The Clinical Significance of Unknown Sequence Variants in BRCA Genes

Abstract
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
| Mutation nomenclature | http://www.hgvs.org/mutnomen/ | |||
| http://www.humgen.nl/mutalyzer/1.0.1/http://research.nhgri.nih.gov/bic | ||||
| Co-occurence in-trans | www.dmubd.net | |||
| Species conservation | www.agvgd.iarc.fr/index.php | |||
| www.ebi.ac.uk/clustalw/index.html | ||||
| In silico analysis | http://www.russell.embl.de/aas/ | |||
| http://coot.embl.de/PolyPhen | ||||
| http://www.agvgd.larc.fr | ||||
| http://blocks.fhcrc.org/sift/SIFT.html | ||||
| http://genetics.bwh.harvard.edu/pph/ | ||||
| http://www.ensembl.org/index.html | ||||
| www.uniprot.org/ | ||||
| www.expasy.ch/prosite/ | ||||
| Splice-site prediction | Berkley Drosophila Genome project | www.fruitfly.org/seq_tools/splice.html | ||
| NetGene2 | www.cbs.dtu.dk/services/NetGene2 | |||
| Alex Dong Li's splice site finder | www.genet.sickkids.on.ca/~ali/splicesitefinder.html | |||
| GeneSplicer Web Interface | www.tigr.org/tdb/GeneSplicer/gene_spl.html | |||
| Splice Sequence Finder(Montpelier) | www.umd.be/SSF | |||
| SNP Database | www.ncbi.nlm.nih.gov/proiects/SNP/ |
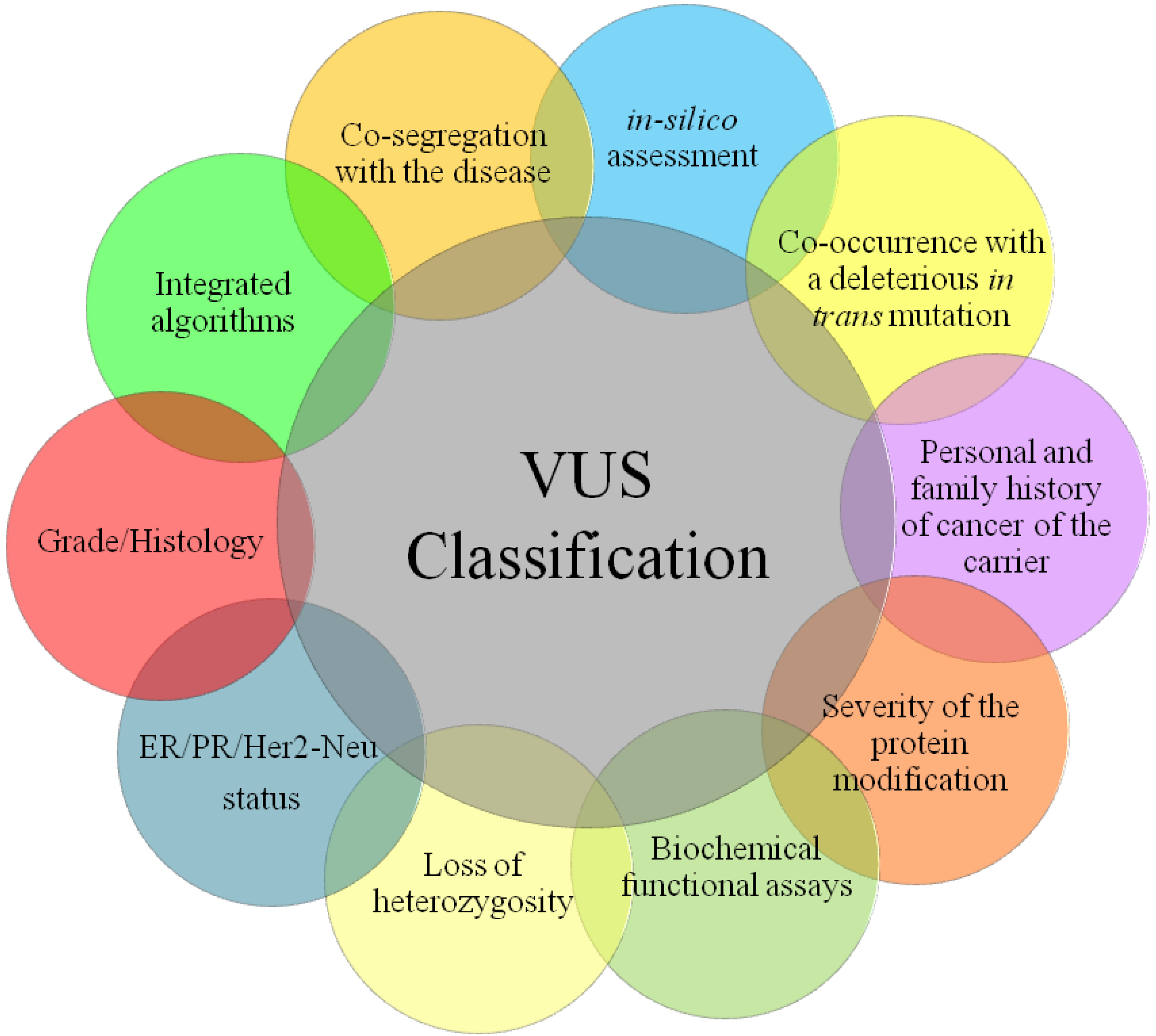
2. Variants of Unknown Significance (VUSs)
3. Principal Methods of VUS Assessment
3.1. Type and Location of VUSs
3.2. Analysis of Control Group
3.3. Co-Segregation with Cancer in Families
3.4. Co-occurrence with Known Deleterious Mutation
3.5. Conservation of Amino Acids across Species
3.6. In silico Analysis of Amino Acid Change
3.7. Loss of Heterozygosity (LOH)
3.8. Biochemical Functional Assays
3.8.1. Protein Assays Related to BRCA1 Function
3.8.2. Protein Assays Related to BRCA2 Function
3.9. Pathological Data
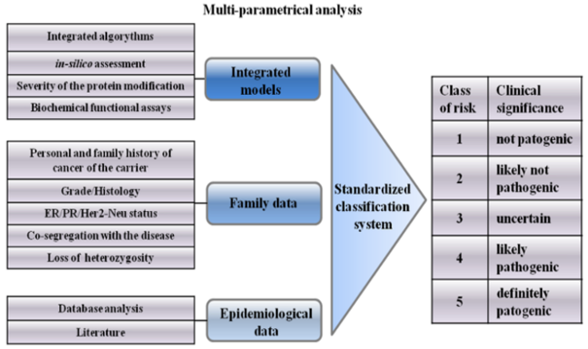
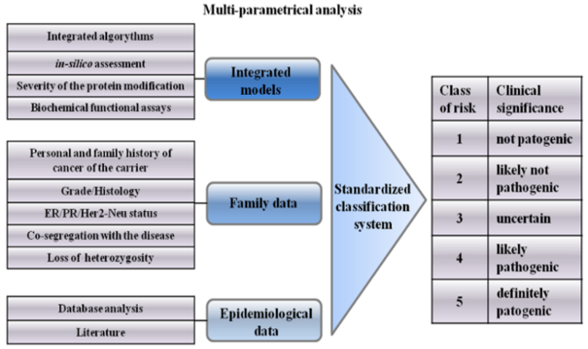
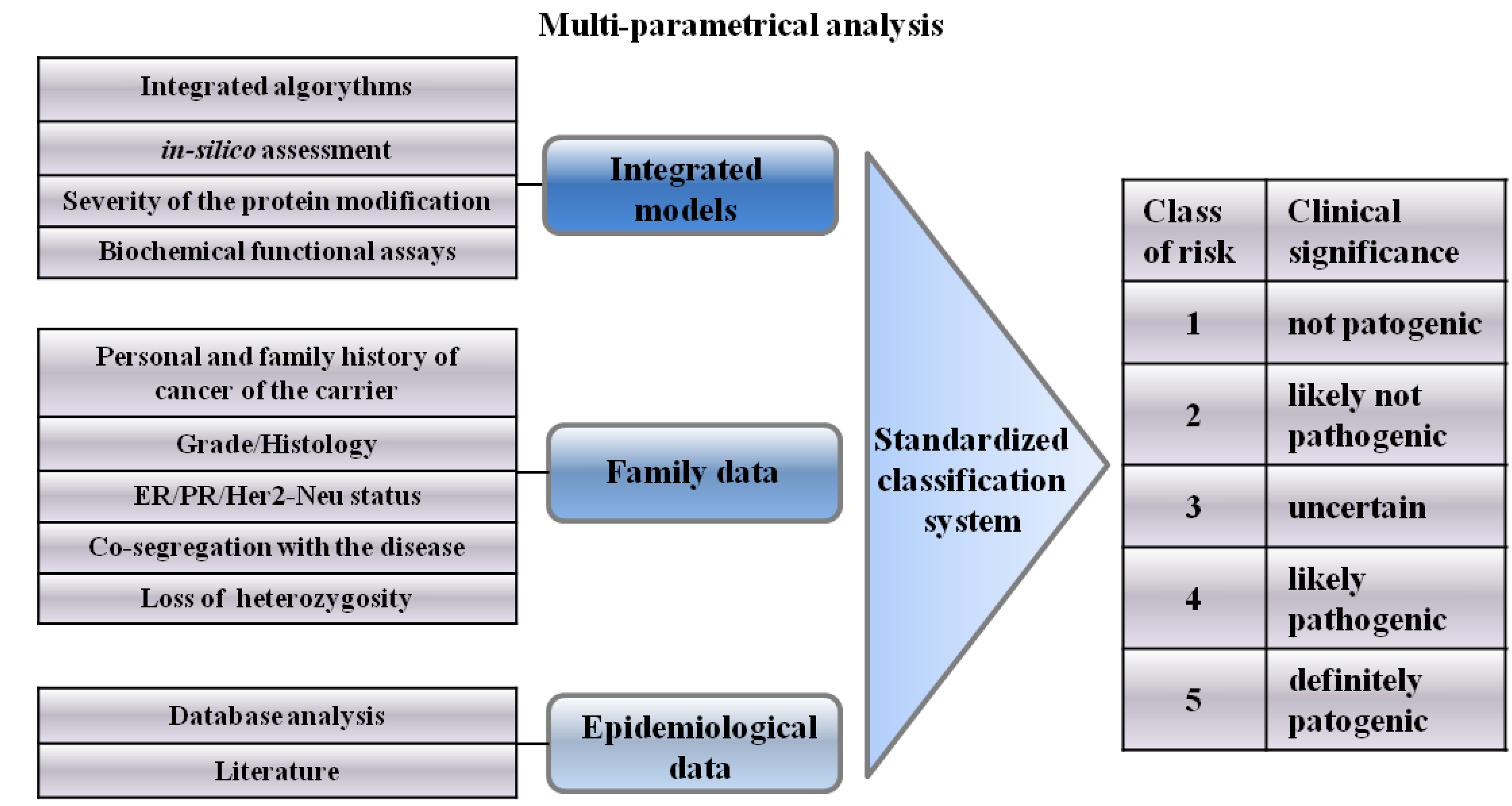
4. Integrated Models for VUS Assessment
5. Psychological Aspects of VUS Carriers
6. Classification System of VUSs
7. Conclusions
Conflict of Interest Statement
References
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene brca1. Science 1994, 266, 66–71. [Google Scholar]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene brca2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef]
- Palma, M.; Ristori, E.; Ricevuto, E.; Giannini, G.; Gulino, A. Brca1 and brca2: The genetic testing and the current management options for mutation carriers. Crit. Rev. Oncol. Hematol. 2006, 57, 1–23. [Google Scholar] [CrossRef]
- Gulati, A.P.; Domchek, S.M. The clinical management of brca1 and brca2 mutation carriers. Curr. Oncol. Rep. 2008, 10, 47–53. [Google Scholar] [CrossRef]
