Paediatric Thyroid Carcinoma: The Genetic Revolution and Its Implications for Therapy and Outcomes

 , , , and
, , , and
Simple Summary
Abstract
1. Introduction
2. Somatic Gene Alterations in Differentiated Thyroid Carcinomas
2.1. RET
2.2. NTRK
2.3. ALK
2.4. BRAF
2.5. TERT
2.6. RAS
2.7. Pax-8/PPAR-Gamma
3. Germline P/LPV Associated with Predisposition to Thyroid Carcinomas
3.1. PTEN Hamartoma Tumour Syndrome
3.2. DICER1 Syndrome
3.3. Carney Complex Type 1
3.4. Familial Adenomatous Polyposis
3.5. Werner Syndrome
3.6. Multiple Endocrine Neoplasia Type 2
4. Germline Molecular Testing for Cancer Predisposition Syndromes
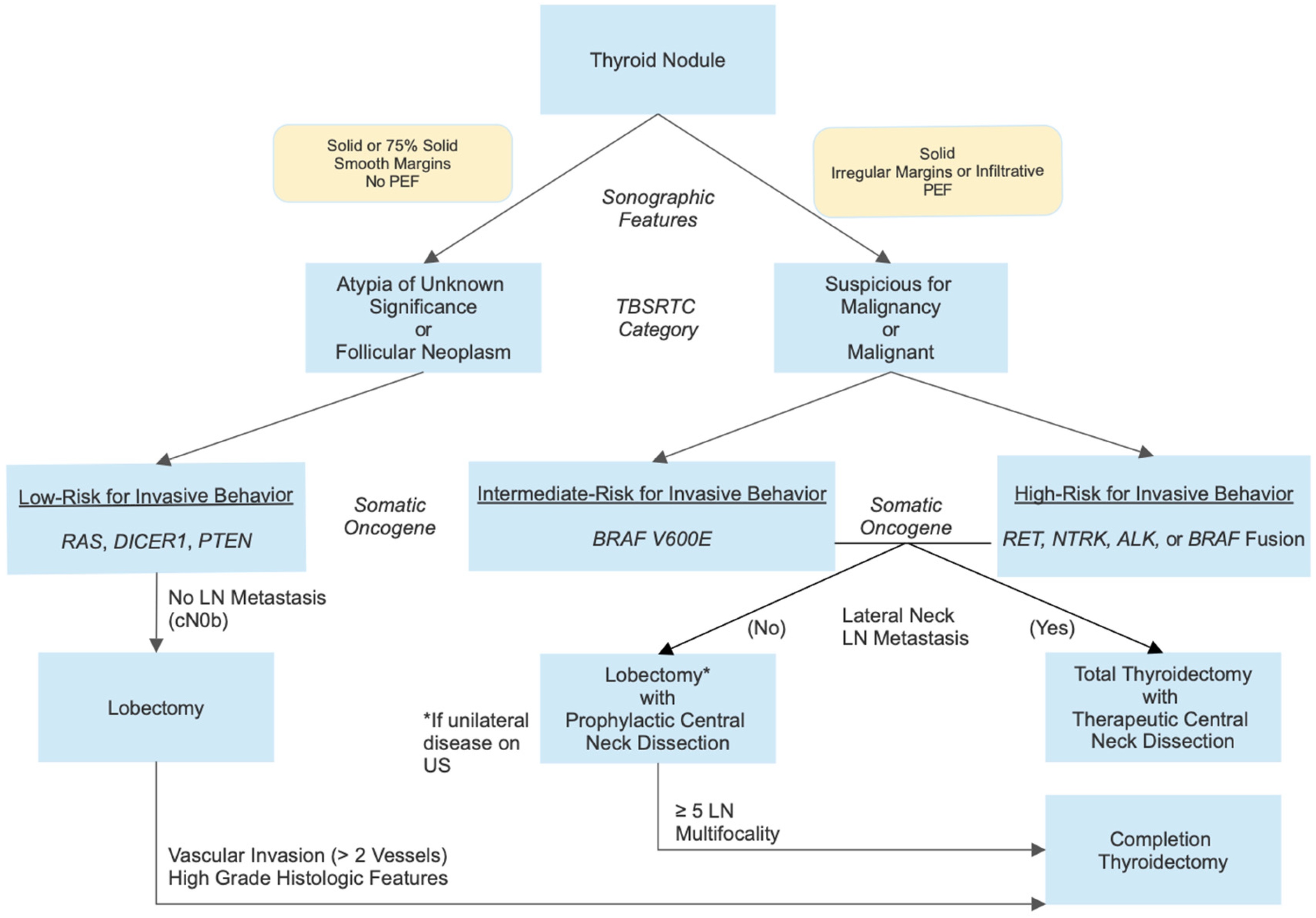
5. Molecular Testing of Thyroid Nodule FNA Samples
6. The Value of a Molecular Diagnosis Post-Operatively
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Richman, D.M.; Benson, C.B.; Doubilet, P.M.; Peters, H.E.; Huang, S.A.; Asch, E.; Wassner, A.J.; Smith, J.R.; Cherella, C.E.; Frates, M.C. Thyroid Nodules in Pediatric Patients: Sonographic Characteristics and Likelihood of Cancer. Radiology 2018, 288, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Lebbink, C.A.; Links, T.P.; Czarniecka, A.; Dias, R.P.; Elisei, R.; Izatt, L.; Krude, H.; Lorenz, K.; Luster, M.; Newbold, K.; et al. 2022 European Thyroid Association Guidelines for the management of pediatric thyroid nodules and differentiated thyroid carcinoma. Eur. Thyroid. J. 2022, 11, e220146. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Ly, S.; Castroneves, L.A.; Frates, M.C.; Benson, C.B.; Feldman, H.A.; Wassner, A.J.; Smith, J.R.; Marqusee, E.; Alexander, E.K.; et al. A standardized assessment of thyroid nodules in children confirms higher cancer prevalence than in adults. J. Clin. Endocrinol. Metab. 2013, 98, 3238–3245. [Google Scholar] [CrossRef] [PubMed]
- Cherella, C.E.; Wassner, A.J. Pediatric thyroid cancer: Recent developments. Best. Pract. Res. Clin. Endocrinol. Metab. 2023, 37, 101715. [Google Scholar] [CrossRef]
- Bauer, A.J. Molecular Genetics of Thyroid Cancer in Children and Adolescents. Endocrinol. Metab. Clin. North. Am. 2017, 46, 389–403. [Google Scholar] [CrossRef]
- Francis, G.L.; Waguespack, S.G.; Bauer, A.J.; Angelos, P.; Benvenga, S.; Cerutti, J.M.; Dinauer, C.A.; Hamilton, J.; Hay, I.D.; Luster, M.; et al. Management Guidelines for Children with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2015, 25, 716–759. [Google Scholar] [CrossRef]
- Handkiewicz-Junak, D.; Niedziela, M.; Lewinski, A.; Bosowski, A.; Chmielik, E.; Czarniecka, A.; Dedecjus, M.; Dembowska-Baginska, B.; Gawlik-Starzyk, A.; Gorecki, W.; et al. Diagnostics and treatment of differentiated thyroid carcinoma in children—Guidelines of the Polish National Scientific Societies, 2024 Update. Endokrynol. Pol. 2024, 75, 565–591. [Google Scholar] [CrossRef]
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef]
- Hensley, S.G.; Hu, M.I.; Bassett, R.L.; Ying, A.K.; Zafereo, M.E.; Perrier, N.D.; Busaidy, N.L.; Hyde, S.M.; Grubbs, E.G.; Waguespack, S.G. Pediatric Medullary Thyroid Carcinoma: Clinical Presentations and Long-Term Outcomes in 144 Patients over 6 Decades. J. Clin. Endocrinol. Metab. 2024, 109, 2256–2268. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef]
- Gallant, J.N.; Chen, S.C.; Ortega, C.A.; Rohde, S.L.; Belcher, R.H.; Netterville, J.L.; Baregamian, N.; Wang, H.; Liang, J.; Ye, F.; et al. Evaluation of the Molecular Landscape of Pediatric Thyroid Nodules and Use of a Multigene Genomic Classifier in Children. JAMA Oncol. 2022, 8, 1323–1327. [Google Scholar] [CrossRef] [PubMed]
- Pekova, B.; Sykorova, V.; Dvorakova, S.; Vaclavikova, E.; Moravcova, J.; Katra, R.; Astl, J.; Vlcek, P.; Kodetova, D.; Vcelak, J.; et al. RET, NTRK, ALK, BRAF, and MET Fusions in a Large Cohort of Pediatric Papillary Thyroid Carcinomas. Thyroid 2020, 30, 1771–1780. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S.; Alswailem, M.; Alswailem, A.A.; Al-Hindi, H.; Goljan, E.; Alsudairy, N.; Abouelhoda, M. Genetic Alterations in Pediatric Thyroid Cancer Using a Comprehensive Childhood Cancer Gene Panel. J. Clin. Endocrinol. Metab. 2020, 105, 3324–3334. [Google Scholar] [CrossRef]
