A Quest for Initiating Cells of Head and Neck Cancer and Their Treatment


Abstract
:1. Introduction
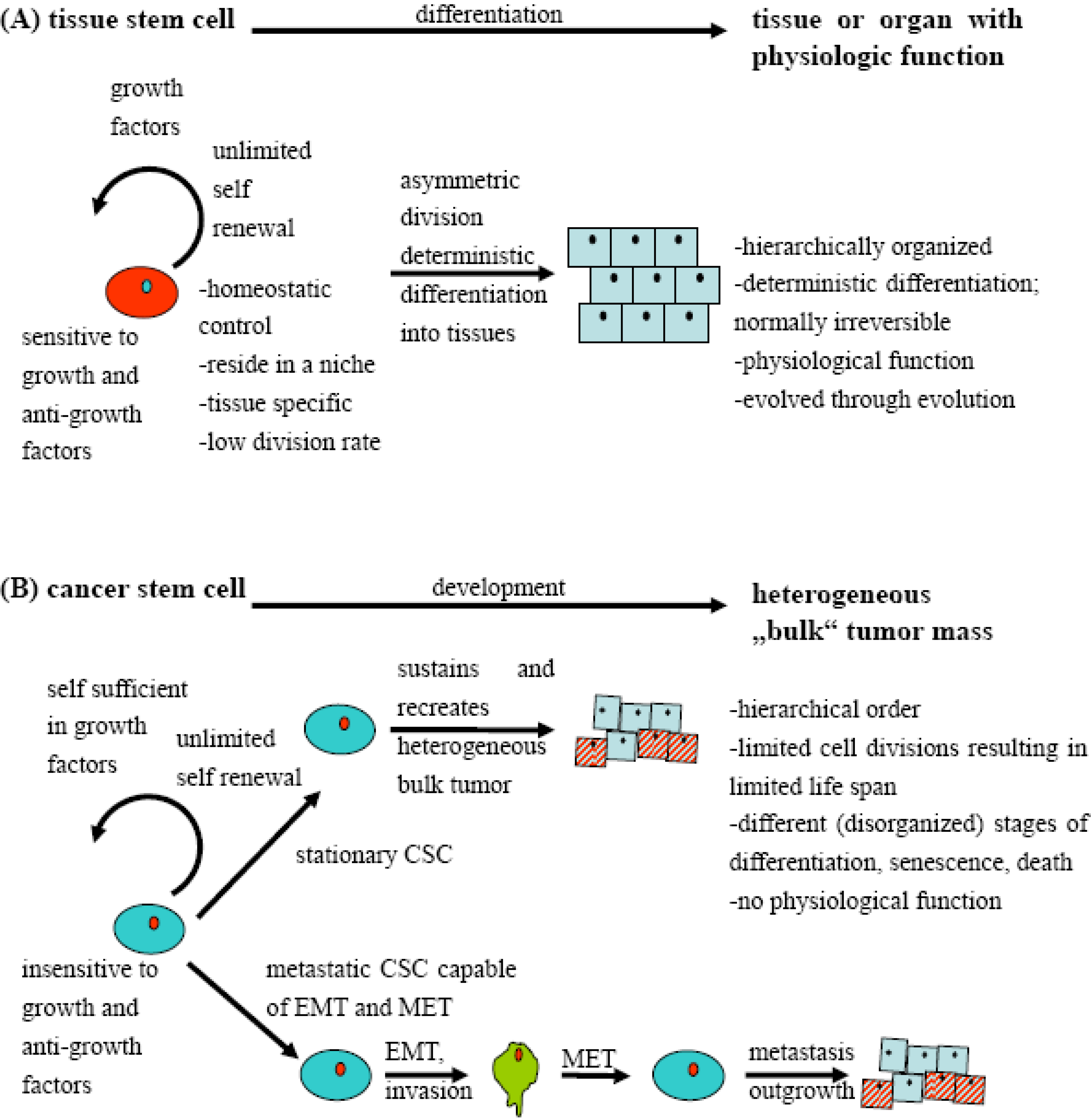
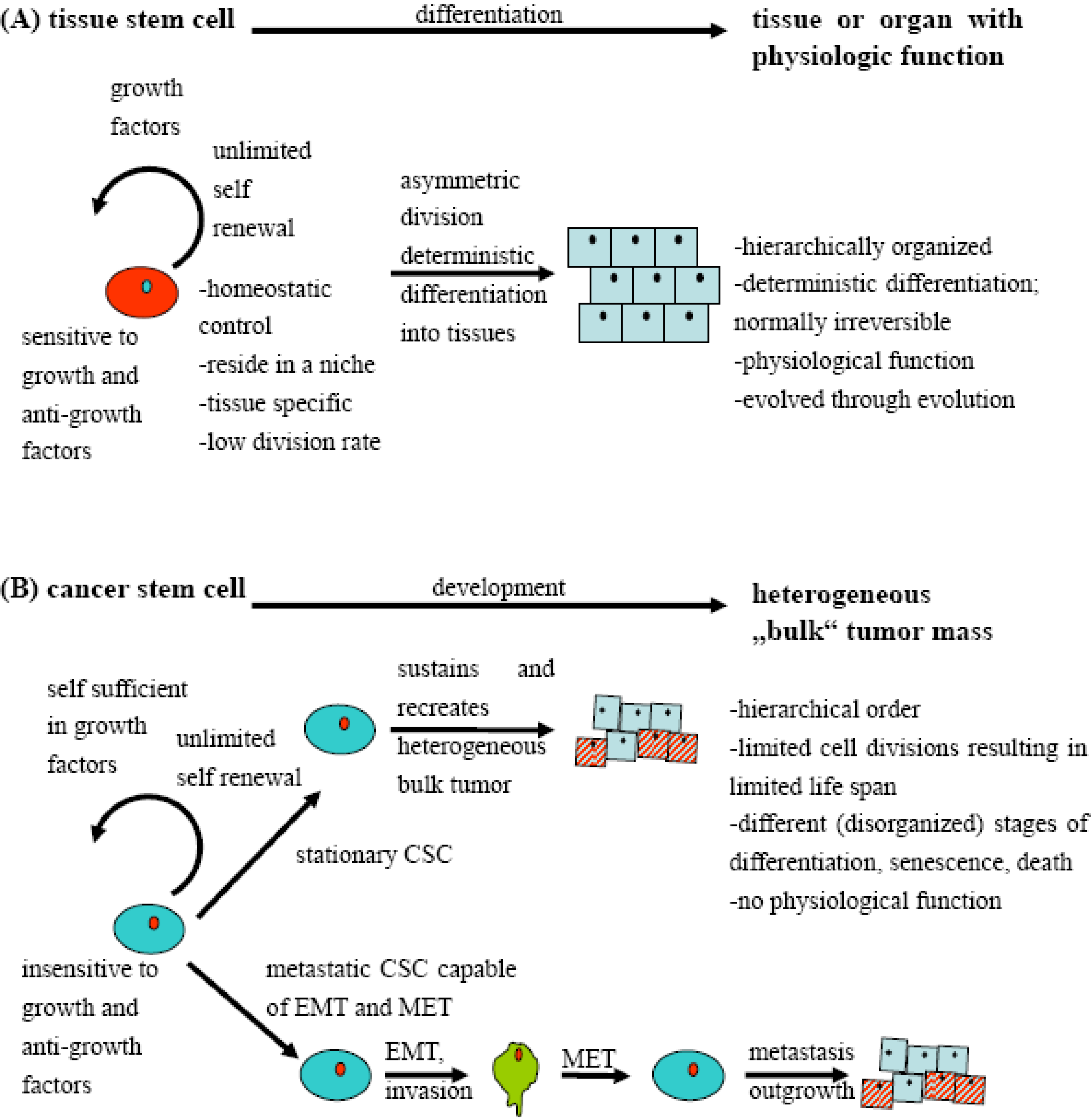
2. Definition of Cancer Stem Cell-Like Cells
3. Dichotomy between Hierarchical and Stochastical Tumor Models?
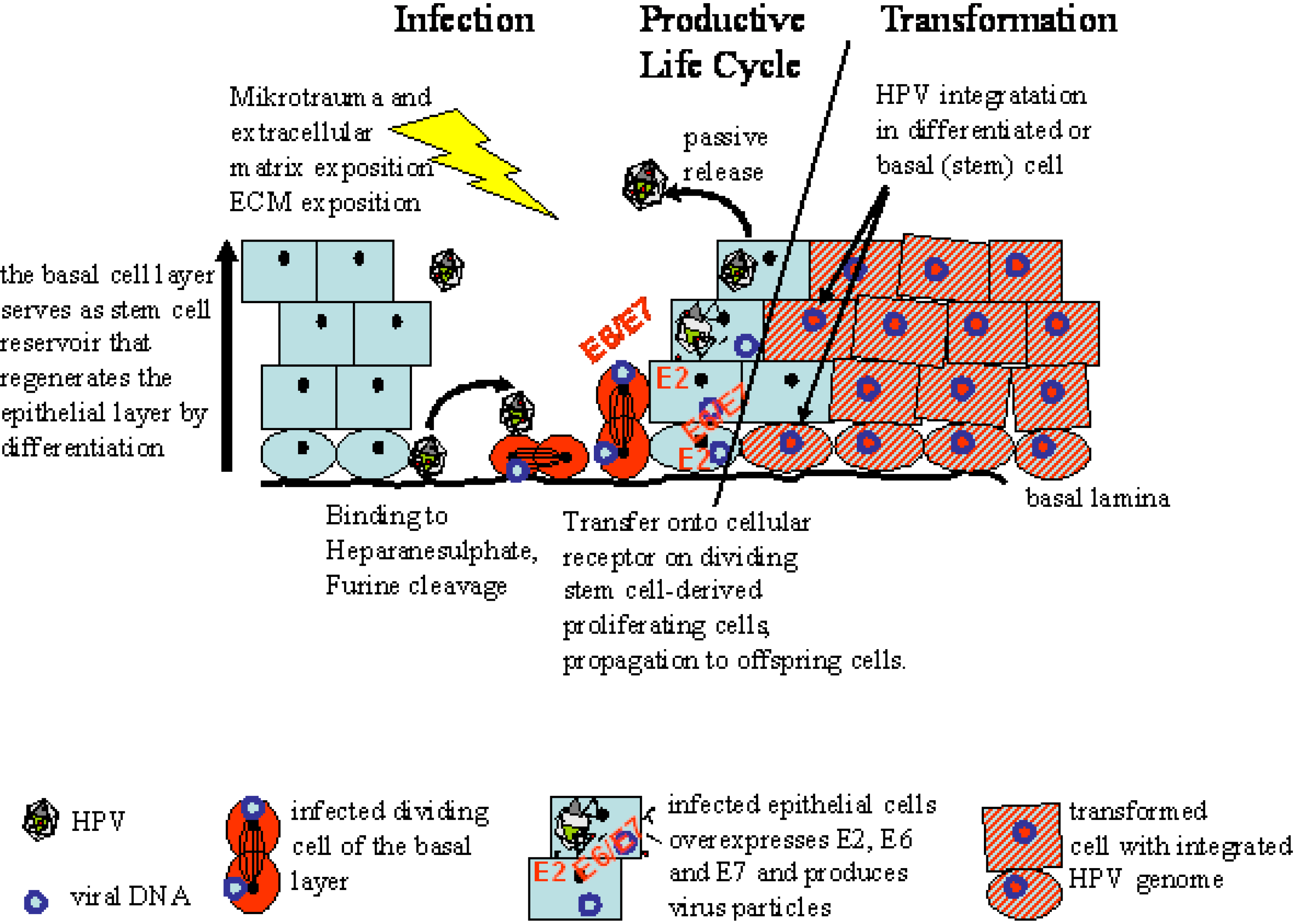
4. Assays for CSC Research
4.1. The Xenotransplant Tumor Initiation Assay
4.2. Genetic Lineage Tracking
4.3. Non-Adherent Sphere Formation Assays
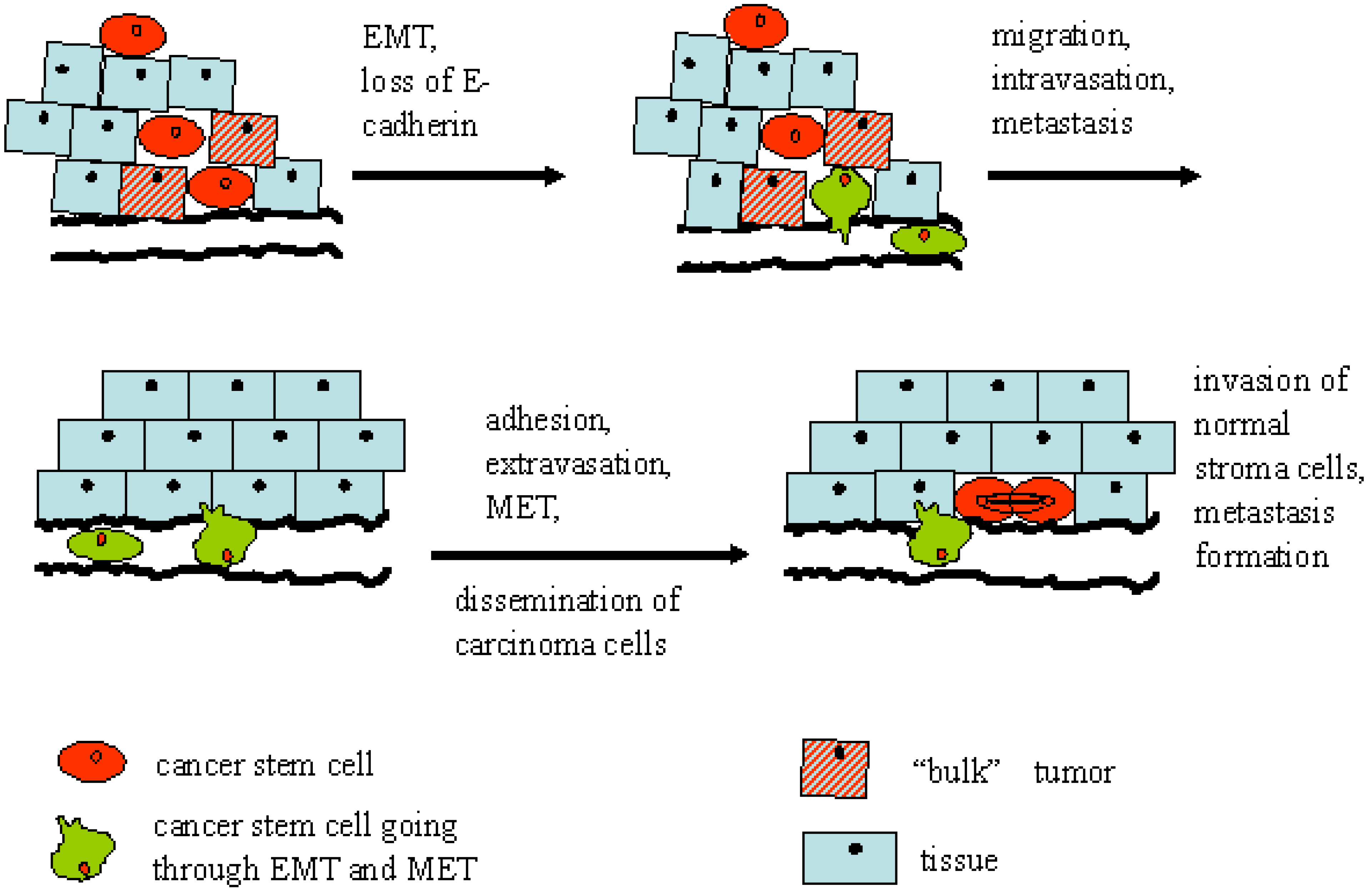
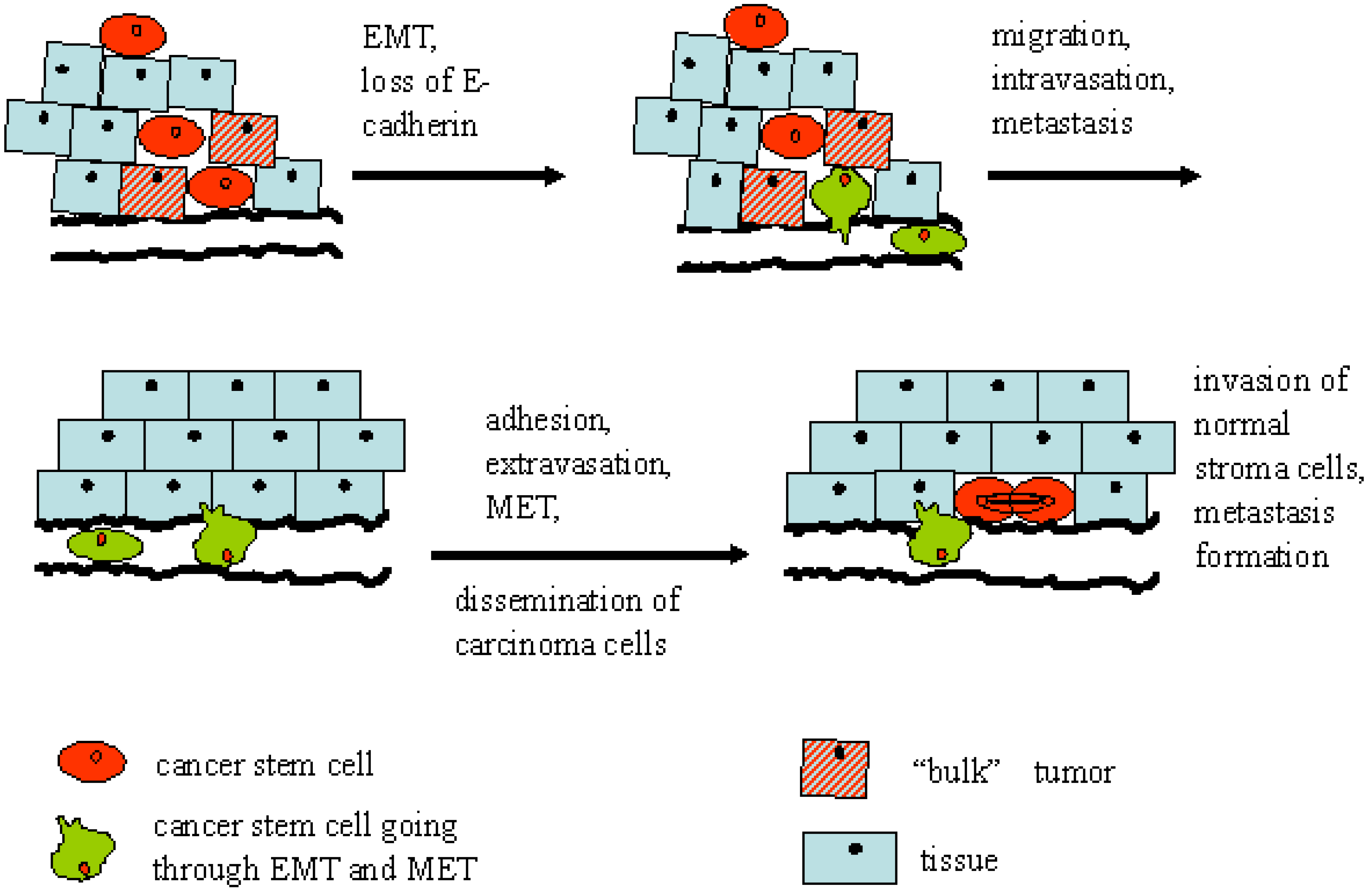
5. The Role of Epithelial-Mesenchymal Transition
6. Potential Markers of CSC in Head and Neck Cancer and Their Relevance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CSC marker | Origin | Function/physiological role | Ref. |
|---|---|---|---|
| CD44+/CD24−/low (ALDH1+) | HNSCC, breast, lung, hepatoma | CD44: A cell-surface glycoprotein involved in cell-cell interaction, cell migration, and adhesion with multiple isoforms that has pleiotropic roles in signaling and homing. The standard form CD44H exhibits a high affinity for hyaluronate; CD44V confers metastatic properties. Several CD44 splice variants are known as being associated with cell transformation. The standard form of CD44 (CD44s) was shown to be part of the signature of cancer stem cells (CSC) in colon, breast, and in head and neck squamous cell carcinomas (HNSCC). This is somewhat in contradiction to previous reports on the expression of CD44s in HNSCC. CD24: A cell adhesion molecule expressed at the surface of most B cells and differentiating neuroblasts. | [14,60,61,62,63,64,65,66] |
| CD44+ Lineage- | HNSCC (controversial) | [64,66] | |
| CD44+/EpCAMhi | Colon | EpCAM: Homophillic Ca2+-independent cell adhesion molecule expressed on the basolateral surfaces of most epithelial cells. | [67] |
| CD44+/CD24−/ESA+ | Pancreas | [68] | |
| ALDH1+ | HNSCC, Breast | ALDH1: A member of the ubiquitous aldehyde dehydrogenase (ALDH) family of cytosolic enzymes that catalyse the oxidation of aliphatic and aromatic aldehydes to carboxylic acids. ALDH1 has a role in the conversion of retinol to retinoic acid, which is important for proliferation, differentiation and survival. Furthermore, ALDH1 enzymatic activity has been identified as responsible for the resistance of progenitor cells to chemotherapeutic agents. | [14,63,69,70] |
| CD133+ | HNSCC, CNS, colon, Ewing’s sarkoma, pancreas, lung, liver | CD133 (Promenin 1): A pentaspan transmembrane glycoprotein domain expressed in several stem cell populations and cancers. Possible role in the organization of plasma membrane topology.Expressed on CD34+ stem and progenitor cells in fetal liver, endothelial precursors, fetal neural stem cells, and developing epithelium. CD133 has been detected by its glycosylated epitope in the majority of studies. Thus, CD133 may be a more reliable cancer stem cell marker | [36,71,72,73,74,75,76] |
| Side population (Hoechst dye) | Mesenchymal | Side population (descriptive term derived from flow-cytometry experiments): Phenotype due to the Hoechst33342 efflux pump present on the plasma membrane in diverse cell types. Activity conferred by the ABC transporter ABCG2. | [77] |
| ABCG5+ | Melanoma | ABCG5+: Member of the ATP binding cassette family, involved in transport of sterol and other lipids. ABCG2 (also known as breast cancer resistance protein) is a multi-drug transporter (see Hoechst SP below). ABCG5 confers doxorubicin resistance. | [37] |
| Snail | HNSCC | Snail: Transcriptional repressor upregulated in EMT and modulated by IL-1beta. Regulates COX-2-dependent E-cadherin expression | [63,78] |