- Russo, A.; Calo, V.; Agnese, V.; Bruno, L.; Corsale, S.; Augello, C.; Gargano, G.; Barbera, F.; Cascio, S.; Intrivici, C.; Rinaldi, G.; Gulotta, G.; Macaluso, M.; Surmacz, E.; Giordano, A.; Gebbia, N.; Bazan, V. Brca1 genetic testing in 106 breast and ovarian cancer families from southern italy (sicily): A mutation analyses. Breast Cancer Res. Treat. 2007, 105, 267–276. [Google Scholar] [CrossRef]
- Calo, V.; Agnese, V.; Gargano, G.; Corsale, S.; Gregorio, V.; Cascio, S.; Cammareri, P.; Bruno, L.; Augello, C.; Gullo, A.; Sisto, P.S.; Badalamenti, G.; Valerio, M.R.; Napoli, L.; Gebbia, N.; Bazan, V.; Russo, A. A new germline mutation in brca1 gene in a sicilian family with ovarian cancer. Breast Cancer Res. Treat. 2006, 96, 97–100. [Google Scholar] [CrossRef]
- King, M.C.; Marks, J.H.; Mandell, J.B. Breast and ovarian cancer risks due to inherited mutations in brca1 and brca2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef]
- Frank, T.S.; Deffenbaugh, A.M.; Reid, J.E.; Hulick, M.; Ward, B.E.; Lingenfelter, B.; Gumpper, K.L.; Scholl, T.; Tavtigian, S.V.; Pruss, D.R.; Critchfield, G.C. Clinical characteristics of individuals with germline mutations in brca1 and brca2: Analysis of 10,000 individuals. J. Clin. Oncol. 2002, 20, 1480–1490. [Google Scholar] [CrossRef]
- Easton, D.F.; Deffenbaugh, A.M.; Pruss, D.; Frye, C.; Wenstrup, R.J.; Allen-Brady, K.; Tavtigian, S.V.; Monteiro, A.N.; Iversen, E.S.; Couch, F.J.; Goldgar, D.E. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the brca1 and brca2 breast cancer-predisposition genes. Am. J. Hum. Genet. 2007, 81, 873–883. [Google Scholar] [CrossRef]
- Augello, C.; Bruno, L.; Bazan, V.; Calo, V.; Agnese, V.; Corsale, S.; Cascio, S.; Gargano, G.; Terrasi, M.; Barbera, F.; Fricano, S.; Adamo, B.; Valerio, M.R.; Colucci, G.; Sumarcz, E.; Russo, A. Y179c, f486l and n550h are brca1 variants that may be associated with breast cancer in a sicilian family: Results of a 5-year goim (gruppo oncologico dell'italia meridionale) prospective study. Ann. Oncol. 2006, 17 (Suppl. 7), 30–33. [Google Scholar]
- Goldgar, D.E.; Easton, D.F.; Deffenbaugh, A.M.; Monteiro, A.N.; Tavtigian, S.V.; Couch, F.J. Integrated evaluation of DNA sequence variants of unknown clinical significance: Application to brca1 and brca2. Am. J. Hum. Genet. 2004, 75, 535–544. [Google Scholar] [CrossRef]
