
Mechanistic Roles of Transcriptional Cyclin-Dependent Kinases in Oncogenesis: Implications for Cancer Therapy
 ,
,  and
and 
Simple Summary
Abstract
1. Introduction
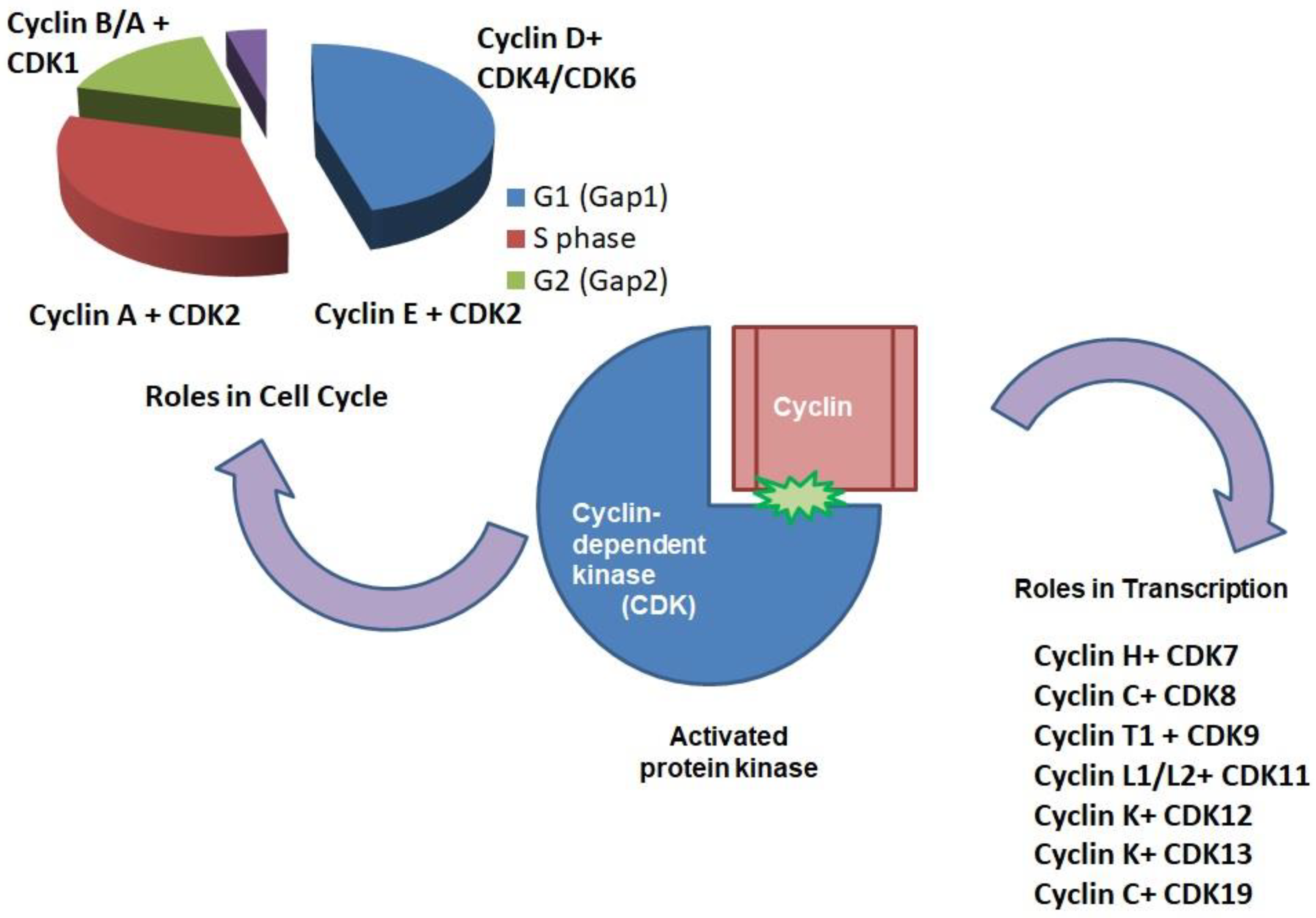
2. CDKs: Roles in Cell Cycle
Cyclin-Dependant Kinase 5
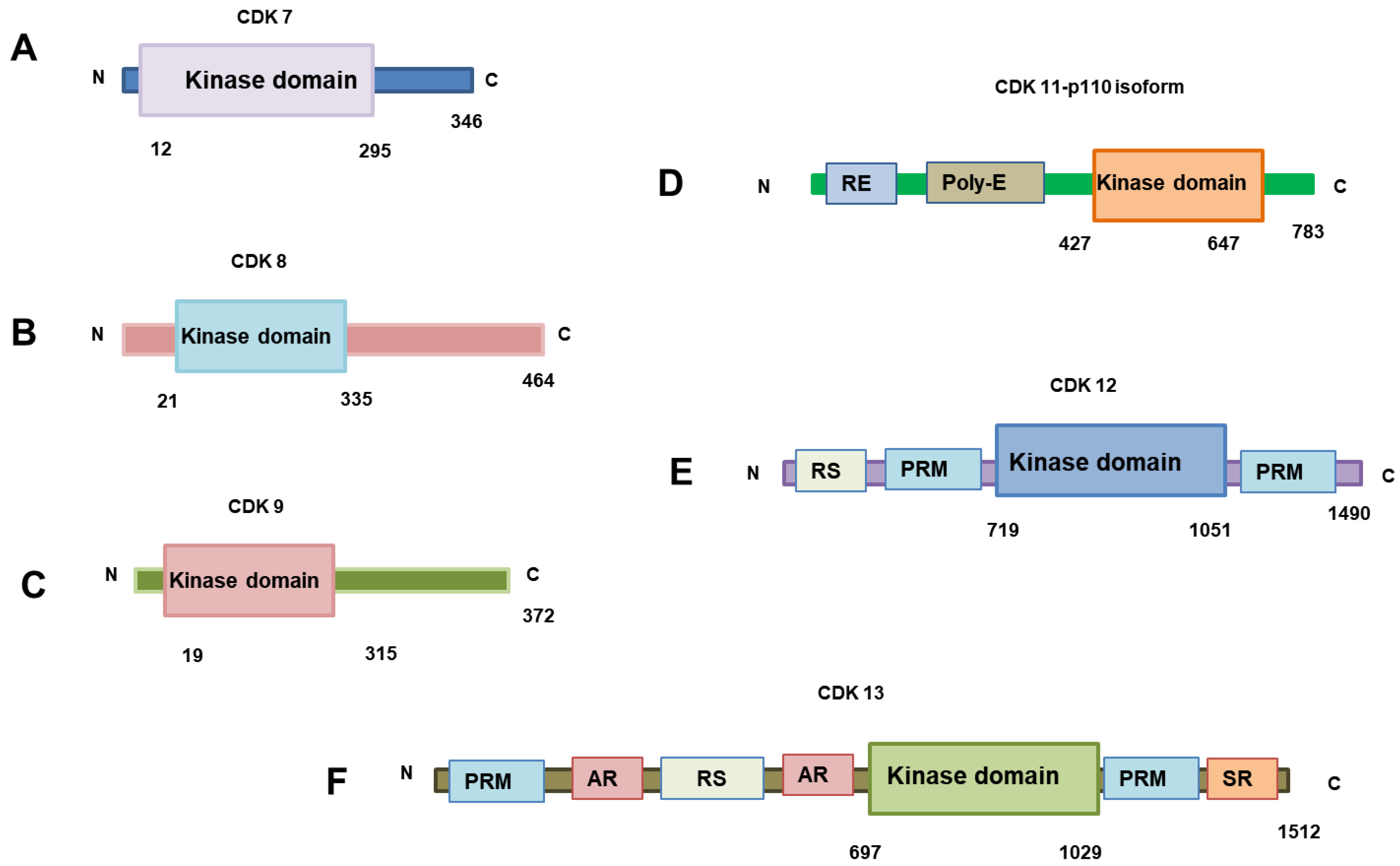
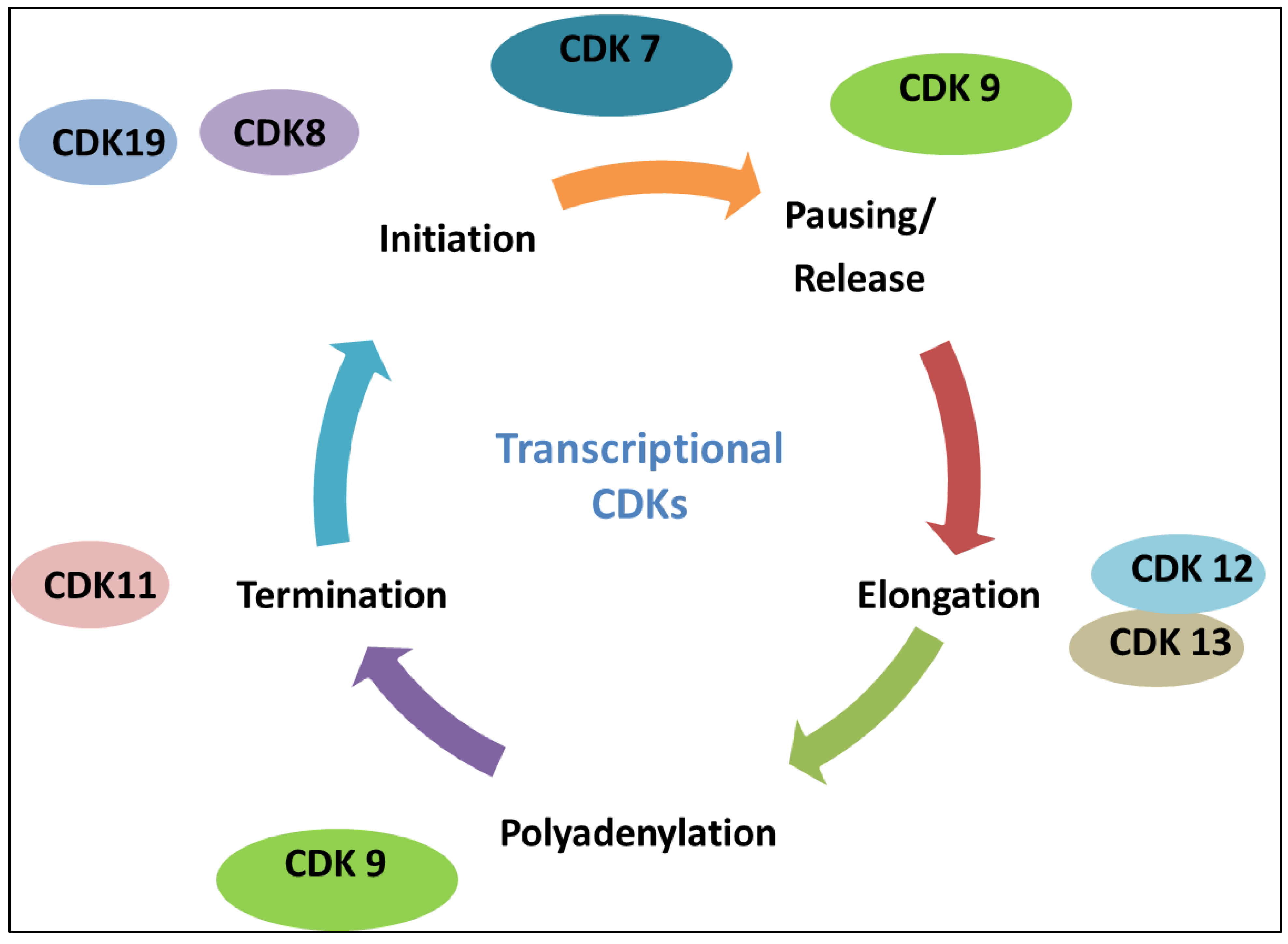
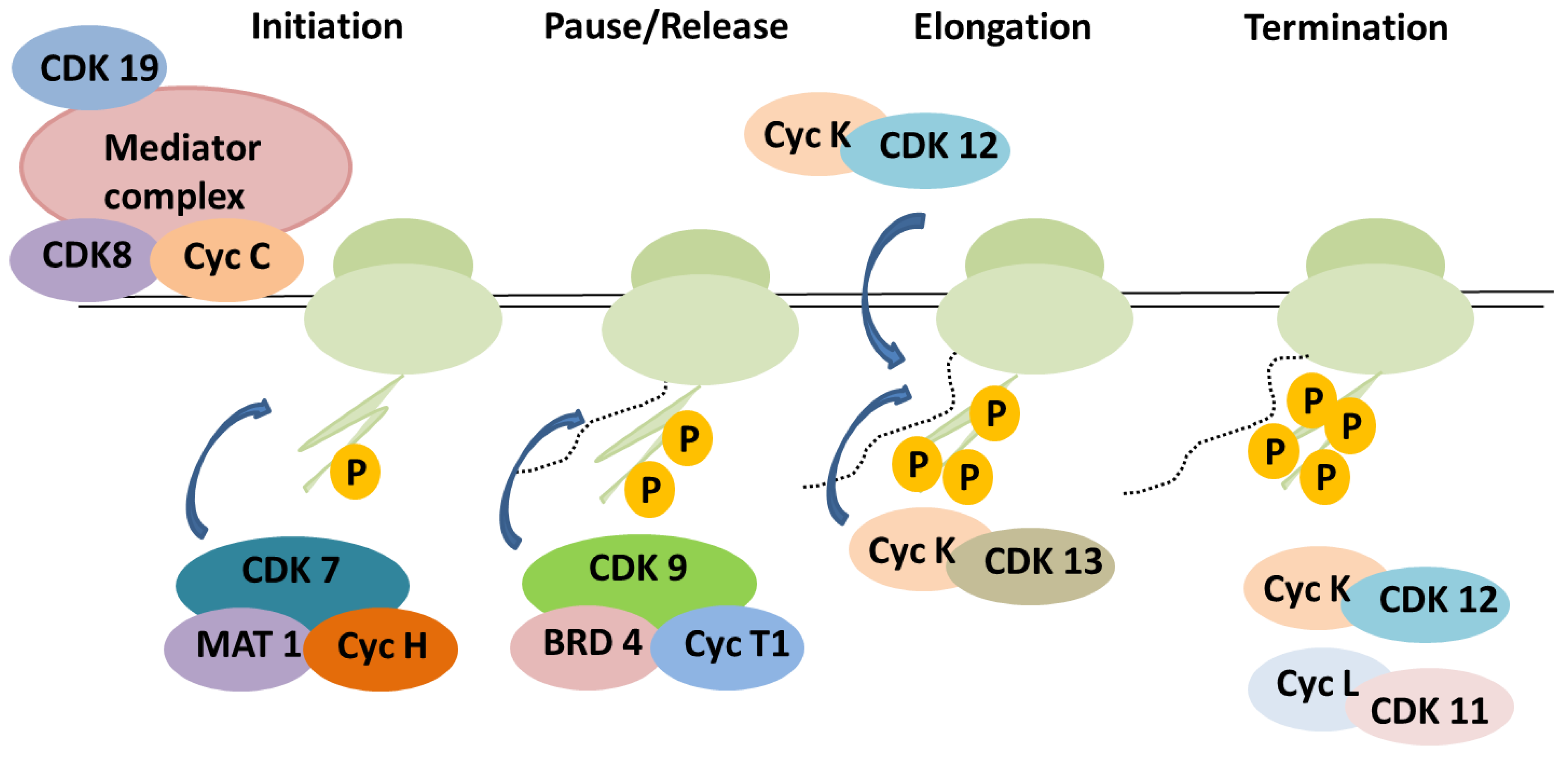
3. The Roles of CDKs in Transcription
4. CDKs as Therapeutic Targets in Cancer Therapeutics
5. CDK7 as a Therapeutic Target in Cancer
5.1. Intrahepatic Cholangiocarcinoma (ICC)
5.2. Epithelial Ovarian Cancer
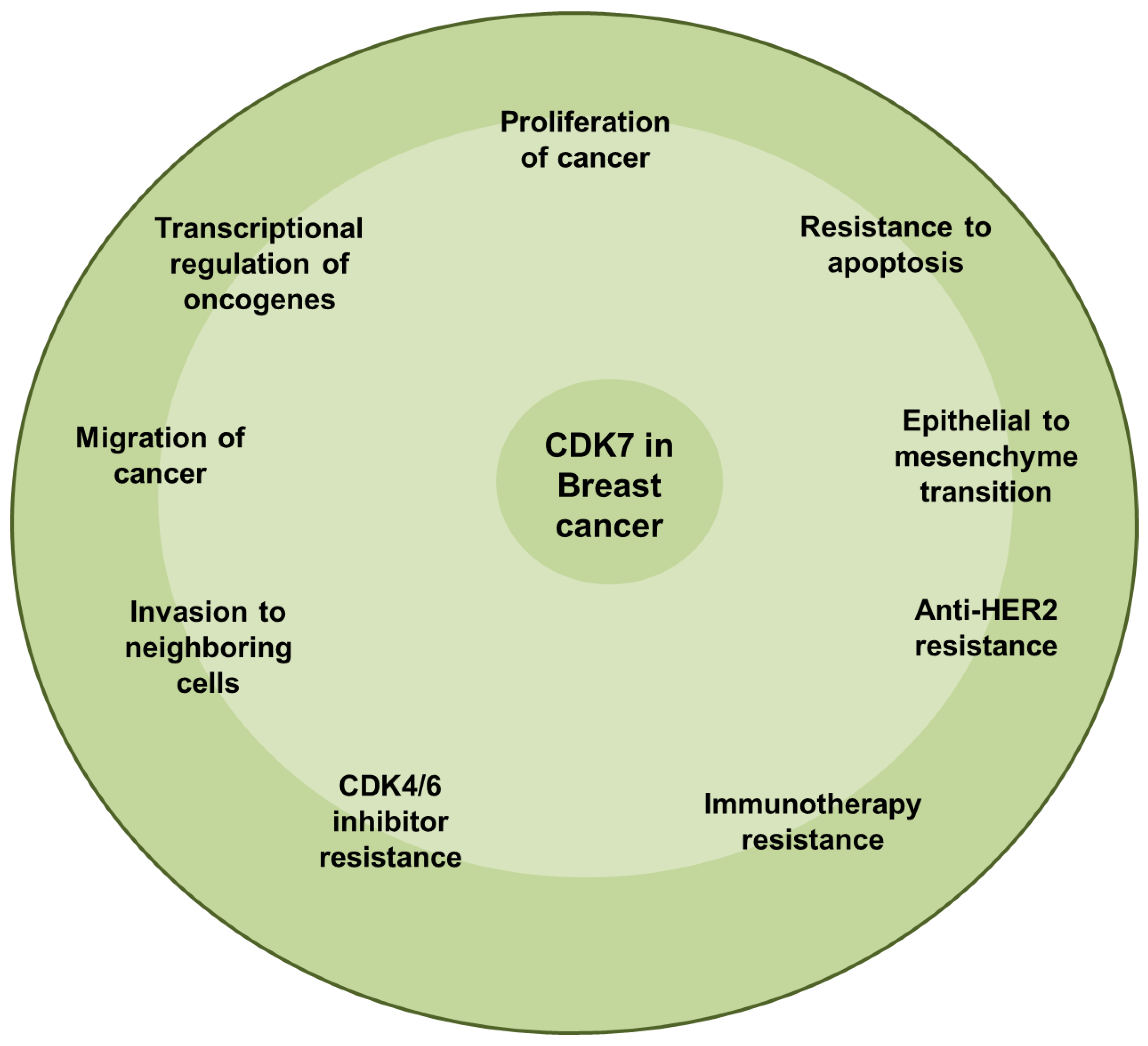
5.3. Breast Cancer
5.4. Pancreatic Ductal Adenocarcinoma
6. Cyclin-Dependent Kinase 8 in Cancer
6.1. Breast Cancer
6.2. Colorectal Cancer β-catenin
7. Cyclin-Dependent Kinase 9 as a Therapeutic Target in Cancer
7.1. Pancreatic Cancer
7.2. Breast Cancer
7.3. Osteosarcoma
7.4. Ovarian Cancer
8. Cyclin-Dependent Kinase 12 in Cancer
8.1. Breast Cancer
8.2. Ovarian Cancer
8.3. Prostate Cancer
9. CDK 12/13 Inhibition as a Therapeutic Option in Cancer Treatment
10. CDK 11
11. CDK 19
Prostate Cancer
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
Correction Statement
References
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Arellano, M.; Moreno, S. Regulation of CDK/cyclin complexes during the cell cycle. Int. J. Biochem. Cell Biol. 1997, 29, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Palmer, N.; Kaldis, P. Less-well known functions of cyclin/CDK complexes. Semin. Cell Dev. Biol. 2020, 107, 54–62. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Jeffrey, P.D.; Russo, A.A.; Polyak, K.; Gibbs, E.; Hurwitz, J.; Massagué, J.; Pavletich, N.P. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 1995, 376, 313–320. [Google Scholar] [CrossRef]
- De Bondt, H.L.; Rosenblatt, J.; Jancarik, J.; Jones, H.D.; Morgan, D.O.; Kim, S.-H. Crystal structure of cyclin-dependent kinase 2. Nature 1993, 363, 595–602. [Google Scholar] [CrossRef]
- Franco, G.; Guzzi, P.H.; Manca, V.; Mazza, T. Mitotic oscillators as MP graphs. In Proceedings of the International Workshop on Membrane Computing, Leiden, The Netherlands, 17–21 July 2006; pp. 382–394. [Google Scholar]
- Gould, K.L.; Moreno, S.; Owen, D.; Sazer, S.; Nurse, P. Phosphorylation at Thr167 is required for Schizosaccharomyces pombe p34cdc2 function. EMBO J. 1991, 10, 3297–3309. [Google Scholar] [CrossRef]
- Scapin, G. Structural biology in drug design: Selective protein kinase inhibitors. Drug Discov. Today 2002, 7, 601–611. [Google Scholar] [CrossRef]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef]
- Harbour, J.W.; Dean, D.C. The Rb/E2F pathway: Expanding roles and emerging paradigms. Genes Dev. 2000, 14, 2393–2409. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, A.S.; Weinberg, R.A. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol. Cell. Biol. 1998, 18, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Luo, R.X.; Dei Santi, A.; Postigo, A.A.; Dean, D.C. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999, 98, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Park, M.-T.; Lee, S.-J. Cell cycle and cancer. BMB Rep. 2003, 36, 60–65. [Google Scholar] [CrossRef]
- Botz, J.; Zerfass-Thome, K.; Spitkovsky, D.; Delius, H.; Vogt, B.; Eilers, M.; Hatzigeorgiou, A.; Jansen-Dürr, P. Cell cycle regulation of the murine cyclin E gene depends on an E2F binding site in the promoter. Mol. Cell. Biol. 1996, 16, 3401–3409. [Google Scholar] [CrossRef]
- You, W.; Henneberg, M. Cancer incidence increasing globally: The role of relaxed natural selection. Evol. Appl. 2018, 11, 140–152. [Google Scholar] [CrossRef]
- Srivastava, S.; Koay, E.J.; Borowsky, A.D.; De Marzo, A.M.; Ghosh, S.; Wagner, P.D.; Kramer, B.S. Cancer overdiagnosis: A biological challenge and clinical dilemma. Nat. Rev. Cancer 2019, 19, 349–358. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell. Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Dalton, S. Linking the cell cycle to cell fate decisions. Trends Cell Biol. 2015, 25, 592–600. [Google Scholar] [CrossRef]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef]
- Liu, J.; Peng, Y.; Wei, W. Cell cycle on the crossroad of tumorigenesis and cancer therapy. Trends Cell Biol. 2022, 31, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Dynlacht, B.D. Regulation of transcription by proteins that control the cell cycle. Nature 1997, 389, 149–152. [Google Scholar] [CrossRef]
- Icard, P.; Fournel, L.; Wu, Z.; Alifano, M.; Lincet, H. Interconnection between metabolism and cell cycle in cancer. Trends Biochem. Sci. 2019, 44, 490–501. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 inhibition in cancer: Beyond cell cycle arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef]
- Nebenfuehr, S.; Kollmann, K.; Sexl, V. The role of CDK6 in cancer. Int. J. Cancer 2020, 147, 2988–2995. [Google Scholar] [CrossRef]
- Sava, G.P.; Fan, H.; Coombes, R.C.; Buluwela, L.; Ali, S. CDK7 inhibitors as anticancer drugs. Cancer Metastasis Rev. 2020, 39, 805–823. [Google Scholar] [CrossRef]
- Bury, M.; Le Calvé, B.; Ferbeyre, G.; Blank, V.; Lessard, F. New insights into CDK regulators: Novel opportunities for cancer therapy. Trends Cell Biol. 2021, 31, 331–344. [Google Scholar] [CrossRef]
- Rugo, H.; Finn, R.; Diéras, V.; Ettl, J.; Lipatov, O.; Joy, A.; Harbeck, N.; Castrellon, A.; Iyer, S.; Lu, D. Palbociclib plus letrozole as first-line therapy in estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer with extended follow-up. Breast Cancer Res. Treat. 2019, 174, 719–729. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Hart, L.; Campone, M.; Petrakova, K.; Winer, E.P.; Janni, W. Overall survival with ribociclib plus letrozole in advanced breast cancer. N. Engl. J. Med. 2022, 386, 942–950. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.S.; Kanani, R.; Roylance, R.; Mittnacht, S. Systematic review of molecular biomarkers predictive of resistance to CDK4/6 inhibition in metastatic breast cancer. JCO Precis. Oncol. 2022, 6, e2100002. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Jiang, B.; Guo, J.; Shao, H.; Del Priore, I.S.; Chang, Q.; Kudo, R.; Li, Z.; Razavi, P.; Liu, B. INK4 tumor suppressor proteins mediate resistance to CDK4/6 kinase inhibitors. Cancer Discov. 2022, 12, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef]