| Twist | HNSCC | Twist: Transcription factor during embryonic development and has recently been found to promote the EMT phenomenon seen during the initial steps of tumor metastasis in various cancers. It regulates the expression of several genes involved in differentiation, adhesion and proliferation. | [59,79,80] |
| Oct-4 | HNSCC, embryonic stem cells (ES), many others | Oct-4: Transcription factor expressed in pluripotent embryonic stem (ES) and germ cells. Oct-4 mRNA is normally found in totipotent and pluripotent stem cells of embryos. Knocking out the Oct-4 gene in mice causes early lethality due to the lack of inner cell mass formation, indicating that Oct-4 has a critical function for self-renewal of ES cells. Oct-4 activates transcription via octamer motifs, and Oct-4 binding sites have been found in various genes fibroblast growth factor 4 and platelet-derived growth factor α receptor. This suggests that Oct-4 functions as a master switch during differentiation by regulating the pluripotent potentials of the stem cell, and Oct-4 plays a pivotal role in mammalian development. | [71,81] |
| SOX2 | ES, many others | The transcription factor SOX2 is essential for maintaining the pluripotent phenotype of ES cells and is a partner of Oct 3/4 in regulating several ES cell-specific genes. Oct 3/4 and SOX2 interact specifically and bind to a composite regulatory element. Activation of this element maintains Oct 3/4 and SOX2 expression in pluripotent cells. | [82] |
| Nanog | HNSCC, ES, many others | Nanog, like Sox2 and Oct4, is a transcription factor essential to maintaining the pluripotent ES cell phenotype. Through a cooperative interaction, Sox2 and Oct4 have been described to drive pluripotent-specific expression of a number of genes. | [71,81,82] |
7. Cancer Stem Cells—adiation and Chemotherapy
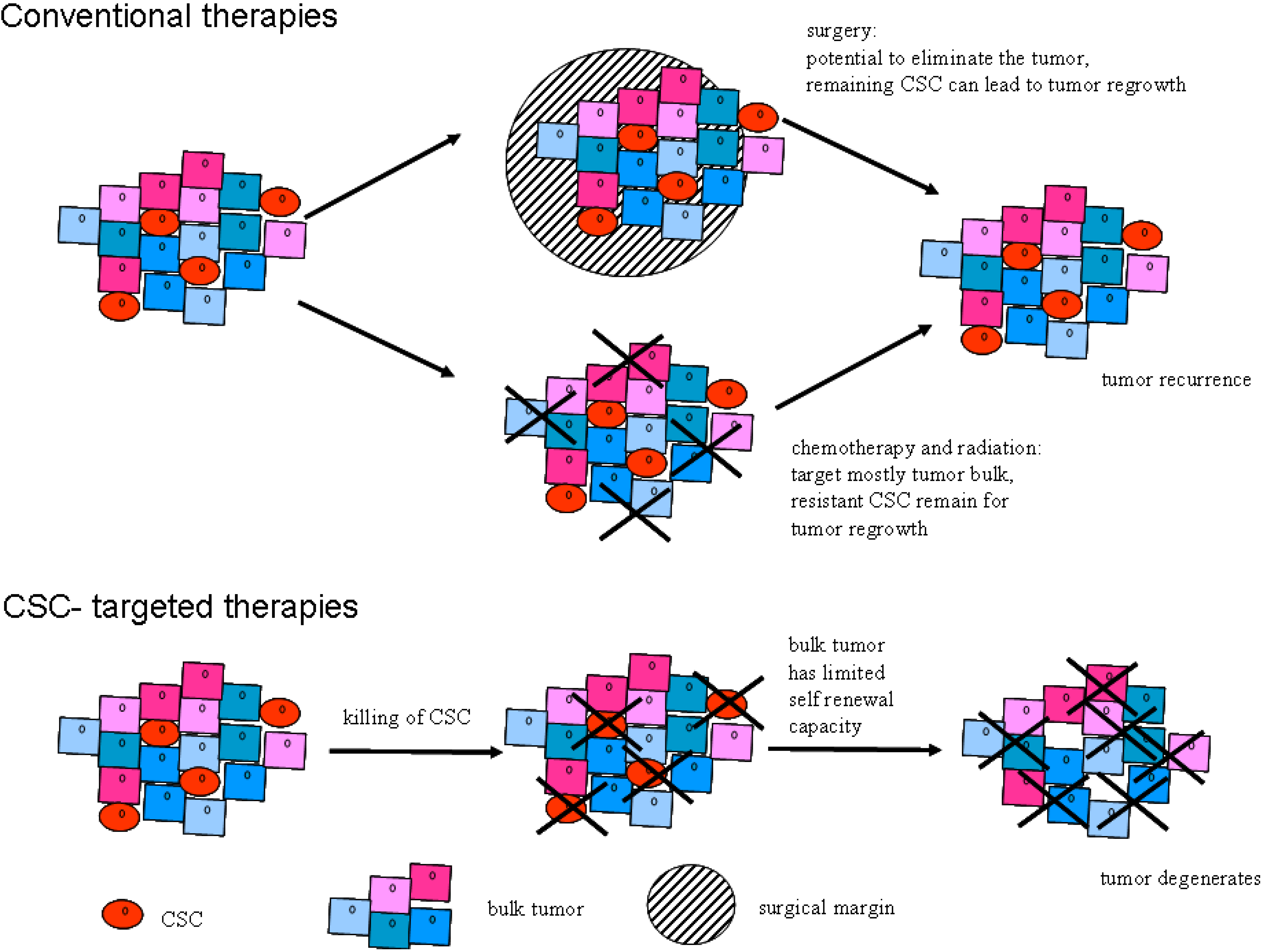
8. Clinical Implications of CSC
| Mode | Target | Tissue | Ref. |
|---|---|---|---|
| Antigen specific immunotherapy | - Dendritic cells loaded with CSC as antigen source - CD8 defined ALDH1-specific epitope | Glioblastoma, HNSCC | [101,102] |
| Knockdown of BMI-1 gene expression by siRNA | Bmi-1, a member of the Polycomb family of transcriptional repressors that mediate gene silencing by regulating chromatin structure. BMI-1 is essential for maintaining the self-renewal abilities of adult stem cells and CSC. | HNSCC | [105] |
| Combined genetic knockdown of Snail and radiochemotherapy | Snail induces EMT, which converts epithelial cells into migratory mesenchymal cells by repressing E-cadherin, desmoplakin, and cytokeratin 18, while its expression is associated with enhanced vimentin and fibronectin production. | HNSCC | [37,63,106,107] |
| Antibody-based target immunotherapy | - Anti-CD133 antibody-drug conjugates (ADCs) -anti-interleukin-3 (IL-3) receptor alpha chain (CD123)-neutralizing antibody - antiCD44a6 - Anti-ABCB5 | - HNSCC hepatocellular and gastric cancers - AML in a SCID mouse model - melanoma | [37,108,109] |
| Modulation of CSC differentiation | Bone morphogenetic proteins (BMPs) induced differentiation of CD133+ brain tumor stem cells, weakening their tumor-forming ability. | glioblastoma | [110] |
9. Conclusions
References
- Greenlee, R.T.; Hill-Harmon, M.B.; Murray, T.; Thun, M. Cancer statistics, 2001. CA Cancer J. Clin. 2001, 51, 15–36. [Google Scholar] [CrossRef]
- Jones, A.S.; Morar, P.; Phillips, D.E.; Field, J.K.; Husband, D.; Helliwell, T.R. Second primary tumors in patients with head and neck squamous cell carcinoma. Cancer 1995, 75, 1343–1353. [Google Scholar] [CrossRef]