- Chenevix-Trench, G.; Healey, S.; Lakhani, S.; Waring, P.; Cummings, M.; Brinkworth, R.; Deffenbaugh, A.M.; Burbidge, L.A.; Pruss, D.; Judkins, T.; Scholl, T.; Bekessy, A.; Marsh, A.; Lovelock, P.; Wong, M.; Tesoriero, A.; Renard, H.; Southey, M.; Hopper, J.L.; Yannoukakos, K.; Brown, M.; Easton, D.; Tavtigian, S.V.; Goldgar, D.; Spurdle, A.B. Genetic and histopathologic evaluation of brca1 and brca2 DNA sequence variants of unknown clinical significance. Cancer Res. 2006, 66, 2019–2027. [Google Scholar] [CrossRef]
- Carvalho, M.A.; Marsillac, S.M.; Karchin, R.; Manoukian, S.; Grist, S.; Swaby, R.F.; Urmenyi, T.P.; Rondinelli, E.; Silva, R.; Gayol, L.; Baumbach, L.; Sutphen, R.; Pickard-Brzosowicz, J.L.; Nathanson, K.L.; Sali, A.; Goldgar, D.; Couch, F.J.; Radice, P.; Monteiro, A.N. Determination of cancer risk associated with germ line brca1 missense variants by functional analysis. Cancer Res. 2007, 67, 1494–1501. [Google Scholar] [CrossRef]
- Couch, F.J.; Rasmussen, L.J.; Hofstra, R.; Monteiro, A.N.; Greenblatt, M.S.; de Wind, N. Assessment of functional effects of unclassified genetic variants. Hum. Mutat. 2008, 29, 1314–1326. [Google Scholar] [CrossRef]
- Nanda, R.; Schumm, L.P.; Cummings, S.; Fackenthal, J.D.; Sveen, L.; Ademuyiwa, F.; Cobleigh, M.; Esserman, L.; Lindor, N.M.; Neuhausen, S.L.; Olopade, O.I. Genetic testing in an ethnically diverse cohort of high-risk women: A comparative analysis of brca1 and brca2 mutations in american families of european and african ancestry. JAMA 2005, 294, 1925–1933. [Google Scholar] [CrossRef]
- Weitzel, J.N.; Lagos, V.; Blazer, K.R.; Nelson, R.; Ricker, C.; Herzog, J.; McGuire, C.; Neuhausen, S. Prevalence of brca mutations and founder effect in high-risk hispanic families. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 1666–1671. [Google Scholar] [CrossRef]
- Judkins, T.; Hendrickson, B.C.; Deffenbaugh, A.M.; Scholl, T. Single nucleotide polymorphisms in clinical genetic testing: The characterization of the clinical significance of genetic variants and their application in clinical research for brca1. Mutat. Res. 2005, 573, 168–179. [Google Scholar] [CrossRef]
- Farrugia, D.J.; Agarwal, M.K.; Pankratz, V.S.; Deffenbaugh, A.M.; Pruss, D.; Frye, C.; Wadum, L.; Johnson, K.; Mentlick, J.; Tavtigian, S.V.; Goldgar, D.E.; Couch, F.J. Functional assays for classification of brca2 variants of uncertain significance. Cancer Res. 2008, 68, 3523–3531. [Google Scholar] [CrossRef]
- Goldgar, D.E.; Easton, D.F.; Byrnes, G.B.; Spurdle, A.B.; Iversen, E.S.; Greenblatt, M.S. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum. Mutat. 2008, 29, 1265–1272. [Google Scholar] [CrossRef]
