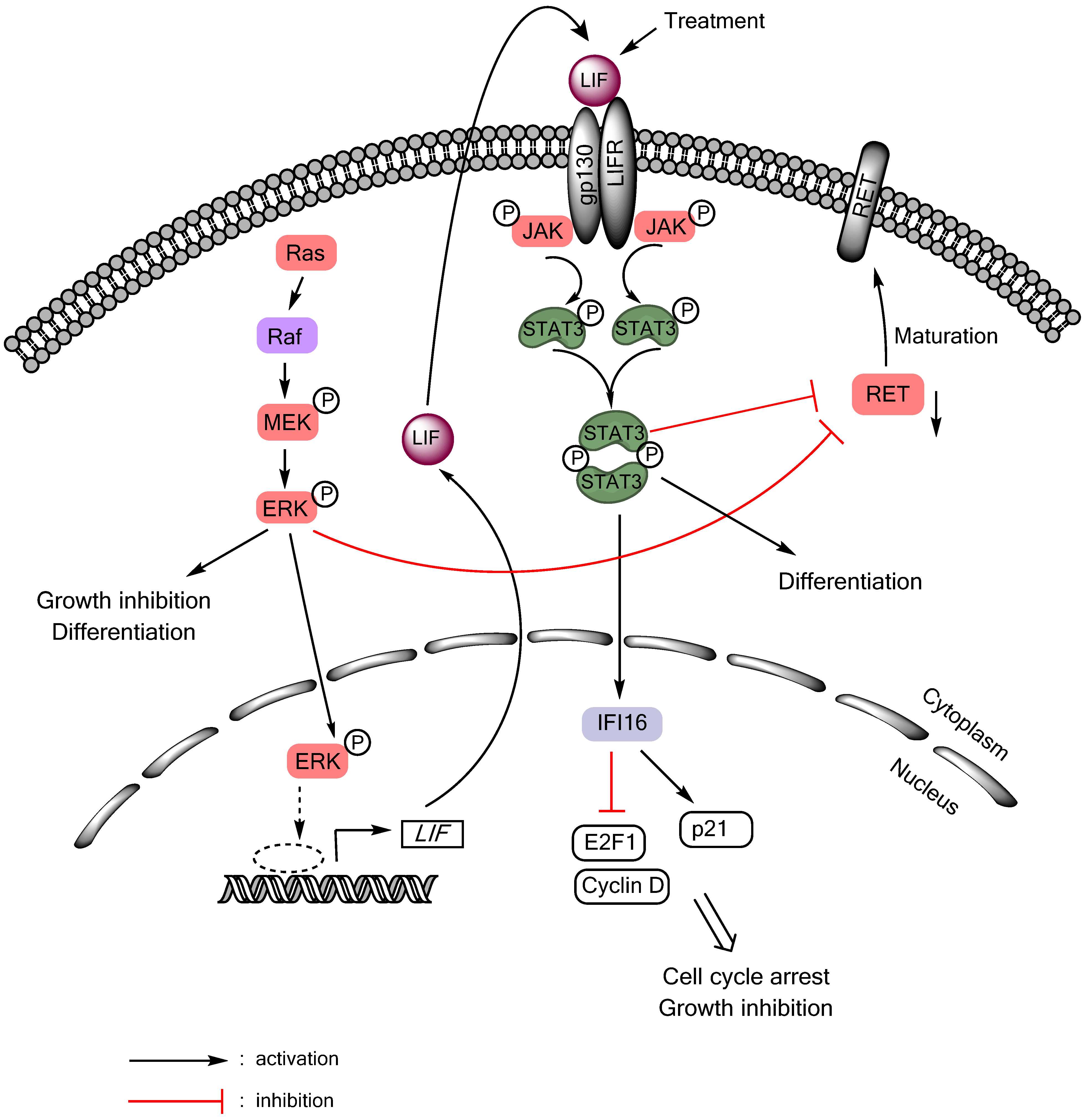
4.2.3. Effects of STAT3 on Thyroid Cancer: Are They Tumor Suppressive?
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef]
- Sprague, B.L.; Warren Andersen, S.; Trentham-Dietz, A. Thyroid cancer incidence and socioeconomic indicators of health care access. Cancer Causes Control 2008, 19, 585–593. [Google Scholar] [CrossRef]
- Cohen, Y.; Xing, M.; Mambo, E.; Guo, Z.; Wu, G.; Trink, B.; Beller, U.; Westra, W.H.; Ladenson, P.W.; Sidransky, D. Braf mutation in papillary thyroid carcinoma. J. Natl. Cancer Inst. 2003, 95, 625–627. [Google Scholar] [CrossRef]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of braf mutations in thyroid cancer: Genetic evidence for constitutive activation of the ret/ptc-ras-braf signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar]
- Hou, P.; Liu, D.; Shan, Y.; Hu, S.; Studeman, K.; Condouris, S.; Wang, Y.; Trink, A.; El-Naggar, A.K.; Tallini, G.; et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/akt pathway in thyroid cancer. Clin. Cancer Res. 2007, 13, 1161–1170. [Google Scholar] [CrossRef]
- Liu, D.; Yang, C.; Bojdani, E.; Murugan, A.K.; Xing, M. Identification of rasal1 as a major tumor suppressor gene in thyroid cancer. J. Natl. Cancer Inst. 2013, 105, 1617–1627. [Google Scholar] [CrossRef]
- Kouvaraki, M.A.; Shapiro, S.E.; Perrier, N.D.; Cote, G.J.; Gagel, R.F.; Hoff, A.O.; Sherman, S.I.; Lee, J.E.; Evans, D.B. Ret proto-oncogene: A review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid 2005, 15, 531–544. [Google Scholar] [CrossRef]
- Ichihara, M.; Murakumo, Y.; Takahashi, M. Ret and neuroendocrine tumors. Cancer Lett. 2004, 204, 197–211. [Google Scholar] [CrossRef]
- Nikiforov, Y.E. Thyroid carcinoma: Molecular pathways and therapeutic targets. Mod. Pathol. 2008, 21, S37–S43. [Google Scholar] [CrossRef]
- Giunti, S.; Antonelli, A.; Amorosi, A.; Santarpia, L. Cellular signaling pathway alterations and potential targeted therapies for medullary thyroid carcinoma. Int. J. Endocrinol. 2013. [Google Scholar] [CrossRef]
- Pinchot, S.N.; Kunnimalaiyaan, M.; Sippel, R.S.; Chen, H. Medullary thyroid carcinoma: Targeted therapies and future directions. J. Oncol. 2009. [Google Scholar] [CrossRef]
- Agrawal, N.; Jiao, Y.; Sausen, M.; Leary, R.; Bettegowda, C.; Roberts, N.J.; Bhan, S.; Ho, A.S.; Khan, Z.; Bishop, J.; et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in ret and ras. J. Clin. Endocrinol. Metab. 2012, 98, E364–E369. [Google Scholar]
- Ciampi, R.; Mian, C.; Fugazzola, L.; Cosci, B.; Romei, C.; Barollo, S.; Cirello, V.; Bottici, V.; Marconcini, G.; Rosa, P.M.; et al. Evidence of a low prevalence of ras mutations in a large medullary thyroid cancer series. Thyroid 2013, 23, 50–57. [Google Scholar] [CrossRef]
- Liu, Z.; Hou, P.; Ji, M.; Guan, H.; Studeman, K.; Jensen, K.; Vasko, V.; El-Naggar, A.K.; Xing, M. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J. Clin. Endocrinol. Metab. 2008, 93, 3106–3116. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Wald, A.I.; Roy, S.; Durso, M.B.; Nikiforov, Y.E. Targeted next-generation sequencing panel (thyroseq) for detection of mutations in thyroid cancer. J. Clin. Endocrinol. Metab. 2013, 98, E1852–E1860. [Google Scholar] [CrossRef]