- Leemans, C.R.; Tiwari, R.; Nauta, J.J.; van der Waal, I.; Snow, G.B. Recurrence at the primary site in head and neck cancer and the significance of neck lymph node metastases as a prognostic factor. Cancer 1994, 73, 187–190. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumors: Accumulating evidence and unresolved questions. Nat. Rev. 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Gillison, M.L.; Shah, K.V. Human papillomavirus-associated head and neck squamous cell carcinoma: Mounting evidence for an etiologic role for human papillomavirus in a subset of head and neck cancers. Curr. Opin. Oncol. 2001, 13, 183–188. [Google Scholar] [CrossRef]
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; Shah, K.V.; Sidransky, D. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst. 2000, 92, 709–720. [Google Scholar] [CrossRef]
- Smith, E.M.; Summersgill, K.F.; Allen, J.; Hoffman, H.T.; McCulloch, T.; Turek, L.P.; Haugen, T.H. Human papillomavirus and risk of laryngeal cancer. Ann. Otol. Rhinol. Laryngol. 2000, 109, 1069–1076. [Google Scholar]
- Hobbs, C.G.; Sterne, J.A.; Bailey, M.; Heyderman, R.S.; Birchall, M.A.; Thomas, S.J. Human papillomavirus and head and neck cancer: A systematic review and meta-analysis. Clin. Otolaryngol. 2006, 31, 259–266. [Google Scholar] [CrossRef]
- Martens, J.E.; Arends, J.; van der Linden, P.J.; De Boer, B.A.; Helmerhorst, T.J. Cytokeratin 17 and p63 are markers of the hpv target cell, the cervical stem cell. Anticancer Res. 2004, 24, 771–775. [Google Scholar]
- zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef]
- Kennedy, J.A.; Barabe, F.; Poeppl, A.G.; Wang, J.C.; Dick, J.E. Comment on "Tumor growth need not be driven by rare cancer stem cells". Science 2007, 318, 1722, author reply 1722. [Google Scholar]
- Ginestier, C.; Min, H.M.; Charafe-Jauffretl, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.L.; Schott, A.; Hayes, D.; Birnbaum, D.; Wicha, M.S.; Dontu, D. Aldh1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar] [CrossRef]
- Bonnet, D.D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into scid mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Schatton, T.; Frank, N.Y.; Frank, M.H. Identification and targeting of cancer stem cells. Bioessays 2009, 31, 1038–1049. [Google Scholar] [CrossRef]
- Fidler, I.J.; Hart, I.R. Biological diversity in metastatic neoplasms: Origins and implications. Science 1982, 217, 998–1003. [Google Scholar]
- Harper, L.J.; Piper, K.; Common, J.; Fortune, F.; Mackenzie, I.C. Stem cell patterns in cell lines derived from head and neck squamous cell carcinoma. J. Oral. Pathol. Med. 2007, 36, 594–603. [Google Scholar] [CrossRef]
- Hamburger, A.W.; Salmon, S.E. Primary bioassay of human tumor stem cells. Science 1977, 197, 461–463. [Google Scholar]
- Sabbath, K.D.; Ball, E.D.; Larcom, P.; Davis, R.B.; Griffin, J.D. Heterogeneity of clonogenic cells in acute myeloblastic leukemia. J. Clin. Invest. 1985, 75, 746–753. [Google Scholar] [CrossRef]
- Griffin, J.D.; Lowenberg, B. Clonogenic cells in acute myeloblastic leukemia. Blood 1986, 68, 1185–1195. [Google Scholar] [Green Version]
- Maenhaut, C.; Dumont, J.E.; Roger, P.; van Staveren, W.C. Cancer stem cells : A reality, a myth, a fuzzy concept or a misnomer? An analysis. Carcinogenesis 2010, 31, 149–158. [Google Scholar] [CrossRef]
- Welte, Y.; Adjaye, J.; Lehrach, H.R.; Regenbrecht, C.R. Cancer stem cells in solid tumors: Elusive or illusive? Cell Commun. Signal. 2010, 8, 6. [Google Scholar] [CrossRef]