- Franco, A.T.; Ricarte-Filho, J.C.; Isaza, A.; Jones, Z.; Jain, N.; Mostoufi-Moab, S.; Surrey, L.; Laetsch, T.W.; Li, M.M.; DeHart, J.C.; et al. Fusion Oncogenes Are Associated with Increased Metastatic Capacity and Persistent Disease in Pediatric Thyroid Cancers. J. Clin. Oncol. 2022, 40, 1081–1090. [Google Scholar] [CrossRef]
- Stosic, A.; Fuligni, F.; Anderson, N.D.; Davidson, S.; de Borja, R.; Acker, M.; Forte, V.; Campisi, P.; Propst, E.J.; Wolter, N.E.; et al. Diverse Oncogenic Fusions and Distinct Gene Expression Patterns Define the Genomic Landscape of Pediatric Papillary Thyroid Carcinoma. Cancer Res. 2021, 81, 5625–5637. [Google Scholar] [CrossRef]
- Guleria, P.; Srinivasan, R.; Rana, C.; Agarwal, S. Molecular Landscape of Pediatric Thyroid Cancer: A Review. Diagnostics 2022, 12, 3136. [Google Scholar] [CrossRef]
- Rangel-Pozzo, A.; Sisdelli, L.; Cordioli, M.I.V.; Vaisman, F.; Caria, P.; Mai, S.; Cerutti, J.M. Genetic Landscape of Papillary Thyroid Carcinoma and Nuclear Architecture: An Overview Comparing Pediatric and Adult Populations. Cancers 2020, 12, 3146. [Google Scholar] [CrossRef]
- Mostoufi-Moab, S.; Labourier, E.; Sullivan, L.; LiVolsi, V.; Li, Y.; Xiao, R.; Beaudenon-Huibregtse, S.; Kazahaya, K.; Adzick, N.S.; Baloch, Z.; et al. Molecular Testing for Oncogenic Gene Alterations in Pediatric Thyroid Lesions. Thyroid 2018, 28, 60–67. [Google Scholar] [CrossRef]
- de Sousa, M.S.A.; Nunes, I.N.; Christiano, Y.P.; Sisdelli, L.; Cerutti, J.M. Genetic alterations landscape in paediatric thyroid tumours and/or differentiated thyroid cancer: Systematic review. Rev. Endocr. Metab. Disord. 2024, 25, 35–51. [Google Scholar] [CrossRef]
- Morton, L.M.; Karyadi, D.M.; Stewart, C.; Bogdanova, T.I.; Dawson, E.T.; Steinberg, M.K.; Dai, J.; Hartley, S.W.; Schonfeld, S.J.; Sampson, J.N.; et al. Radiation-related genomic profile of papillary thyroid carcinoma after the Chernobyl accident. Science 2021, 372, eabg2538. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, Q.; Zhang, K.; Liang, Y.; Ren, F.; Zhang, J.; Kan, C.; Han, F.; Sun, X. NTRK fusions in thyroid cancer: Pathology and clinical aspects. Crit. Rev. Oncol. Hematol. 2023, 184, 103957. [Google Scholar] [CrossRef] [PubMed]
- Ricarte-Filho, J.C.; Halada, S.; O’Neill, A.; Casado-Medrano, V.; Laetsch, T.W.; Franco, A.T.; Bauer, A.J. The clinical aspect of NTRK-fusions in pediatric papillary thyroid cancer. Cancer Genet. 2022, 262, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Nies, M.; Vassilopoulou-Sellin, R.; Bassett, R.L.; Yedururi, S.; Zafereo, M.E.; Cabanillas, M.E.; Sherman, S.I.; Links, T.P.; Waguespack, S.G. Distant Metastases from Childhood Differentiated Thyroid Carcinoma: Clinical Course and Mutational Landscape. J. Clin. Endocrinol. Metab. 2021, 106, e1683–e1697. [Google Scholar] [CrossRef] [PubMed]
- Shih, K.P.; Lee, Y.C.; Tsai, J.J.; Lin, S.H.; Liu, C.Y.; Li, W.S.; Li, C.F.; Hang, J.F. Clinicopathologic Features and Cytologic Correlation of ALK-Rearranged Papillary Thyroid Carcinoma: A Series of Eight Cases. Endocr. Pathol. 2024, 35, 134–146. [Google Scholar] [CrossRef]
- Lee, Y.A.; Lee, H.; Im, S.W.; Song, Y.S.; Oh, D.Y.; Kang, H.J.; Won, J.K.; Jung, K.C.; Kwon, D.; Chung, E.J.; et al. NTRK and RET fusion-directed therapy in pediatric thyroid cancer yields a tumor response and radioiodine uptake. J. Clin. Investig. 2021, 131, e144847. [Google Scholar] [CrossRef]
- Ji, J.H.; Oh, Y.L.; Hong, M.; Yun, J.W.; Lee, H.W.; Kim, D.; Ji, Y.; Kim, D.H.; Park, W.Y.; Shin, H.T.; et al. Identification of Driving ALK Fusion Genes and Genomic Landscape of Medullary Thyroid Cancer. PLoS Genet. 2015, 11, e1005467. [Google Scholar] [CrossRef]
- Hillier, K.; Hughes, A.; Shamberger, R.C.; Shusterman, S.; Perez-Atayde, A.R.; Wassner, A.J.; Iafrate, A.J.; Dubuc, A.; Janeway, K.A.; Rothenberg, S.M.; et al. A Novel ALK Fusion in Pediatric Medullary Thyroid Carcinoma. Thyroid 2019, 29, 1704–1707. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef]