- Boichard, A.; Croux, L.; Al Ghuzlan, A.; Broutin, S.; Dupuy, C.; Leboulleux, S.; Schlumberger, M.; Bidart, J.M.; Lacroix, L. Somatic ras mutations occur in a large proportion of sporadic ret-negative medullary thyroid carcinomas and extend to a previously unidentified exon. J. Clin. Endocrinol. Metab. 2012, 97, E2031–E2035. [Google Scholar] [CrossRef]
- Mohr, A.; Chatain, N.; Domoszlai, T.; Rinis, N.; Sommerauer, M.; Vogt, M.; Muller-Newen, G. Dynamics and non-canonical aspects of jak/stat signalling. Eur. J. Cell Biol. 2012, 91, 524–532. [Google Scholar] [CrossRef]
- Gamero, A.M.; Young, H.A.; Wiltrout, R.H. Inactivation of stat3 in tumor cells: Releasing a brake on immune responses against cancer? Cancer Cell 2004, 5, 111–112. [Google Scholar] [CrossRef]
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C.W. Signaling through the jak/stat pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar]
- Pellegrini, S.; Dusanter-Fourt, I. The structure, regulation and function of the janus kinases (jaks) and the signal transducers and activators of transcription (stats). Eur. J. Biochem. 1997, 248, 615–633. [Google Scholar]
- Levy, D.E.; Kessler, D.S.; Pine, R.; Darnell, J.E., Jr. Cytoplasmic activation of isgf3, the positive regulator of interferon-alpha-stimulated transcription, reconstituted in vitro. Genes Dev. 1989, 3, 1362–1371. [Google Scholar] [CrossRef]
- Levy, D.E.; Kessler, D.S.; Pine, R.; Reich, N.; Darnell, J.E., Jr. Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative transcriptional control. Genes Dev. 1988, 2, 383–393. [Google Scholar] [CrossRef]
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.J.; Wei, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of aprf, a novel ifn-stimulated gene factor 3 p91-related transcription factor involved in the gp130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Zhong, Z.; Wen, Z.; Darnell, J.E., Jr. Stat3: A stat family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994, 264, 95–98. [Google Scholar]
- Stark, G.R.; Darnell, J.E., Jr. The jak-stat pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef]
- Chen, C.L.; Hsieh, F.C.; Lin, J. Systemic evaluation of total stat3 and stat3 tyrosine phosphorylation in normal human tissues. Exp. Mol. Pathol. 2006, 80, 295–305. [Google Scholar] [CrossRef]
- Raz, R.; Lee, C.K.; Cannizzaro, L.A.; d’Eustachio, P.; Levy, D.E. Essential role of stat3 for embryonic stem cell pluripotency. Proc. Natl. Acad. Sci. USA 1999, 96, 2846–2851. [Google Scholar] [CrossRef]
- Takeda, T.; Kurachi, H.; Yamamoto, T.; Homma, H.; Morishige, K.; Miyake, A.; Murata, Y. Participation of jak, stat and unknown proteins in human placental lactogen-induced signaling: A unique signaling pathway different from prolactin and growth hormone. J. Endocrinol. 1997, 153, R1–R3. [Google Scholar] [CrossRef]
- Akira, S. Functional roles of stat family proteins: Lessons from knockout mice. Stem Cells 1999, 17, 138–146. [Google Scholar] [CrossRef]
- Resemann, H.K.; Watson, C.J.; Lloyd-Lewis, B. The stat3 paradox: A killer and an oncogene. Mol. Cell. Endocrinol. 2014, 382, 603–611. [Google Scholar] [CrossRef]
