Butylated Hydroxyanisole Blocks the Occurrence of Tumor Associated Macrophages in Tobacco Smoke Carcinogen-Induced Lung Tumorigenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
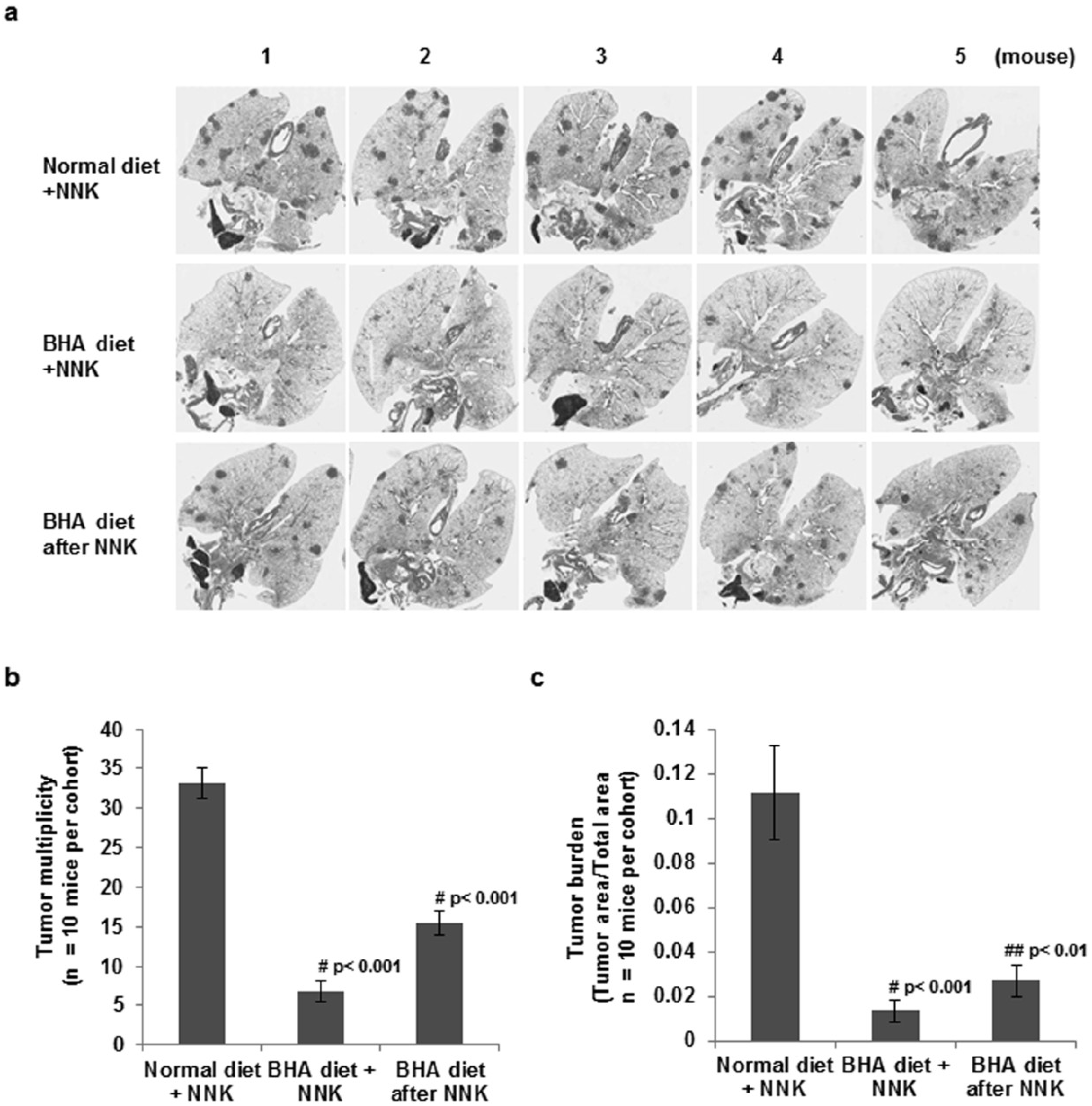
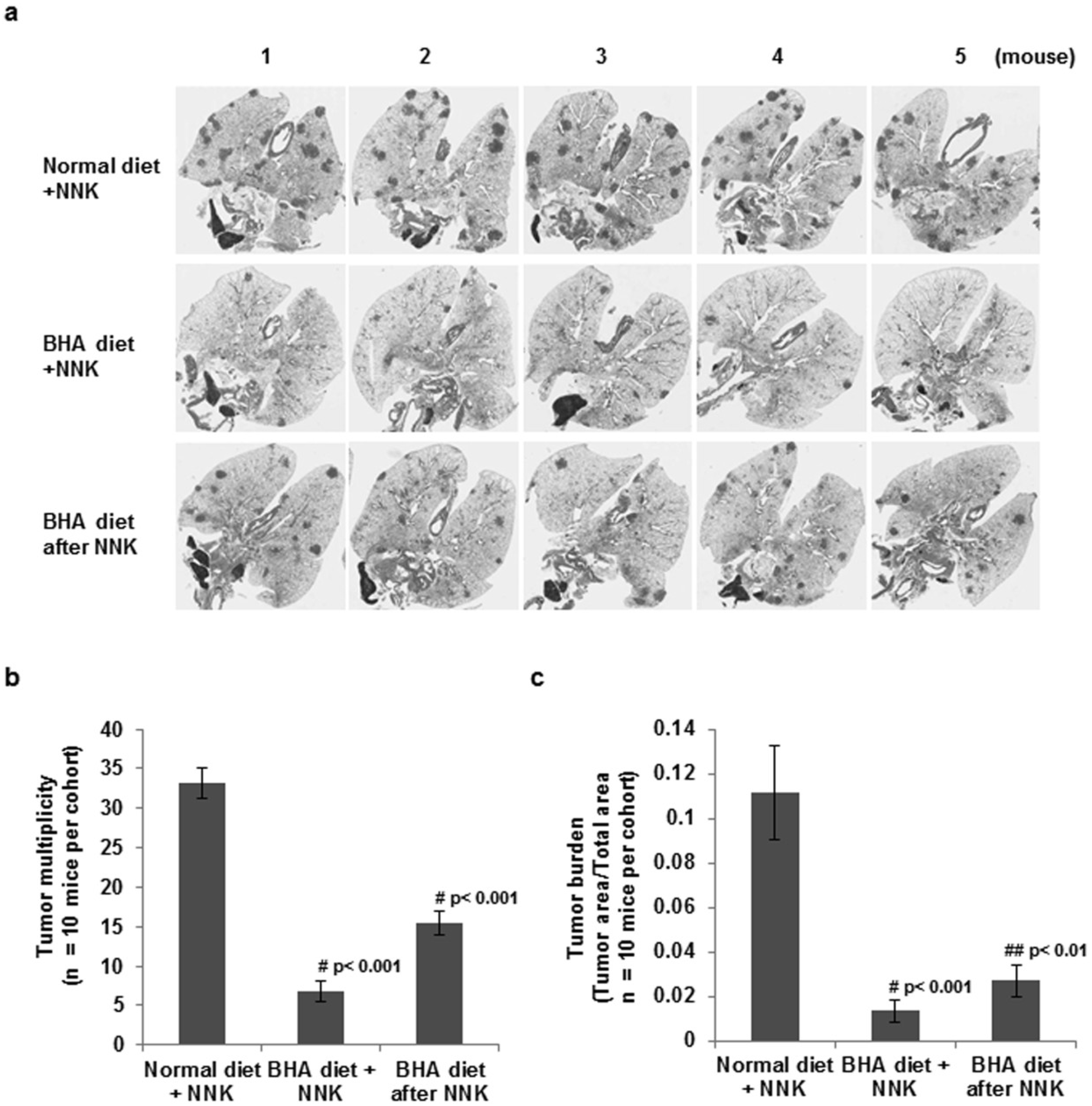
2.1. BHA Suppresses Tobacco Smoke Carcinogen (NNK)-Induced Lung Tumorigenesis
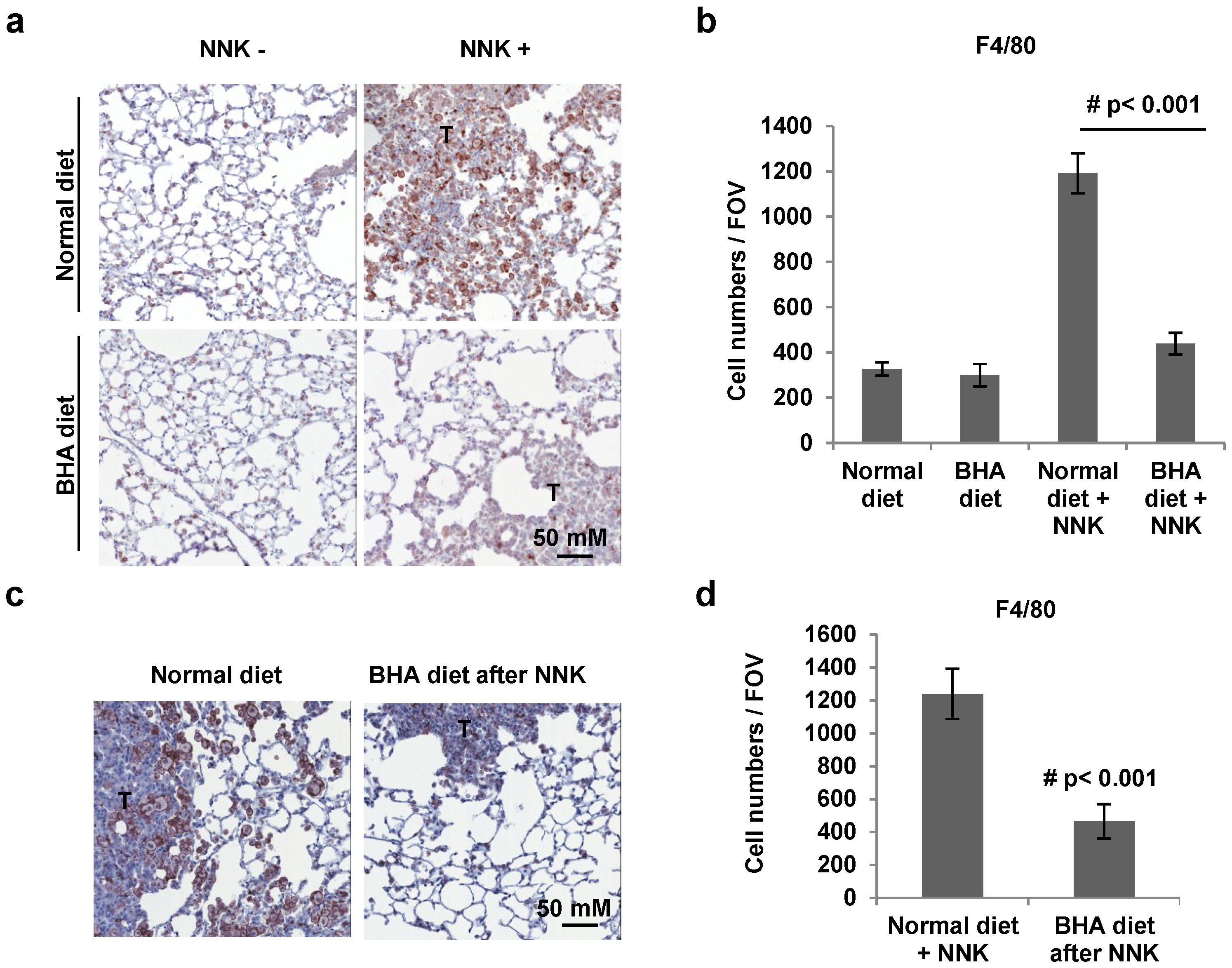
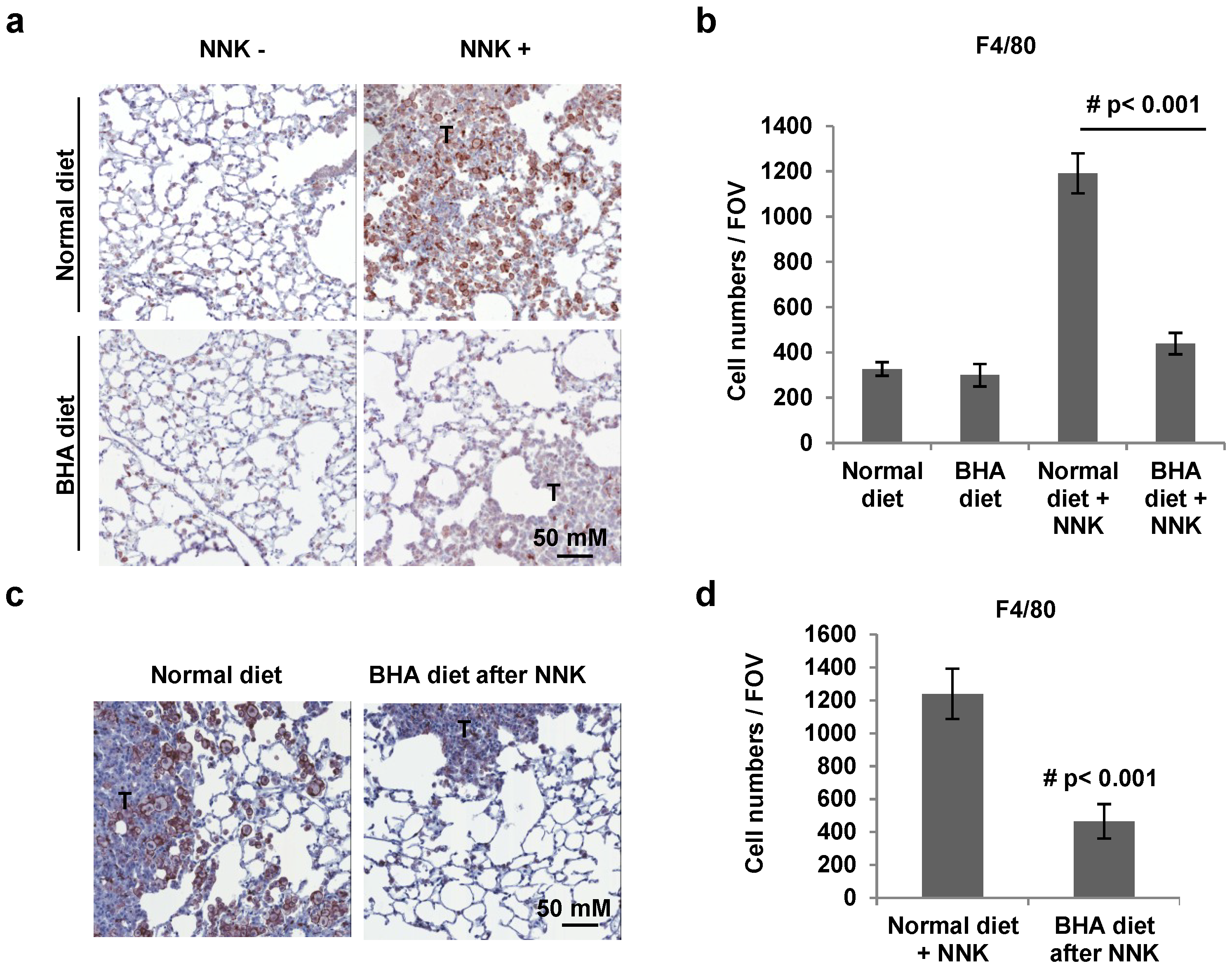
2.2. BHA Blocks NNK-Induced Occurrence of F4/80+ Macrophages in Lungs
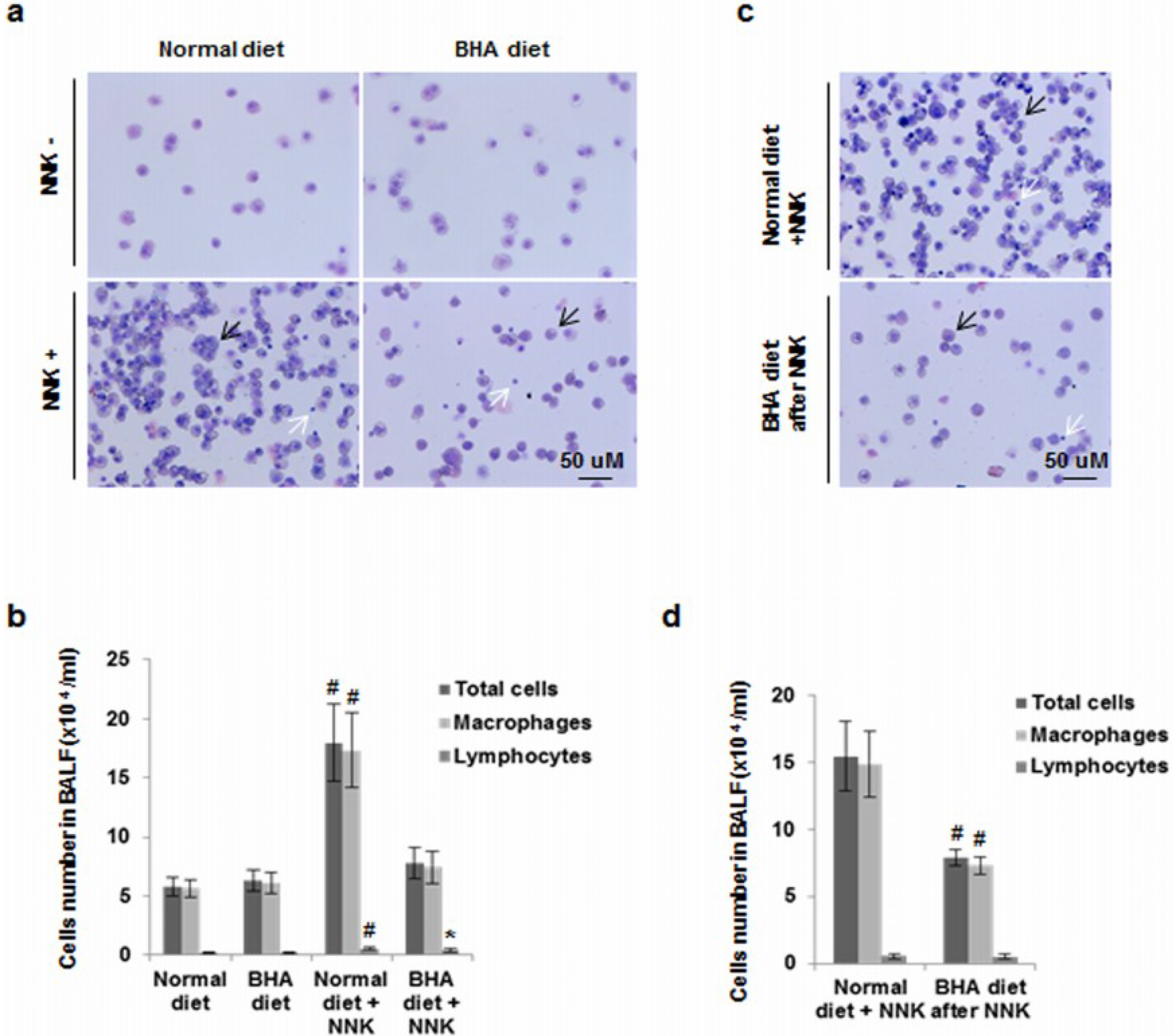
2.3. BHA Blocks NNK-Induced Increase of Macrophages in BALF