- Regenbrecht, C.R.; Lehrach, H.; Adjaye, J. Stemming cancer: Functional genomics of cancer stem cells in solid tumors. Stem Cell Rev. 2008, 4, 319–328. [Google Scholar] [CrossRef]
- Braakhuis, B.J.; Leemans, C.R.; Brakenhoff, R.H. Expanding fields of genetically altered cells in head and neck squamous carcinogenesis. Semin. Cancer Biol. 2005, 15, 113–120. [Google Scholar] [CrossRef]
- Desai, P.C.; Jaglal, M.V.; Gopal, P.; Ghim, S.J.; Miller, D.M.; Farghaly, H.; Jenson, A.B. Human papillomavirus in metastatic squamous carcinoma from unknown primaries in the head and neck: A retrospective 7 year study. Exp. Mol. Pathol. 2009, 87, 94–98. [Google Scholar] [CrossRef]
- Johnson, K.M.; Kines, R.C.; Roberts, J.N.; Lowy, D.R.; Schiller, J.T.; Day, P.M. Role of heparan sulfate in attachment to and infection of the murine female genital tract by human papillomavirus. J. Virol. 2009, 83, 2067–2074. [Google Scholar] [CrossRef]
- Pyeon, D.; Pearce, S.M.; Lank, S.M.; Ahlquist, P.; Lambert, P.F. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 2009, 5, e1000318. [Google Scholar] [CrossRef]
- Vinokurova, S.; Wentzensen, N.; Einenkel, J.; Klaes, R.; Ziegert, C.; Melsheimer, P.; Sartor, H.; Horn, L.C.; Hockel, M.; von Knebel Doeberitz, M. Clonal history of papillomavirus-induced dysplasia in the female lower genital tract. J. Natl. Cancer Inst. 2005, 97, 1816–1821. [Google Scholar] [CrossRef]
- Fidler, I.J.; Kripke, M.L. Metastasis results from preexisting variant cells within a malignant tumor. Science 1977, 197, 893–895. [Google Scholar]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef]
- Dick, J.E. Looking ahead in cancer stem cell research. Nat. Biotechnol. 2009, 27, 44–46. [Google Scholar] [CrossRef]
- Kelly, P.N.; Dakic, A.; Adams, J.M.; Nutt, S.L.; Strasser, A. Tumor growth need not be driven by rare cancer stem cells. Science 2007, 317, 337. [Google Scholar] [CrossRef]
- Jaksch, M.; Munera, J.; Bajpai, R.; Terskikh, A.; Oshima, R.G. Cell cycle-dependent variation of a cd133 epitope in human embryonic stem cell, colon cancer, and melanoma cell lines. Cancer Res. 2008, 68, 7882–7886. [Google Scholar] [CrossRef]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C.; Fuhlbrigge, R.C.; Kupper, T.S.; Sayegh, M.H.; Frank, M.H. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar] [CrossRef]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef]
- Zhu, L.; Gibson, P.; Currle, D.S.; Tong, Y.; Richardson, R.J.; Bayazitov, I.T.; Poppleton, H.; Zakharenko, S.; Ellison, D.W.; Gilbertson, R.J. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 2009, 457, 603–607. [Google Scholar] [CrossRef]
- Vlashi, E.; Kim, K.; Lagadec, C.; Donna, L.D.; McDonald, J.T.; Eghbali, M.; Sayre, J.W.; Stefani, E.; McBride, W.; Pajonk, F. In vivo imaging, tracking, and targeting of cancer stem cells. J. Natl. Cancer Inst. 2009, 101, 350–359. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Tetzlaff, W.; Weiss, S. A multipotent egf-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J. Neurosci. 1992, 12, 4565–4574. [Google Scholar]
- Reynolds, B.A.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992, 255, 1707–1710. [Google Scholar]
- Seaberg, R.M.; van der Kooy, D. Stem and progenitor cells: The premature desertion of rigorous definition. Trends Neurosci. 2003, 26, 125–131. [Google Scholar] [CrossRef]
- Ghods, A.J.; Irvin, D.; Gentao, L.; Yuan, X.; Abdulkadir, I.R.; Tunici, P.; Konda, B.; Wachsmann-Hogiu, S.; Black, K.L.; Yu, J.S. Spheres isolated from 9l gliosarcoma rat cell lines possess chemoresistant and aggressive cancer stem-like cells. Stem Cells 2007, 25, 1645–1653. [Google Scholar] [CrossRef]