- Schindler, C.; Levy, D.E.; Decker, T. Jak-stat signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Kunnumakkara, A.B.; Harikumar, K.B.; Gupta, S.R.; Tharakan, S.T.; Koca, C.; Dey, S.; Sung, B. Signal transducer and activator of transcription-3, inflammation, and cancer: How intimate is the relationship? An. N. Y. Acad. Sci. 2009, 1171, 59–76. [Google Scholar]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase m2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef]
- Chung, J.; Uchida, E.; Grammer, T.C.; Blenis, J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell. Biol. 1997, 17, 6508–6516. [Google Scholar]
- Wen, Z.; Zhong, Z.; Darnell, J.E., Jr. Maximal activation of transcription by stat1 and stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Zabolotny, J.M.; Bence-Hanulec, K.K.; Stricker-Krongrad, A.; Haj, F.; Wang, Y.; Minokoshi, Y.; Kim, Y.B.; Elmquist, J.K.; Tartaglia, L.A.; Kahn, B.B.; et al. Ptp1b regulates leptin signal transduction in vivo. Dev. Cell 2002, 2, 489–495. [Google Scholar] [CrossRef]
- Krebs, D.L.; Hilton, D.J. Socs proteins: Negative regulators of cytokine signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef]
- Chung, C.D.; Liao, J.; Liu, B.; Rao, X.; Jay, P.; Berta, P.; Shuai, K. Specific inhibition of stat3 signal transduction by pias3. Science 1997, 278, 1803–1805. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Huso, D.L.; Nathans, D.; Desiderio, S. Specific ablation of stat3beta distorts the pattern of stat3-responsive gene expression and impairs recovery from endotoxic shock. Cell 2002, 108, 331–344. [Google Scholar] [CrossRef]
- Groner, B.; Lucks, P.; Borghouts, C. The function of stat3 in tumor cells and their microenvironment. Semin. Cell Dev. Biol. 2008, 19, 341–350. [Google Scholar] [CrossRef]
- He, B.; You, L.; Uematsu, K.; Zang, K.; Xu, Z.; Lee, A.Y.; Costello, J.F.; McCormick, F.; Jablons, D.M. Socs-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 14133–14138. [Google Scholar] [CrossRef]
- Chatterjee-Kishore, M.; Wright, K.L.; Ting, J.P.; Stark, G.R. How stat1 mediates constitutive gene expression: A complex of unphosphorylated stat1 and irf1 supports transcription of the lmp2 gene. EMBO J. 2000, 19, 4111–4122. [Google Scholar] [CrossRef]
- Liu, Z.; Hazan-Halevy, I.; Harris, D.M.; Li, P.; Ferrajoli, A.; Faderl, S.; Keating, M.J.; Estrov, Z. Stat-3 activates nf-kappab in chronic lymphocytic leukemia cells. Mol. Cancer Res. 2011, 9, 507–515. [Google Scholar] [CrossRef]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated stat3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated stat3 accumulates in response to il-6 and activates transcription by binding to nfkappab. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef]
- Yang, J.; Stark, G.R. Roles of unphosphorylated stats in signaling. Cell Res. 2008, 18, 443–451. [Google Scholar] [CrossRef]
- Gao, S.P.; Bromberg, J.F. Touched and moved by stat3. Sci. STKE 2006, 2006, pe30. [Google Scholar]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial stat3 supports ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. Stats in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef]
- Ai, T.; Wang, Z.; Zhang, M.; Zhang, L.; Wang, N.; Li, W.; Song, L. Expression and prognostic relevance of stat3 and cyclin d1 in non-small cell lung cancer. Int. J. Biol. Markers 2012, 27, e132–e138. [Google Scholar] [CrossRef]