- Greenblatt, M.S.; Brody, L.C.; Foulkes, W.D.; Genuardi, M.; Hofstra, R.M.; Olivier, M.; Plon, S.E.; Sijmons, R.H.; Sinilnikova, O.; Spurdle, A.B. Locus-specific databases and recommendations to strengthen their contribution to the classification of variants in cancer susceptibility genes. Hum. Mutat. 2008, 29, 1273–1281. [Google Scholar] [CrossRef]
- Hofstra, R.M.; Spurdle, A.B.; Eccles, D.; Foulkes, W.D.; de Wind, N.; Hoogerbrugge, N.; Hogervorst, F.B. Tumor characteristics as an analytic tool for classifying genetic variants of uncertain clinical significance. Hum. Mutat. 2008, 29, 1292–1303. [Google Scholar] [CrossRef]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Couch, F.J.; Hogervorst, F.B.; Radice, P.; Sinilnikova, O.M. Prediction and assessment of splicing alterations: Implications for clinical testing. Hum. Mutat. 2008, 29, 1304–1313. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Greenblatt, M.S.; Lesueur, F.; Byrnes, G.B. In silico analysis of missense substitutions using sequence-alignment based methods. Hum. Mutat. 2008, 29, 1327–1336. [Google Scholar] [CrossRef]
- Sweet, K.; Senter, L.; Pilarski, R.; Wei, L.; Toland, A.E. Characterization of brca1 ring finger variants of uncertain significance. Breast Cancer Res. Treat. 119, 737–743.
- Bonatti, F.; Pepe, C.; Tancredi, M.; Lombardi, G.; Aretini, P.; Sensi, E.; Falaschi, E.; Cipollini, G.; Bevilacqua, G.; Caligo, M.A. Rna-based analysis of brca1 and brca2 gene alterations. Cancer Genet. Cytogenet. 2006, 170, 93–101. [Google Scholar] [CrossRef]
- Ferla, R.; Calo, V.; Cascio, S.; Rinaldi, G.; Badalamenti, G.; Carreca, I.; Surmacz, E.; Colucci, G.; Bazan, V.; Russo, A. Founder mutations in brca1 and brca2 genes. Ann. Oncol. 2007, 18 (Suppl. 6), 93–98. [Google Scholar]
- Russo, A.; Calo, V.; Augello, C.; Bruno, L.; Agnese, V.; Schiro, V.; Barbera, F.; Cascio, S.; Foddai, E.; Badalamenti, G.; Intrivici, C.; Cajozzo, M.; Gulotta, G.; Surmacz, E.; Colucci, G.; Gebbia, N.; Bazan, V. 4843delc of the brca1 gene is a possible founder mutation in southern italy (sicily). Ann. Oncol. 2007, 18 (Suppl. 6), 99–102. [Google Scholar]
- Russo, A.; Calo, V.; Bruno, L.; Schiro, V.; Agnese, V.; Cascio, S.; Foddai, E.; Fanale, D.; Rizzo, S.; Di Gaudio, F.; Gulotta, E.; Surmacz, E.; Di Fede, G.; Bazan, V. Is brca1-5083del19, identified in breast cancer patients of sicilian origin, a calabrian founder mutation? Breast Cancer Re. Treat. 2009, 113, 67–70. [Google Scholar] [CrossRef]
- Tang, T.S.; Solomon, L.J.; Yeh, C.J.; Worden, J.K. The role of cultural variables in breast self-examination and cervical cancer screening behavior in young asian women living in the united states. J. Behav. Med. 1999, 22, 419–436. [Google Scholar] [CrossRef]