- Nikiforov, Y.E. Radiation-induced thyroid cancer: What we have learned from chernobyl. Endocr. Pathol. 2006, 17, 307–317. [Google Scholar] [CrossRef]
- Mitsutake, N.; Saenko, V. Molecular pathogenesis of pediatric thyroid carcinoma. J. Radiat. Res. 2021, 62, i71–i77. [Google Scholar] [CrossRef]
- Givens, D.J.; Buchmann, L.O.; Agarwal, A.M.; Grimmer, J.F.; Hunt, J.P. BRAF V600E does not predict aggressive features of pediatric papillary thyroid carcinoma. Laryngoscope 2014, 124, E389–E393. [Google Scholar] [CrossRef] [PubMed]
- Henke, L.E.; Perkins, S.M.; Pfeifer, J.D.; Ma, C.; Chen, Y.; DeWees, T.; Grigsby, P.W. BRAF V600E mutational status in pediatric thyroid cancer. Pediatr. Blood Cancer 2014, 61, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- Matsuse, M.; Mitsutake, N. TERT promoter mutations in thyroid cancer. Endocr. J. 2023, 70, 1035–1049. [Google Scholar] [CrossRef]
- Yu, P.; Qu, N.; Zhu, R.; Hu, J.; Han, P.; Wu, J.; Tan, L.; Gan, H.; He, C.; Fang, C.; et al. TERT accelerates BRAF mutant-induced thyroid cancer dedifferentiation and progression by regulating ribosome biogenesis. Sci. Adv. 2023, 9, eadg7125. [Google Scholar] [CrossRef]
- Chakraborty, D.; Shakya, S.; Ballal, S.; Agarwal, S.; Bal, C. BRAF V600E and TERT promoter mutations in paediatric and young adult papillary thyroid cancer and clinicopathological correlation. J. Pediatr. Endocrinol. Metab. 2020, 33, 1465–1474. [Google Scholar] [CrossRef]
- Oishi, N.; Kondo, T.; Nakazawa, T.; Mochizuki, K.; Inoue, T.; Kasai, K.; Tahara, I.; Yabuta, T.; Hirokawa, M.; Miyauchi, A.; et al. Frequent BRAF (V600E) and Absence of TERT Promoter Mutations Characterize Sporadic Pediatric Papillary Thyroid Carcinomas in Japan. Endocr. Pathol. 2017, 28, 103–111. [Google Scholar] [CrossRef]
- Geng, J.; Liu, Y.; Guo, Y.; Wang, H.; Tai, J.; Jin, Y.; Zhang, J.; Yu, Y.; Wang, S.; Song, Y.; et al. Correlation between TERT C228T and clinic-pathological features in pediatric papillary thyroid carcinoma. Sci. China Life Sci. 2019, 62, 1563–1571. [Google Scholar] [CrossRef]
- Kondo, T.; Ezzat, S.; Asa, S.L. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 2006, 6, 292–306. [Google Scholar] [CrossRef]
- Raman, P.; Koenig, R.J. Pax-8-PPAR-gamma fusion protein in thyroid carcinoma. Nat. Rev. Endocrinol. 2014, 10, 616–623. [Google Scholar] [CrossRef]
- Vuong, H.G.; Kondo, T.; Oishi, N.; Nakazawa, T.; Mochizuki, K.; Miyauchi, A.; Hirokawa, M.; Katoh, R. Paediatric follicular thyroid carcinoma-indolent cancer with low prevalence of RAS mutations and absence of PAX8-PPARG fusion in a Japanese population. Histopathology 2017, 71, 760–768. [Google Scholar] [CrossRef]
- Paulson, V.A.; Rudzinski, E.R.; Hawkins, D.S. Thyroid Cancer in the Pediatric Population. Genes 2019, 10, 723. [Google Scholar] [CrossRef] [PubMed]
- van der Tuin, K.; Ruano, D.; Knijnenburg, J.; van der Luijt, R.B.; Morreau, H.; Links, T.P.; Hes, F.J.; Dutch Pediatric Thyroid Cancer, C. Clinically Relevant Germline Variants in Children with Nonmedullary Thyroid Cancer. J. Clin. Endocrinol. Metab. 2024, 109, e2214–e2221. [Google Scholar] [CrossRef] [PubMed]
- Balinisteanu, I.; Panzaru, M.C.; Caba, L.; Ungureanu, M.C.; Florea, A.; Grigore, A.M.; Gorduza, E.V. Cancer Predisposition Syndromes and Thyroid Cancer: Keys for a Short Two-Way Street. Biomedicines 2023, 11, 2143. [Google Scholar] [CrossRef] [PubMed]
- Capezzone, M.; Marchisotta, S.; Cantara, S.; Busonero, G.; Brilli, L.; Pazaitou-Panayiotou, K.; Carli, A.F.; Caruso, G.; Toti, P.; Capitani, S.; et al. Familial non-medullary thyroid carcinoma displays the features of clinical anticipation suggestive of a distinct biological entity. Endocr. Relat. Cancer 2008, 15, 1075–1081. [Google Scholar] [CrossRef]