- Turley, E.A.; Veiseh, M.; Radisky, D.C.; Bissell, M.J. Mechanisms of disease: Epithelial mesenchymal transition—does cellular plasticity fuel neoplastic progression? Nat. Clin. Pract. Oncol. 2008, 5, 280–290. [Google Scholar]
- Baum, B.; Settleman, J.; Quinlan, M.P. Transitions between epithelial and mesenchymal states in development and disease. Semin. Cell Dev. Biol. 2008, 19, 294–308. [Google Scholar] [CrossRef]
- Derek, C.R.; LaBarge, M.A. Epithelial-mesenchymal transition and the stem cell phenotype. Cell Stem Cell 2008, 2, 511–512. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Hay, E.D. An overview of epithelio-mesenchymal transformation. Acta Anat. (Basel) 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Perez-Pomares, J.M.; Munoz-Chapuli, R. Epithelial-mesenchymal transitions: A mesodermal cell strategy for evolutive innovation in metazoans. Anat. Rec. 2002, 268, 343–351. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in development and pathologies. Curr. Opin. Cell Biol. 2003, 15, 740–746. [Google Scholar] [CrossRef]
- Savagner, P.; Kusewitt, D.F.; Carver, E.A.; Magnino, F.; Choi, C.; Gridley, T.; Hudson, L.G. Developmental transcription factor slug is required for effective re-epithelialization by adult keratinocytes. J. Cell. Physiol. 2005, 202, 858–866. [Google Scholar] [CrossRef]
- Tsuji, T.; Ibaragi, S.; Shima, K.; Hu, M.G.; Katsurano1, M.; Sasaki, A.; Hu, G.F. Epithelial–mesenchymal transition induced by growth suppressor p12cdk2-ap1 promotes tumor cell local invasion but suppresses distant colony growth. Cancer Res. 2008, 68, 10377–10386. [Google Scholar] [CrossRef]
- Thomson, S.; Buck, E.; Petti, F.; Griffin, G.; Brown, E.; Ramnarine, N.; Iwata, K.K.; Gibson, N.; Haley, J.D. Epithelial to mesenchymal transition is a determinant of sensitivity of non-small-cell lung carcinoma cell lines and xenografts to epidermal growth factor receptor inhibition. Cancer Res. 2005, 65, 9455–9462. [Google Scholar] [CrossRef]
- Frederick, B.A.; Helfrich, B.A.; Coldren, C.D.; Zheng, D.; Chan, D.; Bunn, P.A., Jr.; Raben, D. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol. Cancer Ther. 2007, 6, 1683–1691. [Google Scholar] [CrossRef]
- Black, P.C.; Brown, G.A.; Inamoto, T.; Shrader, M.; Arora, A.; Siefker-Radtke, A.O.; Adam, L.; Theodorescu, D.; Wu, X.F.; Munsell, M.F.; Bar-Eli, M.; McConkey, D.J.; Colin, P.N.; Dinney, C.P.N. Sensitivity to epidermal growth factor receptor inhibitor requires ecadherin expression in urothelial carcinoma cells. Clin. Cancer Res. 2008, 14, 1478–1486. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.J.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; Campbell, L.L.; Polyak, K.; Brisken, C.; Yang, J.; Weinberg, A. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Morel, A.P.; Thomas, C.; George, H.; Ansieau, S.; Alain, P. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, 2888. [Google Scholar] [CrossRef]
- Kupferman, M.E.; Jiffar, T.; El-Naggar, A.; Yilmaz, T.; Zhou, G.; Xie, T.; Feng, L.; Wang, J.; Holsinger, F.C.; Yu, D.; Myers, J.N. Trkb induces emt and has a key role in invasion of head and neck squamous cell carcinoma. Oncogene 2010, 29, 2047–2059. [Google Scholar] [CrossRef]
- Ma, S.C.K.; Lee, T.K.; Tang, K.H.; Wo, J.Y. Aldehyde dehydrogenase discriminates the cd133 liver cancer stem cell populations. Mol. Cancer Res. 2008, 6, 1146–1153. [Google Scholar] [CrossRef]
- Jiang, F.Q.Q.; Khanna, A.; Todd, N.W.; Deepak, J. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol. Cancer Res. 2009, 7, 330–338. [Google Scholar] [CrossRef]