- Boyd, J. Brca1: More than a hereditary breast cancer gene? Nat. Genet. 1995, 9, 335–336. [Google Scholar] [CrossRef]
- Kuschel, B.; Gayther, S.A.; Easton, D.F.; Ponder, B.A.; Pharoah, P.D. Apparent human brca1 knockout caused by mispriming during polymerase chain reaction: Implications for genetic testing. Genes Chromosomes Cancer 2001, 31, 96–98. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Samollow, P.B.; de Silva, D.; Thomas, A. An analysis of unclassified missense substitutions in human brca1. Fam. Cancer 2006, 5, 77–88. [Google Scholar] [CrossRef]
- Judkins, T.; Schwensen, C.; Hendrickson, B.C.; Harpending, H.C.; Barrus, J.; Scholl, T. Automated haplotyping in BRCA1 and localization of polymorphisms on alleles. Am. J. Hum. Genet. 2003, 73, 409. [Google Scholar]
- Fleming, M.A.; Potter, J.D.; Ramirez, C.J.; Ostrander, G.K.; Ostrander, E.A. Understanding missense mutations in the brca1 gene: An evolutionary approach. Proc. Natl. Acad. Sci. USA 2003, 100, 1151–1156. [Google Scholar]
- Ramirez, C.J.; Fleming, M.A.; Potter, J.D.; Ostrander, G.K.; Ostrander, E.A. Marsupial brca1: Conserved regions in mammals and the potential effect of missense changes. Oncogene 2004, 23, 1780–1788. [Google Scholar] [CrossRef]
- Abkevich, V.; Zharkikh, A.; Deffenbaugh, A.M.; Frank, D.; Chen, Y.; Shattuck, D.; Skolnick, M.H.; Gutin, A.; Tavtigian, S.V. Analysis of missense variation in human brca1 in the context of interspecific sequence variation. J. Med. Genet. 2004, 41, 492–507. [Google Scholar] [CrossRef]
- Spearman, A.D.; Sweet, K.; Zhou, X.P.; McLennan, J.; Couch, F.J.; Toland, A.E. Clinically applicable models to characterize brca1 and brca2 variants of uncertain significance. J. Clin. Oncol. 2008, 26, 5393–5400. [Google Scholar] [CrossRef]
- Grantham, R. Amino acid difference formula to help explain protein evolution. Science 1974, 185, 862–864. [Google Scholar]
- Osorio, A.; de la Hoya, M.; Rodriguez-Lopez, R.; Martinez-Ramirez, A.; Cazorla, A.; Granizo, J.J.; Esteller, M.; Rivas, C.; Caldes, T.; Benitez, J. Loss of heterozygosity analysis at the brca loci in tumor samples from patients with familial breast cancer. Int. J. Cancer 2002, 99, 305–309. [Google Scholar] [CrossRef]
- Osorio, A.; Milne, R.L.; Honrado, E.; Barroso, A.; Diez, O.; Salazar, R.; de la Hoya, M.; Vega, A.; Benitez, J. Classification of missense variants of unknown significance in brca1 based on clinical and tumor information. Hum. Mutat. 2007, 28, 477–485. [Google Scholar] [CrossRef]