- Cameselle-Teijeiro, J.M.; Erickson, L.A.; LiVolsi, V.; Nose, V. Endocrine and Neuroendocrine Tumours. In WHO Classification of Tumours, 5th ed.; WHO Classification of Tumours Editorial Board, Ed.; International Agency for Research on Cancer: Lyon, France, 2022; Volume 10, Available online: https://tumourclassification.iarc.who.int/chaptercontent/53/168 (accessed on 3 March 2025).
- Milani, D.; Dolci, A.; Muller, I.; Pavesi, M.A.; Runza, L.; Kuhn, E.; Natacci, F.; Peissel, B.; Ricci, M.T.; Despini, L.; et al. Thyroid findings in pediatric and adult patients with PTEN hamartoma tumor syndrome: A retrospective analysis, and literature review. Endocrine 2023, 81, 98–106. [Google Scholar] [CrossRef]
- Smith, J.R.; Liu, E.; Church, A.J.; Asch, E.; Cherella, C.E.; Srivastava, S.; Kamihara, J.; Wassner, A.J. Natural History of Thyroid Disease in Children with PTEN Hamartoma Tumor Syndrome. J. Clin. Endocrinol. Metab. 2021, 106, e1121–e1130. [Google Scholar] [CrossRef]
- Hendricks, L.A.J.; Hoogerbrugge, N.; Schuurs-Hoeijmakers, J.H.M.; Vos, J.R. A review on age-related cancer risks in PTEN hamartoma tumor syndrome. Clin. Genet. 2021, 99, 219–225. [Google Scholar] [CrossRef]
- Pilarski, R.; Burt, R.; Kohlman, W.; Pho, L.; Shannon, K.M.; Swisher, E. Cowden syndrome and the PTEN hamartoma tumor syndrome: Systematic review and revised diagnostic criteria. J. Natl. Cancer Inst. 2013, 105, 1607–1616. [Google Scholar] [CrossRef]
- Tan, M.H.; Mester, J.; Peterson, C.; Yang, Y.; Chen, J.L.; Rybicki, L.A.; Milas, K.; Pederson, H.; Remzi, B.; Orloff, M.S.; et al. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am. J. Hum. Genet. 2011, 88, 42–56. [Google Scholar] [CrossRef]
- Baran, J.A.; Tsai, S.D.; Isaza, A.; Brodeur, G.M.; MacFarland, S.P.; Zelley, K.; Adams, D.M.; Franco, A.T.; Bauer, A.J. The Clinical Spectrum of PTEN Hamartoma Tumor Syndrome: Exploring the Value of Thyroid Surveillance. Horm. Res. Paediatr. 2020, 93, 634–642. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Colas, C.; Pouwels, S.; Hoogerbrugge, N.; PHTS Guideline Development Group; The European Reference Network, GENTURIS. Cancer Surveillance Guideline for individuals with PTEN hamartoma tumour syndrome. Eur. J. Hum. Genet. 2020, 28, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.A.P.; MacFarland, S.P.; Perrino, M.R.; Mitchell, S.G.; Kamihara, J.; Nelson, A.T.; Mallinger, P.H.R.; Brzezinski, J.J.; Maxwell, K.N.; Woodward, E.R.; et al. Update on Pediatric Surveillance Recommendations for PTEN Hamartoma Tumor Syndrome, DICER1-Related Tumor Predisposition, and Tuberous Sclerosis Complex. Clin. Cancer Res. 2025, 31, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, A.; Baitamouni, S.; Liu, D.; Yehia, L.; Anthony, K.; McCarther, A.; Tischkowitz, M.; MacFarland, S.P.; Ngeow, J.; Hoogerbrugge, N.; et al. Cancer and Overgrowth Manifestations of PTEN Hamartoma Tumour Syndrome: Management Recommendations from the International PHTS Consensus Guidelines Working Group. Clin. Cancer Res. 2025, 31, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.A.P.; Williams, G.M.; Kamihara, J.; Stewart, D.R.; Harris, A.K.; Bauer, A.J.; Turner, J.; Shah, R.; Schneider, K.; Schneider, K.W.; et al. DICER1 and Associated Conditions: Identification of At-risk Individuals and Recommended Surveillance Strategies. Clin. Cancer Res. 2018, 24, 2251–2261. [Google Scholar] [CrossRef]
- Gonzalez, I.A.; Stewart, D.R.; Schultz, K.A.P.; Field, A.P.; Hill, D.A.; Dehner, L.P. DICER1 tumor predisposition syndrome: An evolving story initiated with the pleuropulmonary blastoma. Mod. Pathol. 2022, 35, 4–22. [Google Scholar] [CrossRef]
- Khan, N.E.; Bauer, A.J.; Schultz, K.A.P.; Doros, L.; Decastro, R.M.; Ling, A.; Lodish, M.B.; Harney, L.A.; Kase, R.G.; Carr, A.G.; et al. Quantification of Thyroid Cancer and Multinodular Goiter Risk in the DICER1 Syndrome: A Family-Based Cohort Study. J. Clin. Endocrinol. Metab. 2017, 102, 1614–1622. [Google Scholar] [CrossRef]
- Spaulding, S.L.; Maayah, M.; Dinauer, C.A.; Prasad, M.; Darbinyan, A.; Morotti, R.; Christison-Lagay, E.R. Molecular Genetics Augment Cytopathologic Evaluation and Surgical Planning of Pediatric Thyroid Nodules. J. Pediatr. Surg. 2024, 59, 975–980. [Google Scholar] [CrossRef]