- Tane, S.; Ikenishi, A.; Okayama, H.; Iwamoto, N.; Nakayama, K.I.; Takeuchi, T. CDK inhibitors, p21Cip1 and p27Kip1, participate in cell cycle exit of mammalian cardiomyocytes. Biochem. Biophys. Res. Commun. 2014, 443, 1105–1109. [Google Scholar] [CrossRef]
- Ma, T.; Van Tine, B.A.; Wei, Y.; Garrett, M.D.; Nelson, D.; Adams, P.D.; Wang, J.; Qin, J.; Chow, L.T.; Harper, J.W. Cell cycle–regulated phosphorylation of p220NPAT by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev. 2000, 14, 2298–2313. [Google Scholar] [CrossRef]
- Okuda, M.; Horn, H.F.; Tarapore, P.; Tokuyama, Y.; Smulian, A.G.; Chan, P.-K.; Knudsen, E.S.; Hofmann, I.A.; Snyder, J.D.; Bove, K.E. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell 2000, 103, 127–140. [Google Scholar] [CrossRef]
- Petersen, B.O.; Lukas, J.; Sørensen, C.S.; Bartek, J.; Helin, K. Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO J. 1999, 18, 396–410. [Google Scholar] [CrossRef]
- Welcker, M.; Singer, J.; Loeb, K.R.; Grim, J.; Bloecher, A.; Gurien-West, M.; Clurman, B.E.; Roberts, J.M. Multisite phosphorylation by Cdk2 and GSK3 controls cyclin E degradation. Mol. Cell 2003, 12, 381–392. [Google Scholar] [CrossRef]
- Gavet, O.; Pines, J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 2010, 18, 533–543. [Google Scholar] [CrossRef]
- Santamaría, D.; Barrière, C.; Cerqueira, A.; Hunt, S.; Tardy, C.; Newton, K.; Cáceres, J.F.; Dubus, P.; Malumbres, M.; Barbacid, M. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007, 448, 811–815. [Google Scholar] [CrossRef] [PubMed]
- Timofeev, O.; Cizmecioglu, O.; Settele, F.; Kempf, T.; Hoffmann, I. Cdc25 phosphatases are required for timely assembly of CDK1-cyclin B at the G2/M transition. J. Biol. Chem. 2010, 285, 16978–16990. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell. Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- London, N.; Biggins, S. Signalling dynamics in the spindle checkpoint response. Nat. Rev. Mol. Cell. Biol. 2014, 15, 736–748. [Google Scholar] [CrossRef]
- Ziebold, U.; Bartsch, O.; Marais, R.; Ferrari, S.; Klempnauer, K.-H. Phosphorylation and activation of B-Myb by cyclin A–Cdk2. Curr. Biol. 1997, 7, 253–260. [Google Scholar] [CrossRef]
- Hydbring, P.; Bahram, F.; Su, Y.; Tronnersjö, S.; Högstrand, K.; von der Lehr, N.; Sharifi, H.R.; Lilischkis, R.; Hein, N.; Wu, S. Phosphorylation by Cdk2 is required for Myc to repress Ras-induced senescence in cotransformation. Proc. Natl. Acad. Sci. USA 2010, 107, 58–63. [Google Scholar] [CrossRef]
- Huang, H.; Regan, K.M.; Lou, Z.; Chen, J.; Tindall, D.J. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science 2006, 314, 294–297. [Google Scholar] [CrossRef]
- Yun, J.; Chae, H.-D.; Choi, T.-S.; Kim, E.-H.; Bang, Y.-J.; Chung, J.; Choi, K.-S.; Mantovani, R.; Shin, D.Y. Cdk2-dependent phosphorylation of the NF-Y transcription factor and its involvement in the p53-p21 signaling pathway. J. Biol. Chem. 2003, 278, 36966–36972. [Google Scholar] [CrossRef]
- Ying, M.; Shao, X.; Jing, H.; Liu, Y.; Qi, X.; Cao, J.; Chen, Y.; Xiang, S.; Song, H.; Hu, R. Ubiquitin-dependent degradation of CDK2 drives the therapeutic differentiation of AML by targeting PRDX2. Blood J. Am. Soc. Hematol. 2018, 131, 2698–2711. [Google Scholar] [CrossRef]
- Tadesse, S.; Anshabo, A.T.; Portman, N.; Lim, E.; Tilley, W.; Caldon, C.E.; Wang, S. Targeting CDK2 in cancer: Challenges and opportunities for therapy. Drug Discov. Today 2020, 25, 406–413. [Google Scholar] [CrossRef]
- Grishina, I.; Lattes, B. A novel Cdk2 interactor is phosphorylated by Cdc7 and associates with components of the replication complexes. Cell Cycle 2005, 4, 4120–4126. [Google Scholar] [CrossRef]
- Paglini, G.; Cáceres, A. The role of the Cdk5–p35 kinase in neuronal development. Eur. J. Biochem. 2001, 268, 1528–1533. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, M.; Dudek, H.; Kwon, Y.T.; Ramos, Y.; Tsai, L.-H. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996, 10, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Nakabayashi, T.; Mita, N.; Jin, X.; Aikawa, Y.; Sasamoto, K.; Miyoshi, G.; Miyata, M.; Inoue, T.; Ohshima, T. Involvement of Cdk5 activating subunit p35 in synaptic plasticity in excitatory and inhibitory neurons. Mol. Brain 2022, 15, 37. [Google Scholar] [CrossRef]
- Im, D.S.; Joselin, A.; Svoboda, D.; Takano, T.; Rousseaux, M.W.; Callaghan, S.; Slack, R.S.; Hisanaga, S.-I.; Davis, R.J.; Park, D.S. Cdk5-mediated JIP1 phosphorylation regulates axonal outgrowth through Notch1 inhibition. BMC Biol. 2022, 20, 115. [Google Scholar] [CrossRef]
- Hisanaga, S.I.; Endo, R. Regulation and role of cyclin-dependent kinase activity in neuronal survival and death. J. Neurochem. 2010, 115, 1309–1321. [Google Scholar] [CrossRef]
- Allnutt, A.B.; Waters, A.K.; Kesari, S.; Yenugonda, V.M. Physiological and pathological roles of Cdk5: Potential directions for therapeutic targeting in neurodegenerative disease. ACS Chem. Neurosci. 2020, 11, 1218–1230. [Google Scholar] [CrossRef]
- Cheung, Z.H.; Ip, N.Y. Cdk5: A multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 2012, 22, 169–175. [Google Scholar] [CrossRef]
- Arif, A. Extraneuronal activities and regulatory mechanisms of the atypical cyclin-dependent kinase Cdk5. Biochem. Pharmacol. 2012, 84, 985–993. [Google Scholar] [CrossRef]
- Ozaki, N.; Shibasaki, T.; Kashima, Y.; Miki, T.; Takahashi, K.; Ueno, H.; Sunaga, Y.; Yano, H.; Matsuura, Y.; Iwanaga, T. cAMP-GEFII is a direct target of cAMP in regulated exocytosis. Nat. Cell Biol. 2000, 2, 805–811. [Google Scholar] [CrossRef]