- Emina, H.; Huang, M.J.H.; Zhang, T. Christophe Ginestier, Gabriela Dontu Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (sc) and tracks sc overpopulation during colon tumorigenesis. Cancer Res. 2009, 69, 3382–3389. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Chen, Y.W.; Hsu, H.S.; Tseng, L.M.; Huang, P.I.; Lu, K.H.; Chen, D.T.; Tai, L.K.; Yung, M.C.; Chang, S.C.; Ku, H.H.; Chiou, S.H.; Lo, W.L. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem. Biophys. Res. Commun. 2009, 385, 307–313. [Google Scholar] [CrossRef]
- Mack, B.; Gires, O. Cd44s and cd44v6 expression in head and neck epithelia. PLoS ONE 2008, 3, 3360. [Google Scholar] [CrossRef]
- Prince, M.E.; Ailles, L.E. Cancer stem cells in head and neck squamous cell cancer. J. Clin. Oncol. 2008, 26, 2871–2875. [Google Scholar] [CrossRef]
- Prince, M.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.H.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; Shelton, A.A.; Parmiani, G.; Castelli, C.; Clarke, M.F. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; Schott, A.; Hayes, D.; Birnbaum, D.; Wicha, M.S.; Dontu, G. Aldh1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Douville, J.; Beaulieu, R.; Balicki, D. Aldh1 as a functional marker of cancer stem and progenitor cells. Stem Cells Dev. 2008, 18, 17–25. [Google Scholar] [CrossRef]
- Chiou, S.H.; Yu, C.C.; Huang, C.Y.; Lin, S.C.; Liu, C.J.; Tsai, T.H.; Chou, S.H.; Chien, C.S.; Ku, H.H.; Lo, J.F. Positive correlations of oct-4 and nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin. Cancer Res. 2008, 14, 4085–4095. [Google Scholar] [CrossRef]
- Singh, S.H.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- O'Brien, C.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumor growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Wei, X.D.; Zhou, L.; Cheng, L.; Tian, J.; Jiang, J.J.; Maccallum, J. In vivo investigation of cd133 as a putative marker of cancer stem cells in hep-2 cell line. Head Neck 2009, 31, 94–101. [Google Scholar] [CrossRef]
- Wu, C.; Wei, Q.; Utomo, V.; Nadesan, P.; Whetstone, H. Side population cells isolated from mesenchymal neoplasms have tumor initiating potential. Cancer Res. 2007, 67, 8216–8222. [Google Scholar] [CrossRef]
- St John, M.A.; Dohadwala, M.; Luo, J.; Wang, G.; Lee, G.; Shih, H.; Heinrich, E.; Krysan, K.; Walser, T.; Hazra, S.; et al. Proinflammatory mediators upregulate snail in head and neck squamous cell carcinoma. Clin. Cancer Res. 2009, 15, 6018–6027. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Weinberg, R.A. Exploring a new twist on tumor metastasis. Cancer Res. 2006, 66, 4549–4552. [Google Scholar] [CrossRef]
- Vered, M.; Dayan, D.; Yahalom, R.; Dobriyan, A.; Barshack, I.; Bello, I.O.; Kantola, S.; Salo, T. Cancer-associated fibroblasts and epithelial-mesenchymal transition in metastatic oral tongue squamous cell carcinoma. Int. J. Cancer 2010. [Google Scholar] [CrossRef]
- Kashyap, V.; Rezende, N.C.; Scotland, K.B.; Shaffer, S.M.; Persson, J.L.; Gudas, L.J.; Mongan, N.P. Regulation of stem cell pluripotency and differentiation involves a mutual regulatory circuit of the nanog, oct4, and sox2 pluripotency transcription factors with polycomb repressive complexes and stem cell micrornas. Stem Cells Dev. 2009, 18, 1093–1108. [Google Scholar] [CrossRef]