- Birner, P.; Toumangelova-Uzeir, K.; Natchev, S.; Guentchev, M. Stat3 tyrosine phosphorylation influences survival in glioblastoma. J. Neurooncol. 2010, 100, 339–343. [Google Scholar]
- Liu, X.; He, Z.; Li, C.H.; Huang, G.; Ding, C.; Liu, H. Correlation analysis of jak-stat pathway components on prognosis of patients with prostate cancer. Pathol. Oncol. Res. 2012, 18, 17–23. [Google Scholar] [CrossRef]
- Masuda, M.; Ruan, H.Y.; Ito, A.; Nakashima, T.; Toh, S.; Wakasaki, T.; Yasumatsu, R.; Kutratomi, Y.; Komune, S.; Weinstein, I.B. Signal transducers and activators of transcription 3 up-regulates vascular endothelial growth factor production and tumor angiogenesis in head and neck squamous cell carcinoma. Oral Oncol. 2007, 43, 785–790. [Google Scholar] [CrossRef]
- Min, H.; Wei-hong, Z. Constitutive activation of signal transducer and activator of transcription 3 in epithelial ovarian carcinoma. J. Obstet. Gynaecol. Res. 2009, 35, 918–925. [Google Scholar] [CrossRef]
- Schoppmann, S.F.; Jesch, B.; Friedrich, J.; Jomrich, G.; Maroske, F.; Birner, P. Phosphorylation of signal transducer and activator of transcription 3 (stat3) correlates with her-2 status, carbonic anhydrase 9 expression and prognosis in esophageal cancer. Clin. Exp. Metastasis 2012, 29, 615–624. [Google Scholar] [CrossRef]
- Sheen-Chen, S.M.; Huang, C.C.; Tang, R.P.; Chou, F.F.; Eng, H.L. Prognostic value of signal transducers and activators of transcription 3 in breast cancer. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2286–2290. [Google Scholar] [CrossRef]
- Yakata, Y.; Nakayama, T.; Yoshizaki, A.; Kusaba, T.; Inoue, K.; Sekine, I. Expression of p-stat3 in human gastric carcinoma: Significant correlation in tumour invasion and prognosis. Int. J. Oncol. 2007, 30, 437–442. [Google Scholar]
- Chen, Y.; Wang, J.; Wang, X.; Liu, X.; Li, H.; Lv, Q.; Zhu, J.; Wei, B.; Tang, Y. Stat3, a poor survival predicator, is associated with lymph node metastasis from breast cancer. J. Breast Cancer 2013, 16, 40–49. [Google Scholar] [CrossRef]
- Wei, D.; Le, X.; Zheng, L.; Wang, L.; Frey, J.A.; Gao, A.C.; Peng, Z.; Huang, S.; Xiong, H.Q.; Abbruzzese, J.L.; et al. Stat3 activation regulates the expression of vascular endothelial growth factor and human pancreatic cancer angiogenesis and metastasis. Oncogene 2003, 22, 319–329. [Google Scholar] [CrossRef]
- Scholz, A.; Heinze, S.; Detjen, K.M.; Peters, M.; Welzel, M.; Hauff, P.; Schirner, M.; Wiedenmann, B.; Rosewicz, S. Activated signal transducer and activator of transcription 3 (stat3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology 2003, 125, 891–905. [Google Scholar] [CrossRef]
- Deng, J.; Liang, H.; Zhang, R.; Sun, D.; Pan, Y.; Liu, Y.; Zhang, L.; Hao, X. Stat3 is associated with lymph node metastasis in gastric cancer. Tumour Biol. 2013, 34, 2791–2800. [Google Scholar] [CrossRef]
- James, C.; Ugo, V.; Le Couedic, J.P.; Staerk, J.; Delhommeau, F.; Lacout, C.; Garcon, L.; Raslova, H.; Berger, R.; Bennaceur-Griscelli, A.; et al. A unique clonal jak2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005, 434, 1144–1148. [Google Scholar] [CrossRef]