- Su, S.C.; Tsai, L.-H. Cyclin-dependent kinases in brain development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 465–491. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.-M.; Afshar, M.; Martin, F.; Cavadore, J.-C.; Labbe, J.-C.; Dorée, M. Dual phosphorylation of the T-loop in cdk7: Its role in controlling cyclin H binding and CAK activity. EMBO J. 1997, 16, 343–354. [Google Scholar] [CrossRef]
- Larochelle, S.; Chen, J.; Knights, R.; Pandur, J.; Morcillo, P.; Erdjument-Bromage, H.; Tempst, P.; Suter, B.; Fisher, R.P. T-loop phosphorylation stabilizes the CDK7–cyclin H–MAT1 complex in vivo and regulates its CTD kinase activity. EMBO J. 2001, 20, 3749–3759. [Google Scholar] [CrossRef] [PubMed]
- Serizawa, H.; Mäkelä, T.P.; Conaway, J.W.; Conaway, R.C.; Weinberg, R.A.; Young, R.A. Association of Cdk-activating kinase subunits with transcription factor TFIIH. Nature 1995, 374, 280–282. [Google Scholar] [CrossRef]
- Spangler, L.; Wang, X.; Conaway, J.W.; Conaway, R.C.; Dvir, A. TFIIH action in transcription initiation and promoter escape requires distinct regions of downstream promoter DNA. Proc. Natl. Acad. Sci. USA 2001, 98, 5544–5549. [Google Scholar] [CrossRef]
- Larochelle, S.; Amat, R.; Glover-Cutter, K.; Sansó, M.; Zhang, C.; Allen, J.J.; Shokat, K.M.; Bentley, D.L.; Fisher, R.P. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat. Struct. Mol. Biology 2012, 19, 1108–1115. [Google Scholar] [CrossRef]
- Lu, H.; Fisher, R.P.; Bailey, P.; Levine, A.J. The CDK7-cycH-p36 complex of transcription factor IIH phosphorylates p53, enhancing its sequence-specific DNA binding activity in vitro. Mol. Cell. Biol. 1997, 17, 5923–5934. [Google Scholar] [CrossRef]
- Knuesel, M.T.; Meyer, K.D.; Bernecky, C.; Taatjes, D.J. The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev. 2009, 23, 439–451. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Donner, A.J.; Espinosa, J.M. CDK8: A positive regulator of transcription. Transcription 2010, 1, 4–12. [Google Scholar] [CrossRef]
- Fu, T.-J.; Peng, J.; Lee, G.; Price, D.H.; Flores, O. Cyclin K functions as a CDK9 regulatory subunit and participates in RNA polymerase II transcription. J. Biol. Chem. 1999, 274, 34527–34530. [Google Scholar] [CrossRef]
- Peng, J.; Zhu, Y.; Milton, J.T.; Price, D.H. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998, 12, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, C.H.; Mancini, M.A. The Cdk9 and cyclin T subunits of TAK/P-TEFb localize to splicing factor-rich nuclear speckle regions. J. Cell Sci. 2001, 114, 1491–1503. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.S.; Zhao, R.; Hsu, E.L.; Cayer, J.; Ye, F.; Guo, Y.; Shyr, Y.; Cortez, D. Cyclin-dependent kinase 9–cyclin K functions in the replication stress response. EMBO Rep. 2010, 11, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Tassan, J.-P.; Jaquenoud, M.; Leopold, P.; Schultz, S.J.; Nigg, E.A. Identification of human cyclin-dependent kinase 8, a putative protein kinase partner for cyclin C. Proc. Natl. Acad. Sci. USA 1995, 92, 8871–8875. [Google Scholar] [CrossRef]
- Poss, Z.C.; Ebmeier, C.C.; Odell, A.T.; Tangpeerachaikul, A.; Lee, T.; Pelish, H.E.; Shair, M.D.; Dowell, R.D.; Old, W.M.; Taatjes, D.J. Identification of mediator kinase substrates in human cells using cortistatin A and quantitative phosphoproteomics. Cell Rep. 2016, 15, 436–450. [Google Scholar] [CrossRef]
- Ko, T.K.; Kelly, E.; Pines, J. CrkRS: A novel conserved Cdc2-related protein kinase that colocalises with SC35 speckles. J. Cell Sci. 2001, 114, 2591–2603. [Google Scholar] [CrossRef]
- Juan, H.; Lin, Y.; Chen, H.; Fann, M. Cdk12 is essential for embryonic development and the maintenance of genomic stability. Cell Death Differ. 2016, 23, 1038–1048. [Google Scholar] [CrossRef]
- Bartkowiak, B.; Liu, P.; Phatnani, H.P.; Fuda, N.J.; Cooper, J.J.; Price, D.H.; Adelman, K.; Lis, J.T.; Greenleaf, A.L. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010, 24, 2303–2316. [Google Scholar] [CrossRef]
- Cheng, S.-W.G.; Kuzyk, M.A.; Moradian, A.; Ichu, T.-A.; Chang, V.C.-D.; Tien, J.F.; Vollett, S.E.; Griffith, M.; Marra, M.A.; Morin, G.B. Interaction of cyclin-dependent kinase 12/CrkRS with cyclin K1 is required for the phosphorylation of the C-terminal domain of RNA polymerase II. Mol. Cell. Biol. 2012, 32, 4691–4704. [Google Scholar] [CrossRef]
- Bösken, C.A.; Farnung, L.; Hintermair, C.; Merzel Schachter, M.; Vogel-Bachmayr, K.; Blazek, D.; Anand, K.; Fisher, R.P.; Eick, D.; Geyer, M. The structure and substrate specificity of human Cdk12/Cyclin K. Nat. Commun. 2014, 5, 3505. [Google Scholar] [CrossRef]
- Greifenberg, A.K.; Hönig, D.; Pilarova, K.; Düster, R.; Bartholomeeusen, K.; Bösken, C.A.; Anand, K.; Blazek, D.; Geyer, M. Structural and functional analysis of the Cdk13/Cyclin K complex. Cell Rep. 2016, 14, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Gao, X.; Gilmore, J.M.; Florens, L.; Washburn, M.P.; Smith, E.; Shilatifard, A. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol. Cell. Biol. 2015, 35, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Quigley, D.A.; Robinson, T.M.; Feng, F.Y.; Ashworth, A. Transcription-Associated Cyclin-Dependent Kinases as Targets and Biomarkers for Cancer TherapyTranscription-associated CDKs in Cancer. Cancer Discov. 2020, 10, 351–370. [Google Scholar] [CrossRef] [PubMed]
- Gajdušková, P.; Ruiz de los Mozos, I.; Rájecký, M.; Hluchý, M.; Ule, J.; Blazek, D. CDK11 is required for transcription of replication-dependent histone genes. Nat. Struct. Mol. Biol. 2020, 27, 500–510. [Google Scholar] [CrossRef]
- Loyer, P.; Trembley, J.H. Roles of CDK/Cyclin complexes in transcription and pre-mRNA splicing: Cyclins L and CDK11 at the cross-roads of cell cycle and regulation of gene expression. Semin. Cell Dev. Biol. 2020, 107, 36–45. [Google Scholar] [CrossRef]