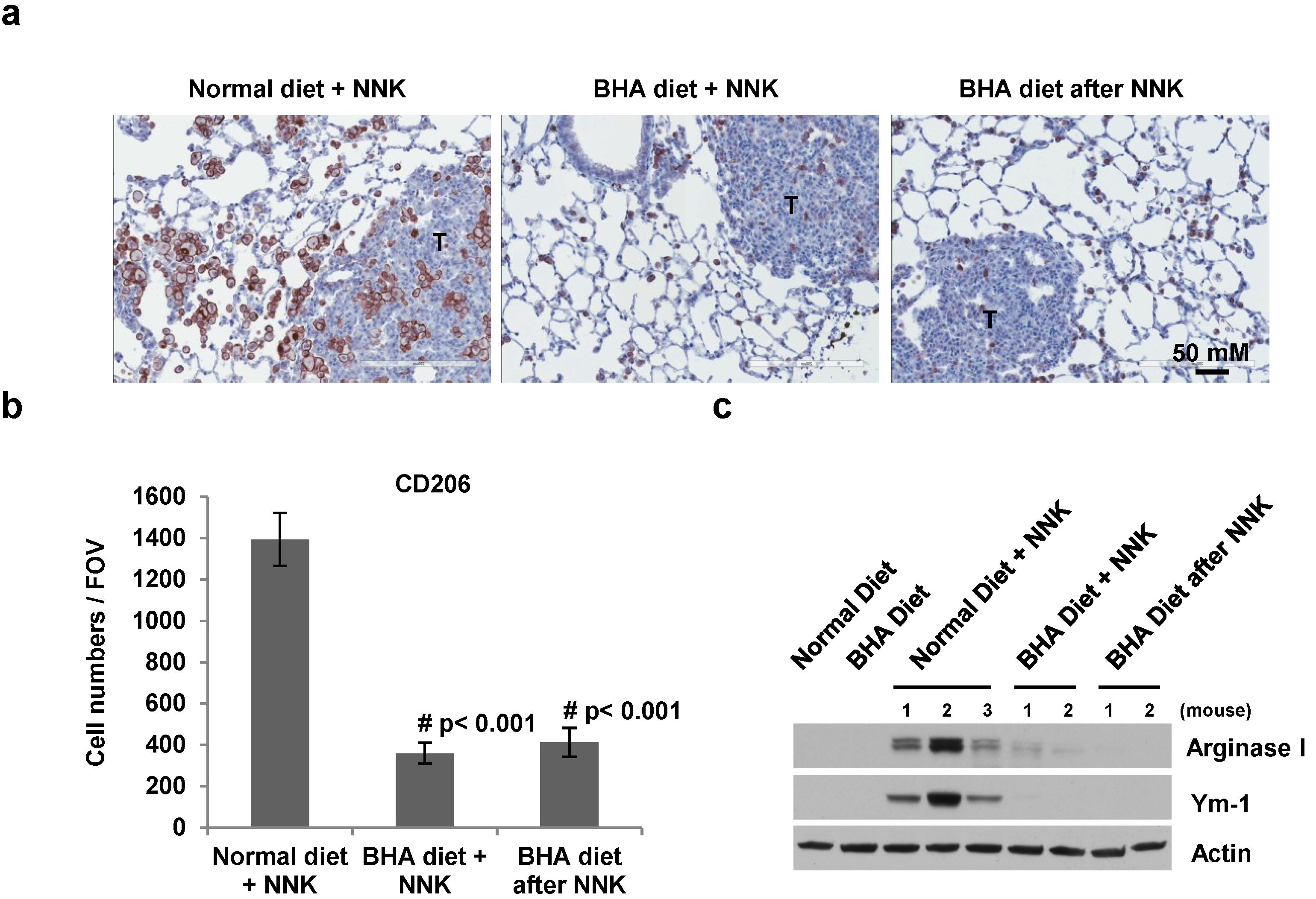
2.4. BHA Specifically Blocks the NNK-Induced Occurrence of TAMs in Lungs
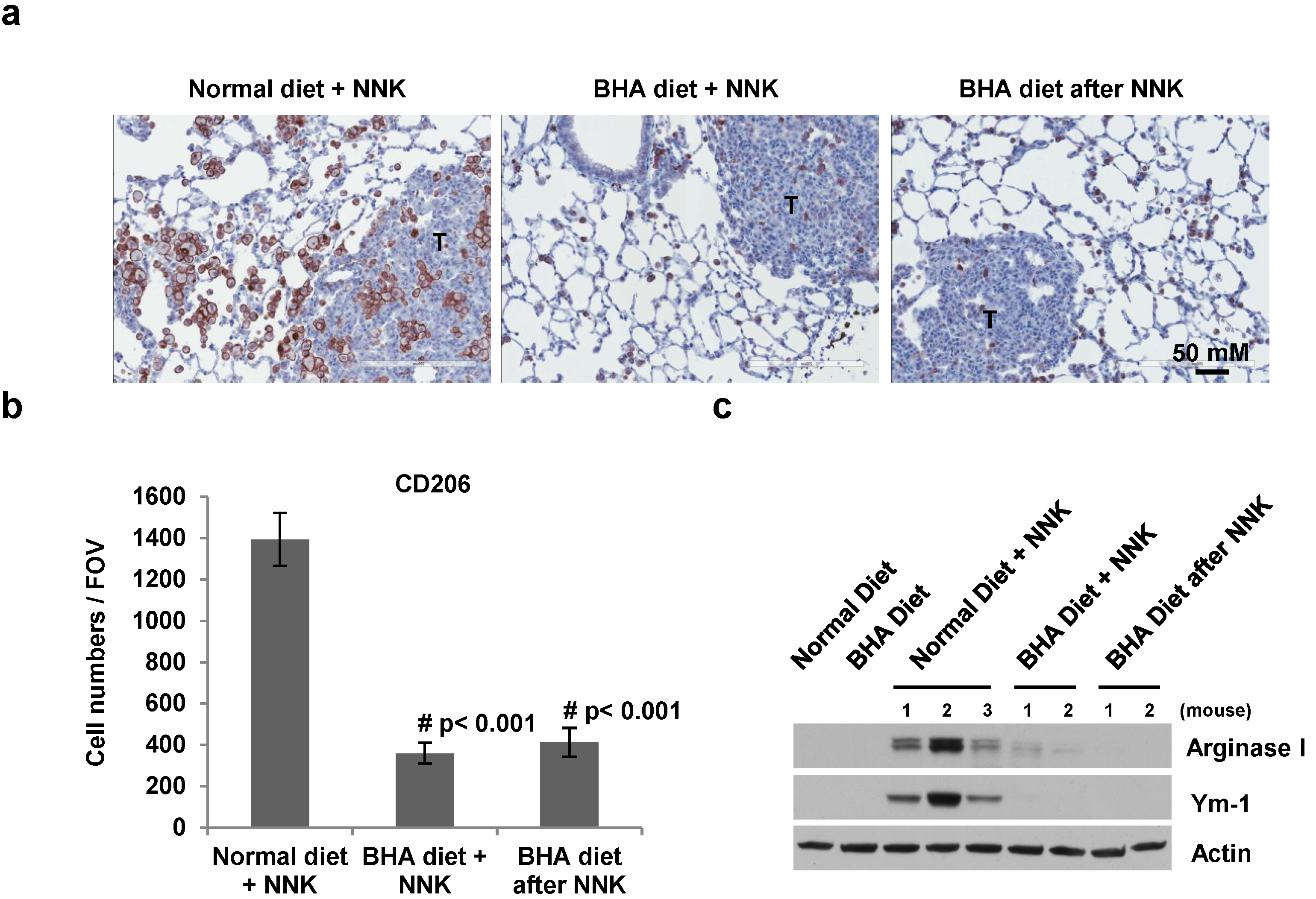
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Reagents and Antibody
4.3. Western Blot Analysis
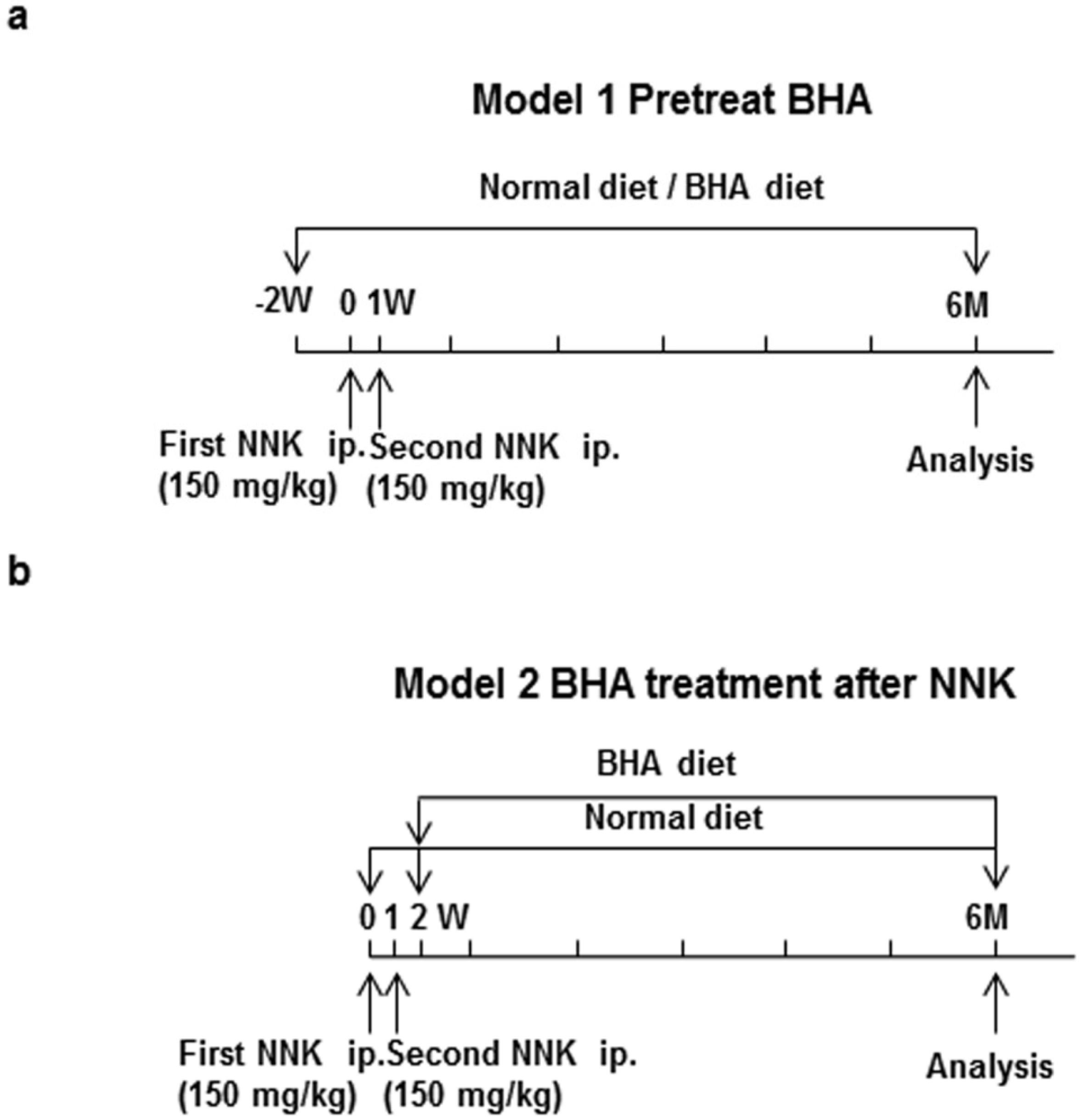
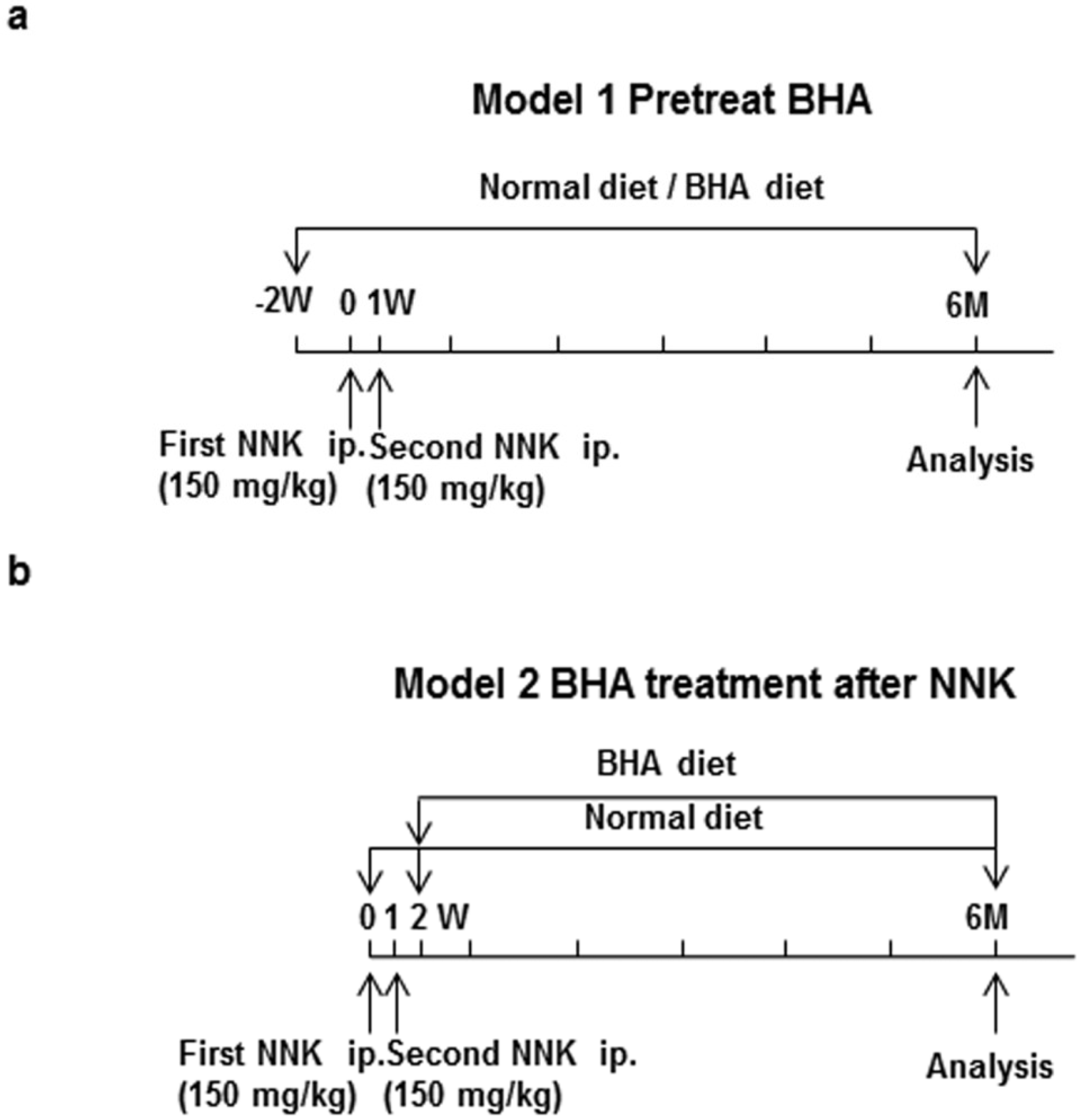
4.4. NNK-Induced Lung Tumor Models
4.5. Evaluation of Lung Tumors
4.6. Immunohistochemical Analysis
4.7. BALF Leukocyte Counts
4.8. Statistical Analysis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Murray, T.; Thun, M.J. Cancer