- Pilati, C.; Amessou, M.; Bihl, M.P.; Balabaud, C.; Nhieu, J.T.; Paradis, V.; Nault, J.C.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic mutations activating stat3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 2011, 208, 1359–1366. [Google Scholar] [CrossRef]
- Rebouissou, S.; Amessou, M.; Couchy, G.; Poussin, K.; Imbeaud, S.; Pilati, C.; Izard, T.; Balabaud, C.; Bioulac-Sage, P.; Zucman-Rossi, J. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature 2009, 457, 200–204. [Google Scholar] [CrossRef]
- Bournazou, E.; Bromberg, J. Targeting the tumor microenvironment: Jak-stat3 signaling. JAKSTAT 2013. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. Stats in cancer inflammation and immunity: A leading role for stat3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef]
- Hedvat, M.; Huszar, D.; Herrmann, A.; Gozgit, J.M.; Schroeder, A.; Sheehy, A.; Buettner, R.; Proia, D.; Kowolik, C.M.; Xin, H.; et al. The jak2 inhibitor azd1480 potently blocks stat3 signaling and oncogenesis in solid tumors. Cancer Cell 2009, 16, 487–497. [Google Scholar] [CrossRef]
- Xin, H.; Herrmann, A.; Reckamp, K.; Zhang, W.; Pal, S.; Hedvat, M.; Zhang, C.; Liang, W.; Scuto, A.; Weng, S.; et al. Antiangiogenic and antimetastatic activity of jak inhibitor azd1480. Cancer Res. 2011, 71, 6601–6610. [Google Scholar] [CrossRef]
- Sonnenblick, A.; Uziely, B.; Nechushtan, H.; Kadouri, L.; Galun, E.; Axelrod, J.H.; Katz, D.; Daum, H.; Hamburger, T.; Maly, B.; et al. Tumor stat3 tyrosine phosphorylation status, as a predictor of benefit from adjuvant chemotherapy for breast cancer. Breast Cancer Res. Treat. 2013, 138, 407–413. [Google Scholar] [CrossRef]
- Dolled-Filhart, M.; Camp, R.L.; Kowalski, D.P.; Smith, B.L.; Rimm, D.L. Tissue microarray analysis of signal transducers and activators of transcription 3 (stat3) and phospho-stat3 (tyr705) in node-negative breast cancer shows nuclear localization is associated with a better prognosis. Clin. Cancer Res. 2003, 9, 594–600. [Google Scholar]
- Pectasides, E.; Egloff, A.M.; Sasaki, C.; Kountourakis, P.; Burtness, B.; Fountzilas, G.; Dafni, U.; Zaramboukas, T.; Rampias, T.; Rimm, D.; et al. Nuclear localization of signal transducer and activator of transcription 3 in head and neck squamous cell carcinoma is associated with a better prognosis. Clin. Cancer Res. 2010, 16, 2427–2434. [Google Scholar] [CrossRef]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the egfr kinase domain mediate stat3 activation via il-6 production in human lung adenocarcinomas. J. Clin. Investig. 2007, 117, 3846–3856. [Google Scholar] [CrossRef]
- Woo, S.; Lee, B.L.; Yoon, J.; Cho, S.J.; Baik, T.K.; Chang, M.S.; Lee, H.E.; Park, J.W.; Kim, Y.H.; Kim, W.H. Constitutive activation of signal transducers and activators of transcription 3 correlates with better prognosis, cell proliferation and hypoxia-inducible factor-1alpha in human gastric cancer. Pathobiology 2011, 78, 295–301. [Google Scholar] [CrossRef]
- Setsu, N.; Kohashi, K.; Endo, M.; Yamamoto, H.; Tamiya, S.; Takahashi, Y.; Yamada, Y.; Ishii, T.; Matsuda, S.; Yokoyama, R.; et al. Phosphorylation of signal transducer and activator of transcription 3 in soft tissue leiomyosarcoma is associated with a better prognosis. Int. J. Cancer 2013, 132, 109–115. [Google Scholar] [CrossRef]