- Bunnell, B.A.; Heath, L.S.; Adams, D.E.; Lahti, J.M.; Kidd, V.J. Increased expression of a 58-kDa protein kinase leads to changes in the CHO cell cycle. Proc. Natl. Acad. Sci. USA 1990, 87, 7467–7471. [Google Scholar] [CrossRef]
- Compe, E.; Egly, J.-M. Nucleotide excision repair and transcriptional regulation: TFIIH and beyond. Annu. Rev. Biochem. 2016, 85, 265–290. [Google Scholar] [CrossRef]
- Tsai, K.-L.; Sato, S.; Tomomori-Sato, C.; Conaway, R.C.; Conaway, J.W.; Asturias, F.J. A conserved Mediator–CDK8 kinase module association regulates Mediator–RNA polymerase II interaction. Nat. Struct. Mol. Biol. 2013, 20, 611–619. [Google Scholar] [CrossRef]
- dos Santos Paparidis, N.F.; Durvale, M.C.; Canduri, F. The emerging picture of CDK9/P-TEFb: More than 20 years of advances since PITALRE. Mol. Biosyst. 2017, 13, 246–276. [Google Scholar] [CrossRef]
- Lui, G.Y.; Grandori, C.; Kemp, C.J. CDK12: An emerging therapeutic target for cancer. J. Clin. Pathol. 2018, 71, 957–962. [Google Scholar] [CrossRef]
- Guen, V.J.; Gamble, C.; Lees, J.A.; Colas, P. The awakening of the CDK10/Cyclin M protein kinase. Oncotarget 2017, 8, 50174. [Google Scholar] [CrossRef] [PubMed]
- dos Santos Paparidis, N.F.; Canduri, F. The emerging picture of CDK11: Genetic, functional and medicinal aspects. Curr. Med. Chem. 2018, 25, 880–888. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; George, R.E. Super-enhancer-driven transcriptional dependencies in cancer. Trends Cancer 2017, 3, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.Y. Targeting super-enhancers for disease treatment and diagnosis. Mol. Cells 2018, 41, 506–514. [Google Scholar]
- Sánchez-Martínez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015–2019). Bioorg. Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef]
- Guarducci, C.; Nardone, A.; Russo, D.; Nagy, Z.; Heraud, C.; Grinshpun, A.; Zhang, Q.; Freelander, A.; Leventhal, M.J.; Feit, A. Selective CDK7 Inhibition Suppresses Cell Cycle Progression and MYC Signaling While Enhancing Apoptosis in Therapy-resistant Estrogen Receptor–positive Breast Cancer. Clin. Cancer Res. 2024, 30, 1889–1905. [Google Scholar] [CrossRef]
- Choi, Y.J.; Lee, H.; Kim, D.-S.; Kim, D.H.; Kang, M.-H.; Cho, Y.-H.; Choi, C.-M.; Yoo, J.; Lee, K.-O.; Choi, E.K. Discovery of a novel CDK7 inhibitor YPN-005 in small cell lung cancer. Eur. J. Pharmacol. 2021, 907, 174298. [Google Scholar] [CrossRef]
- Li, Z.-M.; Liu, G.; Gao, Y.; Zhao, M.-G. Targeting CDK7 in oncology: The avenue forward. Pharmacol. Ther. 2022, 240, 108229. [Google Scholar] [CrossRef]
- Wu, D.; Zhang, Z.; Chen, X.; Yan, Y.; Liu, X. Angel or Devil?-CDK8 as the new drug target. Eur. J. Med. Chem. 2021, 213, 113043. [Google Scholar] [CrossRef]
- Ho, T.-Y.; Sung, T.-Y.; Pan, S.-L.; Huang, W.-J.; Hsu, K.-C.; Hsu, J.-Y.; Lin, T.E.; Hsu, C.-M.; Yang, C.-R. The study of a novel CDK8 inhibitor E966-0530–45418 that inhibits prostate cancer metastasis in vitro and in vivo. Biomed. Pharmacother. 2023, 162, 114667. [Google Scholar] [CrossRef]
- Wang, D.; Zhou, Y.; Hua, L.; Li, J.; Zhu, N.; Liu, Y. CDK3, CDK5 and CDK8 proteins as prognostic and potential biomarkers in colorectal cancer patients. Int. J. Gen. Med. 2022, 15, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Xi, M.; Chen, T.; Wu, C.; Gao, X.; Wu, Y.; Luo, X.; Du, K.; Yu, L.; Cai, T.; Shen, R. CDK8 as a therapeutic target for cancers and recent developments in discovery of CDK8 inhibitors. Eur. J. Med. Chem. 2019, 164, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Morales, F.; Giordano, A. Overview of CDK9 as a target in cancer research. Cell Cycle 2016, 15, 519–527. [Google Scholar] [CrossRef]
- Rahaman, M.H.; Lam, F.; Zhong, L.; Teo, T.; Adams, J.; Yu, M.; Milne, R.W.; Pepper, C.; Lokman, N.A.; Ricciardelli, C. Targeting CDK9 for treatment of colorectal cancer. Mol. Oncol. 2019, 13, 2178–2193. [Google Scholar] [CrossRef]
- Boffo, S.; Damato, A.; Alfano, L.; Giordano, A. CDK9 inhibitors in acute myeloid leukemia. J. Exp. Clin. Cancer Res. 2018, 37, 36. [Google Scholar] [CrossRef]
- McLaughlin, R.P.; He, J.; Van Der Noord, V.E.; Redel, J.; Foekens, J.A.; Martens, J.W.; Smid, M.; Zhang, Y.; Van de Water, B. A kinase inhibitor screen identifies a dual cdc7/CDK9 inhibitor to sensitise triple-negative breast cancer to EGFR-targeted therapy. Breast Cancer Res. 2019, 21, 77. [Google Scholar] [CrossRef]
- Ekumi, K.M.; Paculova, H.; Lenasi, T.; Pospichalova, V.; Bösken, C.A.; Rybarikova, J.; Bryja, V.; Geyer, M.; Blazek, D.; Barboric, M. Ovarian carcinoma CDK12 mutations misregulate expression of DNA repair genes via deficient formation and function of the Cdk12/CycK complex. Nucleic Acids Res. 2015, 43, 2575–2589. [Google Scholar] [CrossRef]
- Liang, J.; Zhou, X.; Yuan, L.; Chen, T.; Wan, Y.; Jiang, Y.; Meng, H.; Xu, M.; Zhang, L.; Cheng, W. Olaparib combined with CDK12-IN-3 to promote genomic instability and cell death in ovarian cancer. Int. J. Biol. Sci. 2024, 20, 4513. [Google Scholar] [CrossRef]
- Rescigno, P.; Gurel, B.; Pereira, R.; Crespo, M.; Rekowski, J.; Rediti, M.; Barrero, M.; Mateo, J.; Bianchini, D.; Messina, C. Characterizing CDK12-mutated prostate cancers. Clin. Cancer Res. 2021, 27, 566–574. [Google Scholar] [CrossRef]
- Quereda, V.; Bayle, S.; Vena, F.; Frydman, S.M.; Monastyrskyi, A.; Roush, W.R.; Duckett, D.R. Therapeutic targeting of CDK12/CDK13 in triple-negative breast cancer. Cancer Cell 2019, 36, 545–558.e7. [Google Scholar] [CrossRef]
- Cheng, L.; Zhou, S.; Zhou, S.; Shi, K.; Cheng, Y.; Cai, M.-C.; Ye, K.; Lin, L.; Zhang, Z.; Jia, C. Dual inhibition of CDK12/CDK13 targets both tumor and immune cells in ovarian cancer. Cancer Res. 2022, 82, 3588–3602. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Duckett, D.R.; Monastyrskyi, A. The promise and current status of CDK12/13 inhibition for the treatment of cancer. Future Med. Chem. 2021, 13, 117–141. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shen, J.K.; Hornicek, F.J.; Kan, Q.; Duan, Z. The emerging roles and therapeutic potential of cyclin-dependent kinase 11 (CDK11) in human cancer. Oncotarget 2016, 7, 40846. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.; Huang, S.; Peng, H.; Liu, M.; Zhao, J.; Shao, Z.; Wu, J. Critical role of CDK11 p58 in human breast cancer growth and angiogenesis. BMC Cancer 2015, 15, 701. [Google Scholar] [CrossRef]