statistics, 2008. CA Cancer J. Clin. 2008, 58, 71–96. [Google Scholar] [CrossRef]
- Hecht, S.S. Cigarette smoking and lung cancer: Chemical mechanisms and approaches to prevention. Lancet Oncol. 2002, 3, 461–469. [Google Scholar] [CrossRef]
- Takahashi, H.; Ogata, H.; Nishigaki, R.; Broide, D.H.; Karin, M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell 2010, 17, 89–97. [Google Scholar] [CrossRef]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef]
- Wells, P.G.; Kim, P.M.; Laposa, R.R.; Nicol, C.J.; Parman, T.; Winn, L.M. Oxidative damage in chemical teratogenesis. Mutat. Res. 1997, 396, 65–78. [Google Scholar] [CrossRef]
- Bowler, R.P.; Crapo, J.D. Oxidative stress in airways: Is there a role for extracellular superoxide dismutase? Am. J. Respir. Crit. Care Med. 2002, 166, S38–S43. [Google Scholar] [CrossRef]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef]
- Sica, A.; Larghi, P.; Mancino, A.; Rubino, L.; Porta, C.; Totaro, M.G.; Rimoldi, M.; Biswas, S.K.; Allavena, P.; Mantovani, A. Macrophage polarization in tumour progression. Semin. Cancer Biol. 2008, 18, 349–355. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef]