- Brzovic, P.S.; Keeffe, J.R.; Nishikawa, H.; Miyamoto, K.; Fox, D., 3rd; Fukuda, M.; Ohta, T.; Klevit, R. Binding and recognition in the assembly of an active brca1/bard1 ubiquitin-ligase complex. Proc. Natl. Acad. Sci. USA 2003, 100, 5646–5651. [Google Scholar] [CrossRef]
- Reid, L.J.; Shakya, R.; Modi, A.P.; Lokshin, M.; Cheng, J.T.; Jasin, M.; Baer, R.; Ludwig, T. E3 ligase activity of brca1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc. Natl. Acad. Sci. USA 2008, 105, 20876–20881. [Google Scholar]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The ring heterodimer brca1-bard1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001, 276, 14537–14540. [Google Scholar]
- Morris, J.R.; Pangon, L.; Boutell, C.; Katagiri, T.; Keep, N.H.; Solomon, E. Genetic analysis of brca1 ubiquitin ligase activity and its relationship to breast cancer susceptibility. Hum. Mol. Genet. 2006, 15, 599–606. [Google Scholar] [CrossRef]
- Hayes, F.; Cayanan, C.; Barilla, D.; Monteiro, A.N. Functional assay for brca1: Mutagenesis of the cooh-terminal region reveals critical residues for transcription activation. Cancer Res. 2000, 60, 2411–2418. [Google Scholar]
- Phelan, C.M.; Dapic, V.; Tice, B.; Favis, R.; Kwan, E.; Barany, F.; Manoukian, S.; Radice, P.; van der Luijt, R.B.; van Nesselrooij, B.P.; Chenevix-Trench, G.; kConFab; Caldes, T.; de la Hoya, M.; Lindquist, S.; Tavtigian, S.V.; Goldgar, D.; Borg, A.; Narod, S.A.; Monteiro, A.N. Classification of brca1 missense variants of unknown clinical significance. J. Med. Genet. 2005, 42, 138–146. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. Brca2 is required for homology-directed repair of chromosomal breaks. Mol. Cell. 2001, 7, 263–272. [Google Scholar] [CrossRef]
- Wu, K.; Hinson, S.R.; Ohashi, A.; Farrugia, D.; Wendt, P.; Tavtigian, S.V.; Deffenbaugh, A.; Goldgar, D.; Couch, F.J. Functional evaluation and cancer risk assessment of brca2 unclassified variants. Cancer Res. 2005, 65, 417–426. [Google Scholar]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of brca2 cellular and clinical function by a nuclear partner, palb2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Stefansson, I.M.; Chappuis, P.O.; Begin, L.R.; Goffin, J.R.; Wong, N.; Trudel, M.; Akslen, L.A. Germline brca1 mutations and a basal epithelial phenotype in breast cancer. J. Natl. Cancer Inst. 2003, 95, 1482–1485. [Google Scholar] [CrossRef]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of 'brcaness' in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef]