- Blazek, D. Therapeutic potential of CDK11 in cancer. Clin. Transl. Med. 2023, 13, e1201. [Google Scholar] [CrossRef]
- Brägelmann, J.; Klümper, N.; Offermann, A.; Von Maessenhausen, A.; Böhm, D.; Deng, M.; Queisser, A.; Sanders, C.; Syring, I.; Merseburger, A.S. Pan-cancer analysis of the mediator complex transcriptome identifies CDK19 and CDK8 as therapeutic targets in advanced prostate cancer. Clin. Cancer Res. 2017, 23, 1829–1840. [Google Scholar] [CrossRef]
- Ortiz Ruiz, M.J.; Popoola, O.; Mallinger, A.; Gowan, S.; Court, W.; Box, G.; Valenti, M.; De Haven Brandon, A.; Te-Poele, R.; Workman, P. Elucidation of the different roles of CDK8 and CDK19 in colorectal cancer (CRC) using CRISPR gene editing technology. Cancer Res. 2016, 76, 4355. [Google Scholar] [CrossRef]
- Hsieh, R.W.; Kuo, A.H.; Scheeren, F.A.; Zarnegar, M.A.; Sikandar, S.S.; Antony, J.; Heitink, L.S.; Periyakoil, D.; Kalisky, T.; Sim, S. CDK19 is a Regulator of Triple-Negative Breast Cancer Growth. BioRxiv 2018, 317776. [Google Scholar] [CrossRef]
- Mayer, I.A.; Abramson, V.G.; Lehmann, B.D.; Pietenpol, J.A. New strategies for triple-negative breast cancer—Deciphering the heterogeneity. Clin. Cancer Res. 2014, 20, 782–790. [Google Scholar] [CrossRef]
- Ganuza, M.; Sáiz-Ladera, C.; Cañamero, M.; Gómez, G.; Schneider, R.; Blasco, M.A.; Pisano, D.; Paramio, J.M.; Santamaría, D.; Barbacid, M. Genetic inactivation of Cdk7 leads to cell cycle arrest and induces premature aging due to adult stem cell exhaustion. EMBO J. 2012, 31, 2498–2510. [Google Scholar] [CrossRef]
- Jagomast, T.; Idel, C.; Klapper, L.; Kuppler, P.; Offermann, A.; Dreyer, E.; Bruchhage, K.-L.; Ribbat-Idel, J.; Perner, S. CDK7 predicts worse outcome in head and neck squamous-cell cancer. Cancers 2022, 14, 492. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, M.; Zhang, X.; Huang, H.; Huang, J.; Ke, J.; Ding, H.; Xiao, J.; Shan, X.; Liu, Q. Upregulation of CDK7 in gastric cancer cell promotes tumor cell proliferation and predicts poor prognosis. Exp. Mol. Pathol. 2016, 100, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Abduljabbar, R.; Lai, C.-F.; Periyasamy, M.; Harrod, A.; Gemma, C.; Steel, J.H.; Patel, N.; Busonero, C.; Jerjees, D. Expression of CDK7, Cyclin H, and MAT1 Is Elevated in Breast Cancer and Is Prognostic in Estrogen Receptor–Positive Breast CancerCDK7 Expression in Breast Cancer. Clin. Cancer Res. 2016, 22, 5929–5938. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Sirica, A.E.; Gores, G.J.; Groopman, J.D.; Selaru, F.M.; Strazzabosco, M.; Wei Wang, X.; Zhu, A.X. Intrahepatic cholangiocarcinoma: Continuing challenges and translational advances. Hepatology 2019, 69, 1803–1815. [Google Scholar] [CrossRef]
- Valle, J.W.; Lamarca, A.; Goyal, L.; Barriuso, J.; Zhu, A.X. New Horizons for Precision Medicine in Biliary Tract Cancers Precision Medicine in BTC. Cancer Discov. 2017, 7, 943–962. [Google Scholar] [CrossRef]
- Chen, H.-D.; Huang, C.-S.; Xu, Q.-C.; Li, F.; Huang, X.-T.; Wang, J.-Q.; Li, S.-J.; Zhao, W.; Yin, X.-Y. Therapeutic targeting of CDK7 suppresses tumor progression in intrahepatic cholangiocarcinoma. Int. J. Biol. Sci. 2020, 16, 1207. [Google Scholar] [CrossRef]
- Jemal, A.; Murray, T.; Ward, E.; Samuels, A.; Tiwari, R.C.; Ghafoor, A.; Feuer, E.J.; Thun, M.J. Cancer Statistics, 2005. CA Cancer J. Clin. 2005, 55, 10–30. [Google Scholar] [CrossRef]
- Robboy, S.J. Robboy’s Pathology of the Female Reproductive Tract; Elsevier Health Sciences: New York, NY, USA, 2009. [Google Scholar]
- Zhou, Q. Targeting cyclin-dependent kinases in ovarian cancer. Cancer Investig. 2017, 35, 367–376. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Bender, H.; Espinosa, J.M. Therapeutic targeting of transcriptional cyclin-dependent kinases. Transcription 2019, 10, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cho, Y.-J.; Ryu, J.-Y.; Hwang, I.; Han, H.D.; Ahn, H.J.; Kim, W.Y.; Cho, H.; Chung, J.-Y.; Hewitt, S.M. CDK7 is a reliable prognostic factor and novel therapeutic target in epithelial ovarian cancer. Gynecol. Oncol. 2020, 156, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Peng, H.; Wang, X.; Yin, X.; Ma, P.; Jing, Y.; Cai, M.-C.; Liu, J.; Zhang, M.; Zhang, S. Preclinical Efficacy and Molecular Mechanism of Targeting CDK7-Dependent Transcriptional Addiction in Ovarian CancerCDK7-Dependent Transcriptional Addiction in Ovarian Cancer. Mol. Cancer Ther. 2017, 16, 1739–1750. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef]
- Sun, B.; Mason, S.; Wilson, R.C.; Hazard, S.E.; Wang, Y.; Fang, R.; Wang, Q.; Yeh, E.S.; Yang, M.; Roberts, T.M. Inhibition of the transcriptional kinase CDK7 overcomes therapeutic resistance in HER2-positive breast cancers. Oncogene 2020, 39, 50–63. [Google Scholar] [CrossRef]
- Attia, Y.M.; Shouman, S.A.; Salama, S.A.; Ivan, C.; Elsayed, A.M.; Amero, P.; Rodriguez-Aguayo, C.; Lopez-Berestein, G. Blockade of CDK7 reverses endocrine therapy resistance in breast cancer. Int. J. Mol. Sci. 2020, 21, 2974. [Google Scholar] [CrossRef]
- Reese, J.M.; Bruinsma, E.S.; Monroe, D.G.; Negron, V.; Suman, V.J.; Ingle, J.N.; Goetz, M.P.; Hawse, J.R. ERβ inhibits cyclin dependent kinases 1 and 7 in triple negative breast cancer. Oncotarget 2017, 8, 96506. [Google Scholar] [CrossRef]
- Peng, J.; Yang, M.; Bi, R.; Wang, Y.; Wang, C.; Wei, X.; Zhang, Z.; Xie, X.; Wei, W. Targeting mutated p53 dependency in triple-negative breast cancer cells through CDK7 inhibition. Front. Oncol. 2021, 11, 664848. [Google Scholar] [CrossRef]