- Pollard, J.W. Trophic macrophages in development and disease. Nat. Rev. Immunol. 2009, 9, 259–270. [Google Scholar] [CrossRef]
- Baay, M.; Brouwer, A.; Pauwels, P.; Peeters, M.; Lardon, F. Tumor cells and tumor-associated macrophages: Secreted proteins as potential targets for therapy. Clin. Dev. Immunol. 2011, 2011, 565187. [Google Scholar]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Karin, M.; Lawrence, T.; Nizet, V. Innate immunity gone awry: Linking microbial infections to chronic inflammation and cancer. Cell 2006, 124, 823–835. [Google Scholar] [CrossRef]
- Fang, L.Y.; Izumi, K.; Lai, K.P.; Liang, L.; Li, L.; Miyamoto, H.; Lin, W.J.; Chang, C. Infiltrating macrophages promote prostate tumorigenesis via modulating androgen receptor-mediated CCL4-STAT3 signaling. Cancer Res. 2013, 73, 5633–5646. [Google Scholar] [CrossRef]
- Zheng, H.C.; Takano, Y. NNK-induced lung tumors: A review of animal model. J. Oncol. 2011, 2011, 635379. [Google Scholar]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar] [CrossRef]
- Castonguay, A.; Pepin, P.; Stoner, G.D. Lung tumorigenicity of NNK given orally to A/J mice: Its application to chemopreventive efficacy studies. Exp. Lung Res. 1991, 17, 485–499. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, Y.; Choksi, S.; Liu, Z.-G. Butylated Hydroxyanisole Blocks the Occurrence of Tumor Associated Macrophages in Tobacco Smoke Carcinogen-Induced Lung Tumorigenesis. Cancers 2013, 5, 1643-1654. https://doi.org/10.3390/cancers5041643
Zhang Y, Choksi S, Liu Z-G. Butylated Hydroxyanisole Blocks the Occurrence of Tumor Associated Macrophages in Tobacco Smoke Carcinogen-Induced Lung Tumorigenesis. Cancers. 2013; 5(4):1643-1654. https://doi.org/10.3390/cancers5041643
Chicago/Turabian StyleZhang, Yan, Swati Choksi, and Zheng-Gang Liu. 2013. "Butylated Hydroxyanisole Blocks the Occurrence of Tumor Associated Macrophages in Tobacco Smoke Carcinogen-Induced Lung Tumorigenesis" Cancers 5, no. 4: 1643-1654. https://doi.org/10.3390/cancers5041643
APA StyleZhang, Y., Choksi, S., & Liu, Z.-G. (2013). Butylated Hydroxyanisole Blocks the Occurrence of Tumor Associated Macrophages in Tobacco Smoke Carcinogen-Induced Lung Tumorigenesis. Cancers, 5(4), 1643-1654. https://doi.org/10.3390/cancers5041643