- Wasserman, J.D.; Sabbaghian, N.; Fahiminiya, S.; Chami, R.; Mete, O.; Acker, M.; Wu, M.K.; Shlien, A.; de Kock, L.; Foulkes, W.D. DICER1 Mutations Are Frequent in Adolescent-Onset Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2018, 103, 2009–2015. [Google Scholar] [CrossRef]
- Lee, Y.A.; Im, S.W.; Jung, K.C.; Chung, E.J.; Shin, C.H.; Kim, J.I.; Park, Y.J. Predominant DICER1 Pathogenic Variants in Pediatric Follicular Thyroid Carcinomas. Thyroid 2020, 30, 1120–1131. [Google Scholar] [CrossRef]
- Sachdev, C.; Gattani, R.G.; Agrawal, J. Carney Complex and Its Association with Thyroid Cancer, Molecular Pathway, and Treatment. Cureus 2023, 15, e48503. [Google Scholar] [CrossRef]
- Carney, J.A.; Lyssikatos, C.; Seethala, R.R.; Lakatos, P.; Perez-Atayde, A.; Lahner, H.; Stratakis, C.A. The Spectrum of Thyroid Gland Pathology in Carney Complex: The Importance of Follicular Carcinoma. Am. J. Surg. Pathol. 2018, 42, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Dinarvand, P.; Davaro, E.P.; Doan, J.V.; Ising, M.E.; Evans, N.R.; Phillips, N.J.; Lai, J.; Guzman, M.A. Familial Adenomatous Polyposis Syndrome: An Update and Review of Extraintestinal Manifestations. Arch. Pathol. Lab. Med. 2019, 143, 1382–1398. [Google Scholar] [CrossRef] [PubMed]
- Herraiz, M.; Barbesino, G.; Faquin, W.; Chan-Smutko, G.; Patel, D.; Shannon, K.M.; Daniels, G.H.; Chung, D.C. Prevalence of thyroid cancer in familial adenomatous polyposis syndrome and the role of screening ultrasound examinations. Clin. Gastroenterol. Hepatol. 2007, 5, 367–373. [Google Scholar] [CrossRef]
- Lam, A.K.; Fridman, M. Characteristics of cribriform morular variant of papillary thyroid carcinoma in post-Chernobyl affected region. Hum. Pathol. 2018, 74, 170–177. [Google Scholar] [CrossRef]
- MacFarland, S.P.; Becktell, K.; Schneider, K.W.; Kuiper, R.P.; Lesmana, H.; Meade, J.; Nichols, K.E.; Porter, C.C.; Savage, S.A.; Schultz, K.A.; et al. Pediatric Cancer Screening in Hereditary Gastrointestinal Cancer Risk Syndromes: An Update from the AACR Childhood Cancer Predisposition Working Group. Clin. Cancer Res. 2024, 30, 4566–4571. [Google Scholar] [CrossRef]
- Oshima, J.; Sidorova, J.M.; Monnat, R.J., Jr. Werner syndrome: Clinical features, pathogenesis and potential therapeutic interventions. Ageing Res. Rev. 2017, 33, 105–114. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Sugano, H.; Matsumoto, T.; Furuichi, Y.; Miller, R.W.; Goto, M. Unusual features of thyroid carcinomas in Japanese patients with Werner syndrome and possible genotype–phenotype relations to cell type and race. Cancer 1999, 85, 1345–1352. [Google Scholar] [CrossRef]
- Mathiesen, J.S.; Effraimidis, G.; Rossing, M.; Rasmussen, A.K.; Hoejberg, L.; Bastholt, L.; Godballe, C.; Oturai, P.; Feldt-Rasmussen, U. Multiple endocrine neoplasia type 2: A review. Semin. Cancer Biol. 2022, 79, 163–179. [Google Scholar] [CrossRef]
- Taylor-Miller, T.; Tucker, K.; Sugo, E.; Anazodo, A.; Mowat, D. Clues for Early Diagnosis of MEN2B Syndrome Before Medullary Thyroid Carcinoma. Pediatrics 2024, 154, e2022059517. [Google Scholar] [CrossRef]
- Castinetti, F.; Waguespack, S.G.; Machens, A.; Uchino, S.; Hasse-Lazar, K.; Sanso, G.; Else, T.; Dvorakova, S.; Qi, X.P.; Elisei, R.; et al. Natural history, treatment, and long-term follow up of patients with multiple endocrine neoplasia type 2B: An international, multicentre, retrospective study. Lancet Diabetes Endocrinol. 2019, 7, 213–220. [Google Scholar] [CrossRef]
- Ali, S.Z.; Baloch, Z.W.; Cochand-Priollet, B.; Schmitt, F.C.; Vielh, P.; VanderLaan, P.A. The 2023 Bethesda System for Reporting Thyroid Cytopathology. Thyroid 2023, 33, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Cherella, C.E.; Hollowell, M.L.; Smith, J.R.; Zendejas, B.; Modi, B.P.; Cibas, E.S.; Wassner, A.J. Subtype of atypia on cytology and risk of malignancy in pediatric thyroid nodules. Cancer Cytopathol. 2022, 130, 330–335. [Google Scholar] [CrossRef]