- Lu, P.; Geng, J.; Zhang, L.; Wang, Y.; Niu, N.; Fang, Y.; Liu, F.; Shi, J.; Zhang, Z.-G.; Sun, Y.-W. THZ1 reveals CDK7-dependent transcriptional addictions in pancreatic cancer. Oncogene 2019, 38, 3932–3945. [Google Scholar] [CrossRef]
- Zeng, S.; Lan, B.; Ren, X.; Zhang, S.; Schreyer, D.; Eckstein, M.; Yang, H.; Britzen-Laurent, N.; Dahl, A.; Mukhopadhyay, D. CDK7 inhibition augments response to multidrug chemotherapy in pancreatic cancer. J. Exp. Clin. Cancer Res. 2022, 41, 241. [Google Scholar] [CrossRef]
- Jing, N.; Tweardy, D.J. Targeting Stat3 in cancer therapy. Anti-Cancer Drugs 2005, 16, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.; Mehanna, H. WEE1 inhibitor: Clinical development. Curr. Oncol. Rep. 2021, 23, 107. [Google Scholar] [CrossRef] [PubMed]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting WEE1 kinase in cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Geenen, J.J.; Schellens, J.H. Molecular Pathways: Targeting the Protein Kinase Wee1 in Cancer Targeting the Protein Kinase Wee1 in Cancer. Clin. Cancer Res. 2017, 23, 4540–4544. [Google Scholar] [CrossRef]
- Osman, S.; Mohammad, E.; Lidschreiber, M.; Stuetzer, A.; Bazsó, F.L.; Maier, K.C.; Urlaub, H.; Cramer, P. The Cdk8 kinase module regulates interaction of the mediator complex with RNA polymerase II. J. Biol. Chem. 2021, 296, 100734. [Google Scholar] [CrossRef]
- Luyties, O.; Taatjes, D.J. The Mediator kinase module: An interface between cell signaling and transcription. Trends Biochem. Sci. 2022, 47, 314–327. [Google Scholar] [CrossRef]
- Broude, E.V.; Gyorffy, B.; Chumanevich, A.A.; Chen, M.; McDermott, M.S.J.; Shtutman, M.; Catroppo, J.F.; Roninson, I. Expression of CDK8 and CDK8-interacting genes as potential biomarkers in breast cancer. Curr. Cancer Drug Targets 2015, 15, 739–749. [Google Scholar] [CrossRef]
- Kapoor, A.; Goldberg, M.S.; Cumberland, L.K.; Ratnakumar, K.; Segura, M.F.; Emanuel, P.O.; Menendez, S.; Vardabasso, C.; LeRoy, G.; Vidal, C.I. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature 2010, 468, 1105–1109. [Google Scholar] [CrossRef]
- Donner, A.J.; Ebmeier, C.C.; Taatjes, D.J.; Espinosa, J.M. CDK8 is a positive regulator of transcriptional elongation within the serum response network. Nat. Struct. Mol. Biol. 2010, 17, 194–201. [Google Scholar] [CrossRef]
- Porter, D.C.; Farmaki, E.; Altilia, S.; Schools, G.P.; West, D.K.; Chen, M.; Chang, B.-D.; Puzyrev, A.T.; Lim, C.-u.; Rokow-Kittell, R. Cyclin-dependent kinase 8 mediates chemotherapy-induced tumor-promoting paracrine activities. Proc. Natl. Acad. Sci. USA 2012, 109, 13799–13804. [Google Scholar] [CrossRef]
- Xu, D.; Li, C.-F.; Zhang, X.; Gong, Z.; Chan, C.-H.; Lee, S.-W.; Jin, G.; Rezaeian, A.-H.; Han, F.; Wang, J. Skp2–MacroH2A1–CDK8 axis orchestrates G2/M transition and tumorigenesis. Nat. Commun. 2015, 6, 6641. [Google Scholar] [CrossRef] [PubMed]
- McDermott, M.S.; Chumanevich, A.A.; Lim, C.-u.; Liang, J.; Chen, M.; Altilia, S.; Oliver, D.; Rae, J.M.; Shtutman, M.; Kiaris, H. Inhibition of CDK8 mediator kinase suppresses estrogen dependent transcription and the growth of estrogen receptor positive breast cancer. Oncotarget 2017, 8, 12558. [Google Scholar] [CrossRef] [PubMed]
- Crown, J. CDK8: A new breast cancer target. Oncotarget 2017, 8, 14269. [Google Scholar] [CrossRef]
- Seo, J.-O.; Han, S.I.; Lim, S.-C. Role of CDK8 and β-catenin in colorectal adenocarcinoma. Oncol. Rep. 2010, 24, 285–291. [Google Scholar]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S. CDK8 is a colorectal cancer oncogene that regulates β-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef]
- Wu, T.; Qin, Z.; Tian, Y.; Wang, J.; Xu, C.; Li, Z.; Bian, J. Recent developments in the biology and medicinal chemistry of CDK9 inhibitors: An update. J. Med. Chem. 2020, 63, 13228–13257. [Google Scholar] [CrossRef]
- Ma, H.; Seebacher, N.A.; Hornicek, F.J.; Duan, Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in osteosarcoma. EBioMedicine 2019, 39, 182–193. [Google Scholar] [CrossRef]
- He, S.; Fang, X.; Xia, X.; Hou, T.; Zhang, T. Targeting CDK9: A novel biomarker in the treatment of endometrial cancer. Oncol. Rep. 2020, 44, 1929–1938. [Google Scholar] [CrossRef]
- Kretz, A.-L.; Schaum, M.; Richter, J.; Kitzig, E.F.; Engler, C.C.; Leithäuser, F.; Henne-Bruns, D.; Knippschild, U.; Lemke, J. CDK9 is a prognostic marker and therapeutic target in pancreatic cancer. Tumor Biol. 2017, 39, 1010428317694304. [Google Scholar] [CrossRef]
- Chen, R.; Wierda, W.G.; Chubb, S.; Hawtin, R.E.; Fox, J.A.; Keating, M.J.; Gandhi, V.; Plunkett, W. Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood J. Am. Soc. Hematol. 2009, 113, 4637–4645. [Google Scholar] [CrossRef]
- Li, X.-Y.; Green, M.R. Tumorigenesis: Transcriptional elongation and cancer. Curr. Biol. 1996, 6, 943–944. [Google Scholar] [CrossRef] [PubMed]
- Guha, M. Cyclin-dependent kinase inhibitors move into Phase III. Nat. Rev. Drug Discov. 2012, 11, 892. [Google Scholar] [CrossRef] [PubMed]
- Schlafstein, A.J.; Withers, A.E.; Rudra, S.; Danelia, D.; Switchenko, J.M.; Mister, D.; Harari, S.; Zhang, H.; Daddacha, W.; Ehdaivand, S. CDK9 expression shows role as a potential prognostic biomarker in breast cancer patients who fail to achieve pathologic complete response after neoadjuvant chemotherapy. Int. J. Breast Cancer 2018, 2018, 6945129. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-m.; Giltnane, J.M.; Balko, J.M.; Schwarz, L.J.; Guerrero-Zotano, A.L.; Hutchinson, K.E.; Nixon, M.J.; Estrada, M.V.; Sánchez, V.; Sanders, M.E. MYC and MCL1 cooperatively promote chemotherapy-resistant breast cancer stem cells via regulation of mitochondrial oxidative phosphorylation. Cell Metab. 2017, 26, 633–647.e7. [Google Scholar] [CrossRef]
- Bolomsky, A.; Vogler, M.; Köse, M.C.; Heckman, C.A.; Ehx, G.; Ludwig, H.; Caers, J. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J. Hematol. Oncol. 2020, 13, 173. [Google Scholar] [CrossRef]