- Monnien, F.; Zaki, H.; Borg, C.; Mougin, C.; Bosset, J.F.; Mercier, M.; Arbez-Gindre, F.; Kantelip, B. Prognostic value of phosphorylated stat3 in advanced rectal cancer: A study from 104 french patients included in the eortc 22921 trial. J. Clin. Pathol. 2010, 63, 873–878. [Google Scholar] [CrossRef]
- Koperek, O.; Aumayr, K.; Schindl, M.; Werba, G.; Soleiman, A.; Schoppmann, S.; Sahora, K.; Birner, P. Phosphorylation of stat3 correlates with her2 status, but not with survival in pancreatic ductal adenocarcinoma. APMIS 2013. [Google Scholar] [CrossRef]
- De la Iglesia, N.; Puram, S.V.; Bonni, A. Stat3 regulation of glioblastoma pathogenesis. Curr. Mol. Med. 2009, 9, 580–590. [Google Scholar] [CrossRef]
- Albi, E.; Curcio, F.; Spelat, R.; Lazzarini, R.; Loreti, E.; Ferri, I.; Ambesi-Impiombato, F.S. The thyroid lobes: The different twins. Arch. Biochem. Biophys. 2012, 518, 16–22. [Google Scholar] [CrossRef]
- Bates, S.H.; Myers, M.G. The role of leptin→stat3 signaling in neuroendocrine function: An integrative perspective. J. Mol. Med. 2004, 82, 12–20. [Google Scholar] [CrossRef]
- Chung, J.; Park, E.S.; Kim, D.; Suh, J.M.; Chung, H.K.; Kim, J.; Kim, H.; Park, S.J.; Kwon, O.Y.; Ro, H.K.; et al. Thyrotropin modulates interferon-gamma-mediated intercellular adhesion molecule-1 gene expression by inhibiting janus kinase-1 and signal transducer and activator of transcription-1 activation in thyroid cells. Endocrinology 2000, 141, 2090–2097. [Google Scholar]
- Park, E.S.; Kim, H.; Suh, J.M.; Park, S.J.; Kwon, O.Y.; Kim, Y.K.; Ro, H.K.; Cho, B.Y.; Chung, J.; Shong, M. Thyrotropin induces socs-1 (suppressor of cytokine signaling-1) and socs-3 in frtl-5 thyroid cells. Mol. Endocrinol. 2000, 14, 440–448. [Google Scholar] [CrossRef]
- Park, E.S.; Kim, H.; Suh, J.M.; Park, S.J.; You, S.H.; Chung, H.K.; Lee, K.W.; Kwon, O.Y.; Cho, B.Y.; Kim, Y.K.; et al. Involvement of jak/stat (janus kinase/signal transducer and activator of transcription) in the thyrotropin signaling pathway. Mol. Endocrinol. 2000, 14, 662–670. [Google Scholar] [CrossRef]
- Kim, H.; Suh, J.M.; Hwang, E.S.; Kim, D.W.; Chung, H.K.; Song, J.H.; Hwang, J.H.; Park, K.C.; Ro, H.K.; Jo, E.K.; et al. Thyrotropin-mediated repression of class ii trans-activator expression in thyroid cells: Involvement of stat3 and suppressor of cytokine signaling. J. Immunol. 2003, 171, 616–627. [Google Scholar]
- Staab, J.; Barth, P.J.; Meyer, T. Cell-type-specific expression of stat transcription factors in tissue samples from patients with lymphocytic thyroiditis. Endocr. Pathol. 2012, 23, 141–150. [Google Scholar] [CrossRef]
- Ardito, G.; Revelli, L.; Boninsegna, A.; Sgambato, A.; Moschella, F.; Marzola, M.C.; Giustozzi, E.; Avenia, N.; Castelli, M.; Rubello, D. Immunohistochemical evaluation of inflammatory and proliferative markers in adjacent normal thyroid tissue in patients undergoing total thyroidectomy: Results of a preliminary study. J. Exp. Clin. Cancer Res. 2010, 29, 77. [Google Scholar] [CrossRef]
- Couto, J.P.; Daly, L.; Almeida, A.; Knauf, J.A.; Fagin, J.A.; Sobrinho-Simoes, M.; Lima, J.; Maximo, V.; Soares, P.; Lyden, D.; et al. Stat3 negatively regulates thyroid tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E2361–E2370. [Google Scholar] [CrossRef]