- Sipos, J.A.; Ringel, M.D. Molecular testing in thyroid cancer diagnosis and management. Best. Pract. Res. Clin. Endocrinol. Metab. 2023, 37, 101680. [Google Scholar] [CrossRef] [PubMed]
- Livhits, M.J.; Zhu, C.Y.; Kuo, E.J.; Nguyen, D.T.; Kim, J.; Tseng, C.H.; Leung, A.M.; Rao, J.; Levin, M.; Douek, M.L.; et al. Effectiveness of Molecular Testing Techniques for Diagnosis of Indeterminate Thyroid Nodules: A Randomized Clinical Trial. JAMA Oncol. 2021, 7, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Steward, D.L.; Carty, S.E.; Sippel, R.S.; Yang, S.P.; Sosa, J.A.; Sipos, J.A.; Figge, J.J.; Mandel, S.; Haugen, B.R.; Burman, K.D.; et al. Performance of a Multigene Genomic Classifier in Thyroid Nodules with Indeterminate Cytology: A Prospective Blinded Multicenter Study. JAMA Oncol. 2019, 5, 204–212. [Google Scholar] [CrossRef]
- Lai, S.T.; Bauer, A.J. Approach to the Pediatric Patient with Thyroid Nodules. J. Clin. Endocrinol. Metab. 2025, 10, dgaf090. [Google Scholar] [CrossRef]
- Alexander, E.K.; Kennedy, G.C.; Baloch, Z.W.; Cibas, E.S.; Chudova, D.; Diggans, J.; Friedman, L.; Kloos, R.T.; LiVolsi, V.A.; Mandel, S.J.; et al. Preoperative diagnosis of benign thyroid nodules with indeterminate cytology. N. Engl. J. Med. 2012, 367, 705–715. [Google Scholar] [CrossRef]
- Patel, K.N.; Angell, T.E.; Babiarz, J.; Barth, N.M.; Blevins, T.; Duh, Q.Y.; Ghossein, R.A.; Harrell, R.M.; Huang, J.; Kennedy, G.C.; et al. Performance of a Genomic Sequencing Classifier for the Preoperative Diagnosis of Cytologically Indeterminate Thyroid Nodules. JAMA Surg. 2018, 153, 817–824. [Google Scholar] [CrossRef]
- Nikiforov, Y.E.; Ohori, N.P.; Hodak, S.P.; Carty, S.E.; LeBeau, S.O.; Ferris, R.L.; Yip, L.; Seethala, R.R.; Tublin, M.E.; Stang, M.T.; et al. Impact of mutational testing on the diagnosis and management of patients with cytologically indeterminate thyroid nodules: A prospective analysis of 1056 FNA samples. J. Clin. Endocrinol. Metab. 2011, 96, 3390–3397. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Wald, A.I.; Roy, S.; Durso, M.B.; Nikiforov, Y.E. Targeted next-generation sequencing panel (ThyroSeq) for detection of mutations in thyroid cancer. J. Clin. Endocrinol. Metab. 2013, 98, E1852–E1860. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Mercurio, S.; Wald, A.I.; Barbi de Moura, M.; Callenberg, K.; Santana-Santos, L.; Gooding, W.E.; Yip, L.; Ferris, R.L.; Nikiforov, Y.E. Analytical performance of the ThyroSeq v3 genomic classifier for cancer diagnosis in thyroid nodules. Cancer 2018, 124, 1682–1690. [Google Scholar] [CrossRef] [PubMed]
- Lupo, M.A.; Walts, A.E.; Sistrunk, J.W.; Giordano, T.J.; Sadow, P.M.; Massoll, N.; Campbell, R.; Jackson, S.A.; Toney, N.; Narick, C.M.; et al. Multiplatform molecular test performance in indeterminate thyroid nodules. Diagn. Cytopathol. 2020, 48, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.T.; Labourier, E.; Ablordeppey, K.K.; Surrey, L.F.; Mostoufi-Moab, S.; Isaza, A.; Adzick, N.S.; Kazahaya, K.; Kumar, G.; Bauer, A.J. miRNA expression can classify pediatric thyroid lesions and increases the diagnostic yield of mutation testing. Pediatr. Blood Cancer 2020, 67, e28276. [Google Scholar] [CrossRef] [PubMed]
- Mollen, K.P.; Shaffer, A.D.; Yip, L.; Monaco, S.E.; Huyett, P.; Viswanathan, P.; Witchel, S.F.; Duvvuri, U.; Simons, J.P. Unique Molecular Signatures Are Associated with Aggressive Histology in Pediatric Differentiated Thyroid Cancer. Thyroid 2022, 32, 236–244. [Google Scholar] [CrossRef]
- Nicholson, K.J.; Roberts, M.S.; McCoy, K.L.; Carty, S.E.; Yip, L. Molecular Testing Versus Diagnostic Lobectomy in Bethesda III/IV Thyroid Nodules: A Cost-Effectiveness Analysis. Thyroid 2019, 29, 1237–1243. [Google Scholar] [CrossRef]
- National Institute for Health and Care. Thyroid Cancer: Assessment and Management. In Evidence Review for Molecular Testing [NICE Guideline No. 230]; NICE: Manchester, UK, 2022; Available online: https://www.nice.org.uk/guidance/ng230 (accessed on 3 March 2025).