- Brisard, D.; Eckerdt, F.; Marsh, L.A.; Blyth, G.T.; Jain, S.; Cristofanilli, M.; Horiuchi, D.; Platanias, L.C. Antineoplastic effects of selective CDK9 inhibition with atuveciclib on cancer stem-like cells in triple-negative breast cancer. Oncotarget 2018, 9, 37305. [Google Scholar] [CrossRef]
- Cheng, S.-S.; Qu, Y.-Q.; Wu, J.; Yang, G.-J.; Liu, H.; Wang, W.; Huang, Q.; Chen, F.; Li, G.; Wong, C.-Y. Inhibition of the CDK9–cyclin T1 protein–protein interaction as a new approach against triple-negative breast cancer. Acta Pharm. Sin. B 2022, 12, 1390–1405. [Google Scholar] [CrossRef]
- Mustafa, E.H.; Laven-Law, G.; Kikhtyak, Z.; Nguyen, V.; Ali, S.; Pace, A.A.; Iggo, R.; Kebede, A.; Noll, B.; Wang, S. Selective inhibition of CDK9 in triple negative breast cancer. Oncogene 2024, 43, 202–215. [Google Scholar] [CrossRef]
- Beird, H.C.; Bielack, S.S.; Flanagan, A.M.; Gill, J.; Heymann, D.; Janeway, K.A.; Livingston, J.A.; Roberts, R.D.; Strauss, S.J.; Gorlick, R. Osteosarcoma. Nat. Rev. Dis. Primers 2022, 8, 77. [Google Scholar] [CrossRef]
- Peyressatre, M.; Prével, C.; Pellerano, M.; Morris, M.C. Targeting cyclin-dependent kinases in human cancers: From small molecules to peptide inhibitors. Cancers 2015, 7, 179–237. [Google Scholar] [CrossRef]
- Walker, R.L.; Hornicek, F.J.; Duan, Z. Transcriptional regulation and therapeutic potential of cyclin-dependent kinase 9 (CDK9) in sarcoma. Biochem. Pharmacol. 2024, 226, 116342. [Google Scholar] [CrossRef] [PubMed]
- Arora, T.; Mullangi, S.; Lekkala, M.R. Ovarian cancer. In StatPearls; StatPearls Publishing LLC: St. Petersburg, FL, USA, 2021. [Google Scholar]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dean, D.C.; Hornicek, F.J.; Shi, H.; Duan, Z. Cyclin-dependent kinase 9 (CDK9) is a novel prognostic marker and therapeutic target in ovarian cancer. FASEB J. 2019, 33, 5990. [Google Scholar] [CrossRef]
- Li, J.; Zhi, X.; Chen, S.; Shen, X.; Chen, C.; Yuan, L.; Guo, J.; Meng, D.; Chen, M.; Yao, L. CDK9 inhibitor CDKI-73 is synergetic lethal with PARP inhibitor olaparib in BRCA1 wide-type ovarian cancer. Am. J. Cancer Res. 2020, 10, 1140. [Google Scholar] [PubMed]
- Liu, H.; Liu, K.; Dong, Z. Targeting CDK12 for cancer therapy: Function, mechanism, and drug discovery. Cancer Res. 2021, 81, 18–26. [Google Scholar] [CrossRef]
- Emadi, F.; Teo, T.; Rahaman, M.H.; Wang, S. CDK12: A potential therapeutic target in cancer. Drug Discov. Today 2020, 25, 2257–2267. [Google Scholar] [CrossRef]
- Eifler, T.T.; Shao, W.; Bartholomeeusen, K.; Fujinaga, K.; Jäger, S.; Johnson, J.R.; Luo, Z.; Krogan, N.J.; Peterlin, B.M. Cyclin-dependent kinase 12 increases 3′ end processing of growth factor-induced c-FOS transcripts. Mol. Cell. Biol. 2015, 35, 468–478. [Google Scholar] [CrossRef]
- Davidson, L.; Muniz, L.; West, S. 3′ end formation of pre-mRNA and phosphorylation of Ser2 on the RNA polymerase II CTD are reciprocally coupled in human cells. Genes Dev. 2014, 28, 342–356. [Google Scholar] [CrossRef]
- Zhang, T.; Kwiatkowski, N.; Olson, C.M.; Dixon-Clarke, S.E.; Abraham, B.J.; Greifenberg, A.K.; Ficarro, S.B.; Elkins, J.M.; Liang, Y.; Hannett, N.M. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol. 2016, 12, 876–884. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Paculová, H.; Kohoutek, J. The emerging roles of CDK12 in tumorigenesis. Cell Div. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Chilà, R.; Guffanti, F.; Damia, G. Role and therapeutic potential of CDK12 in human cancers. Cancer Treat. Rev. 2016, 50, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Pilarova, K.; Herudek, J.; Blazek, D. CDK12: Cellular functions and therapeutic potential of versatile player in cancer. NAR Cancer 2020, 2, zcaa003. [Google Scholar] [CrossRef]
- Peng, F.; Yang, C.; Kong, Y.; Huang, X.; Chen, Y.; Zhou, Y.; Xie, X.; Liu, P. CDK12 promotes breast cancer progression and maintains stemness by activating c-myc/β-catenin signaling. Curr. Cancer Drug Targets 2020, 20, 156–165. [Google Scholar] [CrossRef]
- Choi, H.J.; Jin, S.; Cho, H.; Won, H.Y.; An, H.W.; Jeong, G.Y.; Park, Y.U.; Kim, H.Y.; Park, M.K.; Son, T. CDK 12 drives breast tumor initiation and trastuzumab resistance via WNT and IRS 1-ErbB-PI 3K signaling. EMBO Rep. 2019, 20, e48058. [Google Scholar] [CrossRef]
- Zeng, M.; Kwiatkowski, N.P.; Zhang, T.; Nabet, B.; Xu, M.; Liang, Y.; Quan, C.; Wang, J.; Hao, M.; Palakurthi, S. Targeting MYC dependency in ovarian cancer through inhibition of CDK7 and CDK12/13. eLife 2018, 7, e39030. [Google Scholar] [CrossRef]
- Joshi, P.M.; Sutor, S.L.; Huntoon, C.J.; Karnitz, L.M. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly (ADP-ribose) polymerase inhibitors. J. Biol. Chem. 2014, 289, 9247–9253. [Google Scholar] [CrossRef]
- Reimers, M.A.; Yip, S.M.; Zhang, L.; Cieslik, M.; Dhawan, M.; Montgomery, B.; Wyatt, A.W.; Chi, K.N.; Small, E.J.; Chinnaiyan, A.M. Clinical outcomes in cyclin-dependent kinase 12 mutant advanced prostate cancer. Eur. Urol. 2020, 77, 333–341. [Google Scholar] [CrossRef]
- Wu, Y.-M.; Cieślik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 2018, 173, 1770–1782.e14. [Google Scholar] [CrossRef]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Rodrigues, D.N.; Gurel, B.; Clarke, M. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Investig. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Quigley, D.A.; Dang, H.X.; Zhao, S.G.; Lloyd, P.; Aggarwal, R.; Alumkal, J.J.; Foye, A.; Kothari, V.; Perry, M.D.; Bailey, A.M. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018, 174, 758–769.e9. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Xie, T.; Guo, Y.; Wang, D.; Qiu, M.; Han, R.; Qing, G.; Liang, K.; Liu, H. CDK13 phosphorylates the translation machinery and promotes tumorigenic protein synthesis. Oncogene 2023, 42, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-L.; Zhan, R.; Zhao, G.-M.; Qian, Z.-H.; Gong, C.-C.; Li, Y.-Q. Expression of CDK13 was associated with prognosis and expression of HIF-1α and beclin1 in breast cancer patients. J. Investig. Surg. 2022, 35, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Cesari, E.; Ciucci, A.; Pieraccioli, M.; Caggiano, C.; Nero, C.; Bonvissuto, D.; Sillano, F.; Buttarelli, M.; Piermattei, A.; Loverro, M. Dual inhibition of CDK12 and CDK13 uncovers actionable vulnerabilities in patient-derived ovarian cancer organoids. J. Exp. Clin. Cancer Res. 2023, 42, 126. [Google Scholar] [CrossRef]