- Bane, A.L.; Beck, J.C.; Bleiweiss, I.; Buys, S.S.; Catalano, E.; Daly, M.B.; Giles, G.; Godwin, A.K.; Hibshoosh, H.; Hopper, J.L.; John, E.M.; Layfield, L.; Longacre, T.; Miron, A.; Senie, R.; Southey, M.C.; West, D.W.; Whittemore, A.S.; Wu, H.; Andrulis, I.L.; O'Malley, F.P. Brca2 mutation-associated breast cancers exhibit a distinguishing phenotype based on morphology and molecular profiles from tissue microarrays. Am. J. Surg. Pathol. 2007, 31, 121–128. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Metcalfe, K.; Sun, P.; Hanna, W.M.; Lynch, H.T.; Ghadirian, P.; Tung, N.; Olopade, O.I.; Weber, B.L.; McLennan, J.; Olivotto, I.A.; Begin, L.R.; Narod, S.A. Estrogen receptor status in brca1- and brca2-related breast cancer: The influence of age, grade, and histological type. Clin. Cancer. Res. 2004, 10, 2029–2034. [Google Scholar] [CrossRef]
- Fitch, W.M. Distinguishing homologous from analogous proteins. Syst. Zool. 1970, 19, 99–113. [Google Scholar] [CrossRef]
- Vallon-Christersson, J.; Cayanan, C.; Haraldsson, K.; Loman, N.; Bergthorsson, J.T.; Brondum-Nielsen, K.; Gerdes, A.M.; Moller, P.; Kristoffersson, U.; Olsson, H.; Borg, A.; Monteiro, A.N. Functional analysis of brca1 c-terminal missense mutations identified in breast and ovarian cancer families. Hum. Mol. Genet. 2001, 10, 353–360. [Google Scholar] [CrossRef]
- Capriotti, E.; Arbiza, L.; Casadio, R.; Dopazo, J.; Dopazo, H.; Marti-Renom, M.A. Use of estimated evolutionary strength at the codon level improves the prediction of disease-related protein mutations in humans. Hum. Mutat. 2008, 29, 198–204. [Google Scholar] [CrossRef]
- Chan, P.A.; Duraisamy, S.; Miller, P.J.; Newell, J.A.; McBride, C.; Bond, J.P.; Raevaara, T.; Ollila, S.; Nystrom, M.; Grimm, A.J.; Christodoulou, J.; Oetting, W.S.; Greenblatt, M.S. Interpreting missense variants: Comparing computational methods in human disease genes cdkn2a, mlh1, msh2, mecp2, and tyrosinase (tyr). Hum. Mutat. 2007, 28, 683–693. [Google Scholar] [CrossRef]
- Tavtigian, S.V.; Byrnes, G.B.; Goldgar, D.E.; Thomas, A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum. Mutat. 2008, 29, 1342–1354. [Google Scholar] [CrossRef]
- Yang. Likelihood ratio tests for detecting positive selection and application to primate lysozyme evolution. Mol. Biol. Evol. 1998, 15, 568–573. [Google Scholar] [CrossRef]
- Lakhani, S.R.; Gusterson, B.A.; Jacquemier, J.; Sloane, J.P.; Anderson, T.J.; van de Vijver, M.J.; Venter, D.; Freeman, A.; Antoniou, A.; McGuffog, L.; Smyth, E.; Steel, C.M.; Haites, N.; Scott, R.J.; Goldgar, D.; Neuhausen, S.; Daly, P.A.; Ormiston, W.; McManus, R.; Scherneck, S.; Ponder, B.A.; Futreal, P.A.; Peto, J.; Stoppa-Lyonnet, D.; Bignon, Y.J.; Stratton, M.R. The pathology of familial breast cancer: Histological features of cancers in families not attributable to mutations in brca1 or brca2. Clin. Cancer Res. 2000, 6, 782–789. [Google Scholar]