- Lau, L.M.S.; Khuong-Quang, D.A.; Mayoh, C.; Wong, M.; Barahona, P.; Ajuyah, P.; Senapati, A.; Nagabushan, S.; Sherstyuk, A.; Altekoester, A.K.; et al. Precision-guided treatment in high-risk pediatric cancers. Nat. Med. 2024, 30, 1913–1922. [Google Scholar] [CrossRef]
- Yang, A.T.; Lai, S.T.; Laetsch, T.W.; Bhatti, T.; Baloch, Z.; Surrey, L.F.; Franco, A.T.; Ricarte-Filho, J.C.M.; Mostoufi-Moab, S.; Adzick, N.S.; et al. Molecular landscape and therapeutic strategies in pediatric differentiated thyroid carcinoma. Endocr. Rev. 2025, 10, bnaf003. [Google Scholar] [CrossRef]
- Wirth, L.J.; Sherman, E.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.; Worden, F.; Brose, M.; Patel, J.; Leboulleux, S.; et al. Efficacy of Selpercatinib in RET-Altered Thyroid Cancers. N. Engl. J. Med. 2020, 383, 825–835. [Google Scholar] [CrossRef]
- Subbiah, V.; Hu, M.I.; Wirth, L.J.; Schuler, M.; Mansfield, A.S.; Curigliano, G.; Brose, M.S.; Zhu, V.W.; Leboulleux, S.; Bowles, D.W.; et al. Pralsetinib for patients with advanced or metastatic RET-altered thyroid cancer (ARROW): A multi-cohort, open-label, registrational, phase 1/2 study. Lancet Diabetes Endocrinol. 2021, 9, 491–501. [Google Scholar] [CrossRef]
- Morgenstern, D.A.; Casanova, M.; van Tilburg, C.M.; Ziegler, D.S.; Campbell, M.; Watt, T.C.; Pappo, A.S.; Laetsch, T.W.; Liu, D.; Liming, K.; et al. Safety and efficacy of selpercatinib in pediatric patients with RET-altered solid tumors: Updated results from LIBRETTO-121. J. Clin. Oncol. 2024, 42, 10022. [Google Scholar] [CrossRef]
- Kazahaya, K.; Prickett, K.K.; Paulson, V.A.; Dahl, J.P.; Manning, S.C.; Rudzinski, E.R.; Rastatter, J.C.; Parikh, S.R.; Hawkins, D.S.; Brose, M.S.; et al. Targeted Oncogene Therapy Before Surgery in Pediatric Patients with Advanced Invasive Thyroid Cancer at Initial Presentation: Is It Time for a Paradigm Shift? JAMA Otolaryngol. Head. Neck Surg. 2020, 146, 748–753. [Google Scholar] [CrossRef] [PubMed]
- FDA. RETEVMO® (Selpercatinib) Capsules, for Oral Use. U.S. Patent; Updated 12/2024. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2024/218160s004lbl.pdf (accessed on 25 April 2025).
- Waguespack, S.G.; Drilon, A.; Lin, J.J.; Brose, M.S.; McDermott, R.; Almubarak, M.; Bauman, J.; Casanova, M.; Krishnamurthy, A.; Kummar, S.; et al. Efficacy and safety of larotrectinib in patients with TRK fusion-positive thyroid carcinoma. Eur. J. Endocrinol. 2022, 186, 631–643. [Google Scholar] [CrossRef] [PubMed]
- FDA. VITRAKVI® (Larotrectinib) Capsules, for Oral Use. U.S. Patent; Updated 11/2023. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/211710s009lbl.pdf (accessed on 25 April 2025).
- Bowles, D.W.; Bazhenova, L.; Hescot, S.; Folprecht, G.; Daga, H.; Massarelli, E.; Conley, A.P.; Lamartina, L.; Lin, J.; Ahn, E.; et al. Entrectinib in patients with ntrk fusion-positive (ntrk-fp) thyroid cancer: Updated data from startrk-2. Endocr. Abstr. 2022, 84, OP03-14. [Google Scholar] [CrossRef]
- Gouda, M.A.; Subbiah, V. Expanding the Benefit: Dabrafenib/Trametinib as Tissue-Agnostic Therapy for BRAF V600E-Positive Adult and Pediatric Solid Tumors. Am. Soc. Clin. Oncol. Educ. Book. 2023, 43, e404770. [Google Scholar] [CrossRef]
- Subbiah, V.; Kreitman, R.J.; Wainberg, Z.A.; Cho, J.Y.; Schellens, J.H.M.; Soria, J.C.; Wen, P.Y.; Zielinski, C.C.; Cabanillas, M.E.; Boran, A.; et al. Dabrafenib plus trametinib in patients with BRAF V600E-mutant anaplastic thyroid cancer: Updated analysis from the phase II ROAR basket study. Ann. Oncol. 2022, 33, 406–415. [Google Scholar] [CrossRef]
- Van Tilburg, C.; Albert, C.M.; Bielack, S.; DuBois, S.G.; Federman, N.; Georger, B.; RNagasubramanian, R.; Pappo, A.S.; Norenburg, R.; Dima, L.; et al. Abstract: Efficacy and safety of larotrectinib in pediatric patients with non-central nervous system tropomysin receptor kinase fusion-positive cancer: And expanded dataset. Pediatr. Blood Cancer 2021, 68 (Suppl. S5), e29349. [Google Scholar] [CrossRef]
- Mascarenhas, L.; van Tilburg, C.M.; Doz, F.; Zwaan, C.M.; Albert, C.M.; Blattman, C.; Geoerger, B.; DuBois, S.G.; Federman, N.; Nagasubramanian, R.; et al. Efficacy and safety of larotrectinib in pediatric patients with tropomyosin receptor kinase (TRK) fusion-positive cancer: An expanded dataset. J. Clin. Oncol. 2022, 40, 10030. [Google Scholar] [CrossRef]