- Rodda, D.J.; Chew, J.L.; Lim, L.H.; Loh, Y.H.; Wang, B.; Ng, H.H.; Robson, P. Transcriptional regulation of nanog by oct4 and sox2. J. Biol. Chem. 2005, 280, 24731–24737. [Google Scholar]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospectivve identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Patrick, C.; Stephan, L.H.; Tanja, H.; Alexandra, A.; Joachim, W.E. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Bozec, A.; Gros, F.X.; Penault-Llorca, F.; Formento, P.; Cayre, A.; Dental, C.; Etienne-Grimaldi, M.C.; Fischel, J.L.; Milano, G. Vertical vegf targeting: A combination of ligand blockade with receptor tyrosine kinase inhibition. Eur. J. Cancer 2008, 44, 1922–1930. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of cd133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef]
- Nakai, E.; Park, K.; Yawata, T.; Chihara, T.; Kumazawa, A.; Nakabayashi, H.; Shimizu, K. Enhanced mdr1 expression and chemoresistance of cancer stem cells derived from glioblastoma. Cancer Invest. 2009, 27, 901–908. [Google Scholar] [CrossRef]
- Ropolo, M.; Daga, A.; Griffero, F.; Foresta, M.; Casartelli, G.; Zunino, A.; Poggi, A.; Cappelli, E.; Zona, G.; Spaziante, R.; Corte, G.; Frosina, G. Comparative analysis of DNA repair in stem and nonstem glioma cell cultures. Mol. Cancer Res. 2009, 7, 383–392. [Google Scholar] [CrossRef]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of cd24(-/low)/cd44+ breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; Wong, H.; Rosen, J.; Chang, J.C. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef]
- Hong, S.P.; Wen, J.; Bang, S.; Park, S.; Song, S.Y. Cd44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int. J. Cancer 2009, 125, 2323–2331. [Google Scholar] [CrossRef]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. Cd133+ hcc cancer stem cells confer chemoresistance by preferential expression of the akt/pkb survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef]
- Todaro, M.; Alea, M.P.; Di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; Stassi, G. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 2007, 1, 389–402. [Google Scholar] [CrossRef]
- Frank, N.Y.; Margaryan, A.; Huang, Y.; Schatton, T.; Waaga-Gasser, A.M.; Gasser, M.; Sayegh, M.H.; Sadee, W.; Frank, M.H. Abcb5-mediated doxorubicin transport and chemoresistance in human malignant melanoma. Cancer Res. 2005, 65, 4320–4333. [Google Scholar] [CrossRef]
- Al-Assar, O.; Muschel, R.J.; Mantoni, T.S.; McKenna, W.G.; Brunner, T.B. Radiation response of cancer stem-like cells from established human cell lines after sorting for surface markers. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 1216–1225. [Google Scholar] [CrossRef]
- Costello, R.T.; Mallet, F.; Gaugler, B.; Sainty, D.; Arnoulet, C.; Gastaut, J.A.; Olive, D. Human acute myeloid leukemia cd34+/cd38- progenitor cells have decreased sensitivity to chemotherapy and fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer Res. 2000, 60, 4403–4411. [Google Scholar]
- de Grouw, E.P.; Raaijmakers, M.H.; Boezeman, J.B.; van der Reijden, B.A.; van de Locht, L.T.; de Witte, T.J.; Jansen, J.H.; Raymakers, R.A. Preferential expression of a high number of atp binding cassette transporters in both normal and leukemic cd34+cd38- cells. Leukemia 2006, 20, 750–754. [Google Scholar] [CrossRef]
- Wulf, G.G.; Wang, R.Y.; Kuehnle, I.; Weidner, D.; Marini, F.; Brenner, M.K.; Andreeff, M.; Goodell, M.A. A leukemic stem cell with intrinsic drug efflux capacity in acute myeloid leukemia. Blood 2001, 98, 1166–1173. [Google Scholar] [CrossRef]
- Visus, C.; Ito, D.; Amoscato, A.; Maciejewska-Franczak, M.; Abdelsalem, A.; Dhir, R.; Shin, D.M.; Donnenberg, V.S.; Whiteside, T.L.; DeLeo, A.B. Identification of human aldehyde dehydrogenase 1 family member a1 as a novel cd8+ t-cell-defined tumor antigen in squamous cell carcinoma of the head and neck. Cancer Res. 2007, 67, 10538–10545. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, G.; Yuan, X.; Xu, M.; Wang, H.; Ji, J.; Konda, B.; Black, K.L.; Yu, J.S. Antigen-specific t-cell response from dendritic cell vaccination using cancer stem-like cell-associated antigens. Stem Cells 2009, 27, 1734–1740. [Google Scholar] [CrossRef]