- Liu, X.; Gao, Y.; Shen, J.; Yang, W.; Choy, E.; Mankin, H.; Hornicek, F.J.; Duan, Z. Cyclin-dependent kinase 11 (CDK11) is required for ovarian cancer cell growth in vitro and in vivo, and its inhibition causes apoptosis and sensitizes cells to paclitaxel. Mol. Cancer Ther. 2016, 15, 1691–1701. [Google Scholar] [CrossRef]
- Jia, B.; Choy, E.; Cote, G.; Harmon, D.; Ye, S.; Kan, Q.; Mankin, H.; Hornicek, F.; Duan, Z. Cyclin-dependent kinase 11 (CDK11) is crucial in the growth of liposarcoma cells. Cancer Lett. 2014, 342, 104–112. [Google Scholar] [CrossRef]
- Kelso, S.; O’Brien, S.; Kurinov, I.; Angers, S.; Sicheri, F. Crystal structure of the CDK11 kinase domain bound to the small-molecule inhibitor OTS964. Structure 2022, 30, 1615–1625.e4. [Google Scholar] [CrossRef]
- Kren, B.T.; Unger, G.M.; Abedin, M.J.; Vogel, R.I.; Henzler, C.M.; Ahmed, K.; Trembley, J.H. Preclinical evaluation of cyclin dependent kinase 11 and casein kinase 2 survival kinases as RNA interference targets for triple negative breast cancer therapy. Breast Cancer Res. 2015, 17, 19. [Google Scholar] [CrossRef]
- Zhou, Y.; Han, C.; Li, D.; Yu, Z.; Li, F.; Li, F.; An, Q.; Bai, H.; Zhang, X.; Duan, Z. Cyclin-dependent kinase 11p110 (CDK11p110) is crucial for human breast cancer cell proliferation and growth. Sci. Rep. 2015, 5, 10433. [Google Scholar] [CrossRef]
- Borowczak, J.; Szczerbowski, K.; Ahmadi, N.; Szylberg, Ł. CDK9 inhibitors in multiple myeloma: A review of progress and perspectives. Med. Oncol. 2022, 39, 39. [Google Scholar] [CrossRef]
- Tiedemann, R.E.; Zhu, Y.X.; Schmidt, J.; Shi, C.X.; Sereduk, C.; Yin, H.; Mousses, S.; Stewart, A.K. Identification of molecular vulnerabilities in human multiple myeloma cells by RNA interference lethality screening of the druggable genome. Cancer Res. 2012, 72, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Sassi, S.; Shen, J.K.; Yang, X.; Gao, Y.; Osaka, E.; Zhang, J.; Yang, S.; Yang, C.; Mankin, H.J. Targeting Cdk11 in osteosarcoma cells using the CRISPR-cas9 system. J. Orthop. Res. 2015, 33, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Zhang, J.; Choy, E.; Harmon, D.; Liu, X.; Nielsen, P.; Mankin, H.; Gray, N.S.; Hornicek, F.J. Systematic kinome shRNA screening identifies CDK11 (PITSLRE) kinase expression is critical for osteosarcoma cell growth and proliferation. Clin. Cancer Res. 2012, 18, 4580–4588. [Google Scholar] [CrossRef] [PubMed]
- Fant, C.B.; Taatjes, D.J. Regulatory functions of the Mediator kinases CDK8 and CDK19. Transcription 2019, 10, 76–90. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CDK Name | Functions | Expression in Cancer | References |
|---|---|---|---|
| CDK7 |
|
| [28,97,98,99] |
| CDK8 |
|
| [100,101,102,103] |
| CDK9 |
|
| [104,105,106,107] |
| CDK12 |
|
| [108,109,110] |
| CDK13 |
|
| [111,112,113] |
| CDK11 |
|
| [114,115,116] |
| CDK19 |
|
| [117,118,119] |
| Cancer Type | Expression and Association | Effects of CDK11 Knockdown | Therapeutic Implications | References |
|---|---|---|---|---|
| Breast Cancer (BC) | Overexpressed in BC cell lines and tumor tissues; associated with poor prognosis, advanced TNM stage, and poor differentiation. There is 100% staining and high expression in TNBC in patient tissues. | Reduces viability and clonal survival; causes apoptosis; and inhibits growth and migration. Tumors are reduced by in vivo treatment. | Promising target for BC, particularly TNBC. | [201,202] |
| Multiple Myeloma (MM) | Relative to normal tissues, overexpressed in initial myeloma tissues. Determined using RNAi lethality screening to be a survival gene. | Its function as a survival gene is supported by inhibition, which lowers viability. | Represents a novel therapeutic strategy for myeloma. | [203,204] |
| Osteosarcoma | High expression in osteosarcoma cells is linked to a lower survival rate for patients. | Causes apoptosis and cell death; inhibits growth, migration, and invasion. The findings of CRISPR-Cas9 silencing are comparable. | The development of CDK11 inhibitors may help treat osteosarcoma. | [205,206] |
| Liposarcoma | Compared with benign lipomas, overexpressed in liposarcoma tissues. | Increases doxorubicin cytotoxicity while decreasing proliferation and inducing apoptosis. | Additional pathway research is necessary to determine whether CDK11 is a viable therapeutic target. | [199] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alrouji, M.; Alshammari, M.S.; Anwar, S.; Venkatesan, K.; Shamsi, A. Mechanistic Roles of Transcriptional Cyclin-Dependent Kinases in Oncogenesis: Implications for Cancer Therapy. Cancers 2025, 17, 1554. https://doi.org/10.3390/cancers17091554
Alrouji M, Alshammari MS, Anwar S, Venkatesan K, Shamsi A. Mechanistic Roles of Transcriptional Cyclin-Dependent Kinases in Oncogenesis: Implications for Cancer Therapy. Cancers. 2025; 17(9):1554. https://doi.org/10.3390/cancers17091554
Chicago/Turabian StyleAlrouji, Mohammed, Mohammed S. Alshammari, Saleha Anwar, Kumar Venkatesan, and Anas Shamsi. 2025. "Mechanistic Roles of Transcriptional Cyclin-Dependent Kinases in Oncogenesis: Implications for Cancer Therapy" Cancers 17, no. 9: 1554. https://doi.org/10.3390/cancers17091554
APA StyleAlrouji, M., Alshammari, M. S., Anwar, S., Venkatesan, K., & Shamsi, A. (2025). Mechanistic Roles of Transcriptional Cyclin-Dependent Kinases in Oncogenesis: Implications for Cancer Therapy. Cancers, 17(9), 1554. https://doi.org/10.3390/cancers17091554