- Park, J.I.; Strock, C.J.; Ball, D.W.; Nelkin, B.D. The ras/raf/mek/extracellular signal-regulated kinase pathway induces autocrine-paracrine growth inhibition via the leukemia inhibitory factor/jak/stat pathway. Mol. Cell. Biol. 2003, 23, 543–554. [Google Scholar] [CrossRef]
- Park, J.I.; Strock, C.J.; Ball, D.W.; Nelkin, B.D. Interleukin-1beta can mediate growth arrest and differentiation via the leukemia inhibitory factor/jak/stat pathway in medullary thyroid carcinoma cells. Cytokine 2005, 29, 125–134. [Google Scholar] [CrossRef]
- Vindrieux, D.; Augert, A.; Girard, C.A.; Gitenay, D.; Lallet-Daher, H.; Wiel, C.; Le Calve, B.; Gras, B.; Ferrand, M.; Verbeke, S.; et al. Pla2r1 mediates tumor suppression by activating jak2. Cancer Res. 2013, 73, 6334–6345. [Google Scholar] [CrossRef]
- Lu, C.; Kerbel, R.S. Interleukin-6 undergoes transition from paracrine growth inhibitor to autocrine stimulator during human melanoma progression. J. Cell Biol. 1993, 120, 1281–1288. [Google Scholar] [CrossRef]
- Zhang, J.; Gill, A.; Atmore, B.; Johns, A.; Delbridge, L.; Lai, R.; McMullen, T. Upregulation of the signal transducers and activators of transcription 3 (stat3) pathway in lymphatic metastases of papillary thyroid cancer. Int. J. Clin. Exp. Pathol. 2011, 4, 356–362. [Google Scholar]
- Trovato, M.; Grosso, M.; Vitarelli, E.; Ruggeri, R.M.; Alesci, S.; Trimarchi, F.; Barresi, G.; Benvenga, S. Distinctive expression of stat3 in papillary thyroid carcinomas and a subset of follicular adenomas. Histol. Histopathol. 2003, 18, 393–399. [Google Scholar]
- Kim, W.G.; Choi, H.J.; Kim, W.B.; Kim, E.Y.; Yim, J.H.; Kim, T.Y.; Gong, G.; Kim, S.Y.; Chung, N.; Shong, Y.K. Basal stat3 activities are negatively correlated with tumor size in papillary thyroid carcinomas. J. Endocrinol. Investig. 2012, 35, 413–418. [Google Scholar]
- Hwang, J.H.; Kim, D.W.; Suh, J.M.; Kim, H.; Song, J.H.; Hwang, E.S.; Park, K.C.; Chung, H.K.; Kim, J.M.; Lee, T.H.; et al. Activation of signal transducer and activator of transcription 3 by oncogenic ret/ptc (rearranged in transformation/papillary thyroid carcinoma) tyrosine kinase: Roles in specific gene regulation and cellular transformation. Mol. Endocrinol. 2003, 17, 1155–1166. [Google Scholar] [CrossRef]
- Lin, H.; Chen, M.C.; Chiu, C.Y.; Song, Y.M.; Lin, S.Y. Cdk5 regulates stat3 activation and cell proliferation in medullary thyroid carcinoma cells. J. Biol. Chem. 2007, 282, 2776–2784. [Google Scholar] [CrossRef]
- Kim, T.H.; Lee, S.Y.; Rho, J.H.; Jeong, N.Y.; Soung, Y.H.; Jo, W.S.; Kang, D.Y.; Kim, S.H.; Yoo, Y.H. Mutant p53 (g199v) gains antiapoptotic function through signal transducer and activator of transcription 3 in anaplastic thyroid cancer cells. Mol. Cancer Res. 2009, 7, 1645–1654. [Google Scholar] [CrossRef]
- Masago, K.; Asato, R.; Fujita, S.; Hirano, S.; Tamura, Y.; Kanda, T.; Mio, T.; Katakami, N.; Mishima, M.; Ito, J. Epidermal growth factor receptor gene mutations in papillary thyroid carcinoma. Int. J. Cancer 2009, 124, 2744–2749. [Google Scholar] [CrossRef]
- Mooi, W.J.; Peeper, D.S. Oncogene-induced cell senescence—Halting on the road to cancer. N. Engl. J. Med. 2006, 355, 1037–1046. [Google Scholar] [CrossRef]