- Mellodew, K.; Suhr, R.; Uwanogho, D.A.; Reuter, I.; Lendahl, U.; Hodges, H.; Price, J. Nestin expression is lost in a neural stem cell line through a mechanism involving the proteasome and notch signaling. Brain Res. Dev. Brain Res. 2004, 151, 13–23. [Google Scholar] [CrossRef]
- Ben-Saadon, R.; Zaaroor, D.; Ziv, T.; Ciechanover, A. The polycomb protein ring1b generates self atypical mixed ubiquitin chains required for its in vitro histone h2a ligase activity. Mol. Cell 2006, 24, 701–711. [Google Scholar] [CrossRef]
- Chen, Y.C.; Chang, C.J.; Hsu, H.S.; Chen, Y.W.; Tai, L.K.; Tseng, L.M.; Chiou, G.Y.; Chang, S.C.; Kao, S.Y.; Chiou, S.H.; Lo, W.L. Inhibition of tumorigenicity and enhancement of radiochemosensitivity in head and neck squamous cell cancer-derived aldh1-positive cells by knockdown of bmi-1. Oral. Oncol. 2010, 46, 158–165. [Google Scholar] [CrossRef]
- Barrallo-Gimeno, A.; Nieto, M.A. The snail genes as inducers of cell movement and survival: Implications in development and cancer. Development 2005, 132, 3151–3161. [Google Scholar] [CrossRef]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Smith, L.M.; Nesterova, A.; Ryan, M.C.; Duniho, S.; Jonas, M.; Anderson, M.; Zabinski, R.F.; Sutherland, M.K.; Gerber, H.P.; van Orden, K.L.; Moore, P.A.; Ruben, S.M.; Carter, P.J. Cd133/prominin-1 is a potential therapeutic target for antibody-drug conjugates in hepatocellular and gastric cancers. Br. J. Cancer 2008, 99, 100–109. [Google Scholar] [CrossRef]
- Jin, L.; Lee, E.M.; Ramshaw, H.S.; Busfield, S.J.; Peoppl, A.G.; Wilkinson, L.; Guthridge, M.A.; Thomas, D.; Barry, E.F.; Boyd, A.; Gearing, D.P.; Vairo, G.; Lopez, A.F.; Dick, J.E.; Lock, R.B. Monoclonal antibody-mediated targeting of cd123, il-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell 2009, 5, 31–42. [Google Scholar] [CrossRef]
- Piccirillo, S.G.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; Dimeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761–765. [Google Scholar] [CrossRef]
- Park, C.Y.; Tseng, D.; Weissman, I.L. Cancer stem cell-directed therapies: Recent data from the laboratory and clinic. Mol. Ther. 2009, 17, 219–230. [Google Scholar] [CrossRef]
- Chen, J.S.; Pardo, F.S.; Wang-Rodriguez, J.; Chu, T.S.; Lopez, J.P.; Aguilera, J.; Altuna, X.; Weisman, R.A.; Ongkeko, W.M. Egfr regulates the side population in head and neck squamous cell carcinoma. Laryngoscope 2006, 116, 401–406. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, C.; Köberle, B.; Kaufmann, A.M.; Albers, A.E. A Quest for Initiating Cells of Head and Neck Cancer and Their Treatment. Cancers 2010, 2, 1528-1554. https://doi.org/10.3390/cancers2031528
Chen C, Köberle B, Kaufmann AM, Albers AE. A Quest for Initiating Cells of Head and Neck Cancer and Their Treatment. Cancers. 2010; 2(3):1528-1554. https://doi.org/10.3390/cancers2031528
Chicago/Turabian StyleChen, Chao, Beate Köberle, Andreas M. Kaufmann, and Andreas E. Albers. 2010. "A Quest for Initiating Cells of Head and Neck Cancer and Their Treatment" Cancers 2, no. 3: 1528-1554. https://doi.org/10.3390/cancers2031528
APA StyleChen, C., Köberle, B., Kaufmann, A. M., & Albers, A. E. (2010). A Quest for Initiating Cells of Head and Neck Cancer and Their Treatment. Cancers, 2(3), 1528-1554. https://doi.org/10.3390/cancers2031528