- Lakhani, S.R.; Reis-Filho, J.S.; Fulford, L.; Penault-Llorca, F.; van der Vijver, M.; Parry, S.; Bishop, T.; Benitez, J.; Rivas, C.; Bignon, Y.J.; Chang-Claude, J.; Hamann, U.; Cornelisse, C.J.; Devilee, P.; Beckmann, M.W.; Nestle-Kramling, C.; Daly, P.A.; Haites, N.; Varley, J.; Lalloo, F.; Evans, G.; Maugard, C.; Meijers-Heijboer, H.; Klijn, J.G.; Olah, E.; Gusterson, B.A.; Pilotti, S.; Radice, P.; Scherneck, S.; Sobol, H.; Jacquemier, J.; Wagner, T.; Peto, J.; Stratton, M.R.; McGuffog, L.; Easton, D.F. Prediction of brca1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin. Cancer Res. 2005, 11, 5175–5180. [Google Scholar] [CrossRef]
- Roussi, P.; Sherman, K.A.; Miller, S.; Buzaglo, J.; Daly, M.; Taylor, A.; Ross, E.; Godwin, A. Enhanced counselling for women undergoing brca1/2 testing: Impact on knowledge and psychological distress–results from a randomised clinical trial. Psychol. Health 2009, 25, 401–415. [Google Scholar]
- Granader, E.J.; Dwamena, B.; Carlos, R.C. Mri and mammography surveillance of women at increased risk for breast cancer: Recommendations using an evidence-based approach. Acad. Radiol. 2008, 15, 1590–1595. [Google Scholar] [CrossRef]
- Metcalfe, K.A.; Finch, A.; Poll, A.; Horsman, D.; Kim-Sing, C.; Scott, J.; Royer, R.; Sun, P.; Narod, S.A. Breast cancer risks in women with a family history of breast or ovarian cancer who have tested negative for a brca1 or brca2 mutation. Br. J. Cancer 2009, 100, 421–425. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Calò, V.; Bruno, L.; La Paglia, L.; Perez, M.; Margarese, N.; Di Gaudio, F.; Russo, A. The Clinical Significance of Unknown Sequence Variants in BRCA Genes. Cancers 2010, 2, 1644-1660. https://doi.org/10.3390/cancers2031644
Calò V, Bruno L, La Paglia L, Perez M, Margarese N, Di Gaudio F, Russo A. The Clinical Significance of Unknown Sequence Variants in BRCA Genes. Cancers. 2010; 2(3):1644-1660. https://doi.org/10.3390/cancers2031644
Chicago/Turabian StyleCalò, Valentina, Loredana Bruno, Laura La Paglia, Marco Perez, Naomi Margarese, Francesca Di Gaudio, and Antonio Russo. 2010. "The Clinical Significance of Unknown Sequence Variants in BRCA Genes" Cancers 2, no. 3: 1644-1660. https://doi.org/10.3390/cancers2031644
APA StyleCalò, V., Bruno, L., La Paglia, L., Perez, M., Margarese, N., Di Gaudio, F., & Russo, A. (2010). The Clinical Significance of Unknown Sequence Variants in BRCA Genes. Cancers, 2(3), 1644-1660. https://doi.org/10.3390/cancers2031644