- Courtois-Cox, S.; Jones, S.L.; Cichowski, K. Many roads lead to oncogene-induced senescence. Oncogene 2008, 27, 2801–2809. [Google Scholar] [CrossRef]
- Nakagawa, T.; Mabry, M.; de Bustros, A.; Ihle, J.N.; Nelkin, B.D.; Baylin, S.B. Introduction of v-ha-ras oncogene induces differentiation of cultured human medullary thyroid carcinoma cells. Proc. Natl. Acad. Sci. USA 1987, 84, 5923–5927. [Google Scholar] [CrossRef]
- Wood, K.W.; Qi, H.; D’Arcangelo, G.; Armstrong, R.C.; Roberts, T.M.; Halegoua, S. The cytoplasmic raf oncogene induces a neuronal phenotype in pc12 cells: A potential role for cellular raf kinases in neuronal growth factor signal transduction. Proc. Natl. Acad. Sci. USA 1993, 90, 5016–5020. [Google Scholar] [CrossRef]
- Carson, E.B.; McMahon, M.; Baylin, S.B.; Nelkin, B.D. Ret gene silencing is associated with raf-1-induced medullary thyroid carcinoma cell differentiation. Cancer Res. 1995, 55, 2048–2052. [Google Scholar]
- Park, J.I.; Powers, J.F.; Tischler, A.S.; Strock, C.J.; Ball, D.W.; Nelkin, B.D. Gdnf-induced leukemia inhibitory factor can mediate differentiation via the mek/erk pathway in pheochromocytoma cells derived from nf1-heterozygous knockout mice. Exp. Cell Res. 2005, 303, 79–88. [Google Scholar]
- Carson-Walter, E.B.; Smith, D.P.; Ponder, B.A.; Baylin, S.B.; Nelkin, B.D. Post-transcriptional silencing of ret occurs, but is not required, during raf-1 mediated differentiation of medullary thyroid carcinoma cells. Oncogene 1998, 17, 367–376. [Google Scholar]
- Arthan, D.; Hong, S.K.; Park, J.I. Leukemia inhibitory factor can mediate ras/raf/mek/erk-induced growth inhibitory signaling in medullary thyroid cancer cells. Cancer Lett. 2010, 297, 31–41. [Google Scholar] [CrossRef]
- Kim, E.J.; Park, J.I.; Nelkin, B.D. Ifi16 is an essential mediator of growth inhibition, but not differentiation, induced by the leukemia inhibitory factor/jak/stat pathway in medullary thyroid carcinoma cells. J. Biol. Chem. 2005, 280, 4913–4920. [Google Scholar]
- Starenki, D.; Singh, N.K.; Jensen, D.R.; Peterson, F.C.; Park, J.I. Recombinant leukemia inhibitory factor suppresses human medullary thyroid carcinoma cell line xenografts in mice. Cancer Lett. 2013, 339, 144–151. [Google Scholar] [CrossRef]
- Couto, J.P.; Almeida, A.; Daly, L.; Sobrinho-Simoes, M.; Bromberg, J.F.; Soares, P. Azd1480 blocks growth and tumorigenesis of ret- activated thyroid cancer cell lines. PLoS One 2012, 7, e46869. [Google Scholar]
- Cichowski, K.; Hahn, W.C. Unexpected pieces to the senescence puzzle. Cell 2008, 133, 958–961. [Google Scholar] [CrossRef]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: Sms-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sosonkina, N.; Starenki, D.; Park, J.-I. The Role of STAT3 in Thyroid Cancer. Cancers 2014, 6, 526-544. https://doi.org/10.3390/cancers6010526
Sosonkina N, Starenki D, Park J-I. The Role of STAT3 in Thyroid Cancer. Cancers. 2014; 6(1):526-544. https://doi.org/10.3390/cancers6010526
Chicago/Turabian StyleSosonkina, Nadiya, Dmytro Starenki, and Jong-In Park. 2014. "The Role of STAT3 in Thyroid Cancer" Cancers 6, no. 1: 526-544. https://doi.org/10.3390/cancers6010526
APA StyleSosonkina, N., Starenki, D., & Park, J.-I. (2014). The Role of STAT3 in Thyroid Cancer. Cancers, 6(1), 526-544. https://doi.org/10.3390/cancers6010526