- Chuk, M.K.; Widemann, B.C.; Minard, C.G.; Liu, X.; Kim, A.; Bernhardt, M.B.; Kudgus, R.A.; Reid, J.M.; Voss, S.D.; Blaney, S.; et al. A phase 1 study of cabozantinib in children and adolescents with recurrent or refractory solid tumors, including CNS tumors: Trial ADVL1211, a report from the Children’s Oncology Group. Pediatr. Blood Cancer 2018, 65, e27077. [Google Scholar] [CrossRef]
- Chesover, A.D.; Vali, R.; Hemmati, S.H.; Wasserman, J.D. Lung Metastasis in Children with Differentiated Thyroid Cancer: Factors Associated with Diagnosis and Outcomes of Therapy. Thyroid 2021, 31, 50–60. [Google Scholar] [CrossRef]
- Sugino, K.; Nagahama, M.; Kitagawa, W.; Ohkuwa, K.; Uruno, T.; Matsuzu, K.; Suzuki, A.; Tomoda, C.; Hames, K.Y.; Akaishi, J.; et al. Distant Metastasis in Pediatric and Adolescent Differentiated Thyroid Cancer: Clinical Outcomes and Risk Factor Analyses. J. Clin. Endocrinol. Metab. 2020, 105, e3981–e3988. [Google Scholar] [CrossRef]
- Alzahrani, A.S.; Alswailem, M.; Moria, Y.; Almutairi, R.; Alotaibi, M.; Murugan, A.K.; Qasem, E.; Alghamdi, B.; Al-Hindi, H. Lung Metastasis in Pediatric Thyroid Cancer: Radiological Pattern, Molecular Genetics, Response to Therapy, and Outcome. J. Clin. Endocrinol. Metab. 2019, 104, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Waguespack, S.G.; Tewari, S.O.; Busaidy, N.L.; Zafereo, M.E. Larotrectinib Before Initial Radioactive Iodine Therapy in Pediatric TRK Fusion-Positive Papillary Thyroid Carcinoma: Time to Reconsider the Treatment Paradigm for Distantly Metastatic Disease? JCO Precis. Oncol. 2022, 6, e2100467. [Google Scholar] [CrossRef] [PubMed]
- Groussin, L.; Theodon, H.; Bessiene, L.; Bricaire, L.; Bonnet-Serrano, F.; Cochand-Priollet, B.; Leroy, K.; Garinet, S.; Pasmant, E.; Zerbit, J.; et al. Redifferentiating Effect of Larotrectinib in NTRK-Rearranged Advanced Radioactive-Iodine Refractory Thyroid Cancer. Thyroid 2022, 32, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Werner, R.A.; Sayehli, C.; Hanscheid, H.; Higuchi, T.; Serfling, S.E.; Fassnacht, M.; Goebeler, M.E.; Buck, A.K.; Kroiss, M. Successful combination of selpercatinib and radioiodine after pretherapeutic dose estimation in RET-altered thyroid carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2023, 50, 1833–1834. [Google Scholar] [CrossRef]
- Syed, A.R.; Gorana, A.; Nohr, E.; Yuan, X.K.; Amin, M.P.; Ghaznavi, S.; Lamb, D.; McIntyre, J.; Eszlinger, M.; Paschke, R. Predictors of radioiodine (RAI)-avidity restoration for NTRK fusion-positive RAI-resistant metastatic thyroid cancers. Eur. Thyroid. J. 2024, 13, e230227. [Google Scholar] [CrossRef]
- Weiler, D.; Perez Lago, M.D.S. Successful radioiodine redifferentiation with selpercatinib in RET fusion-positive papillary thyroid carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2024, 51, 3467–3468. [Google Scholar] [CrossRef]

{kind=link}
| Diagnostic Category | ROM Mean (Range) | |
|---|---|---|
| I | Nondiagnostic | 14% (0–33) |
| II | Benign a | 6% (0–27) |
| III | Atypia of undetermined significance | 28% (11–54) |
| IV | Follicular neoplasm b | 50% (28–100) |
| V | Suspicious for malignancy | 81% (40–100) |
| VI | Malignant | 98% (86–100) |
| Tier | Molecular Alteration | Risk of Invasive Disease | Surgical Approach | |
|---|---|---|---|---|
| 1 | Point mutations
| Low | Lobectomy without central neck dissection provided no evidence of LN metastasis on US | Completion thyroidectomy if >2 vessel vascular invasion or high-grade histologic features |
| 2 | Point mutations
| Intermediate | Total thyroidectomy with prophylactic/therapeutic central neck dissection. Consider lobectomy with prophylactic central neck dissection if no evidence of LN metastasis on US. | Completion thyroidectomy if multifocality or ≥5 positive LNs |
| 3 | Fusions
| High | ||
| Agent | Molecular Target(s) | Histology | n | Age (Years) | Response | Citation |
|---|---|---|---|---|---|---|
| Selpercatinib | RET | MTC | 14 | 2–20 2–20 | ORR 83.3% | [92] |
| PTC | 10 | ORR 100% | ||||
| 2-year PFS 92.4% | ||||||
| Larotrectinib | NTRK | Agnostic | 78 | 0.1–17.8 | ORR 88% | [100] |
| 94 | 0–18 | ORR 84% | [101] | |||
| PTC | 2 | 6–13 | PR 50%, CR 50% | [95] | ||
| Cabozantinib | VEGFR2, RET, MET, FLT3, NTRK, AXL | MTC | 5 | 1 | PR 40% | [102] |
| Agent | Molecular Target(s) | Histology | Age (Years) | Location(s) | Status | ClinicalTrials.gov ID |
|---|---|---|---|---|---|---|
| Larotrectinib | NTRK | DTC | ≥1 | USA | Recruiting | NCT05783323 |
| Repotrectinib | ALK, ROS1, NTRK | Agnostic | ≥12 | International | Recruiting | NCT03093116 |
| Pralsetinib | RET | Agnostic | 0.5–21 | International | Active, not recruiting | NCT03899792 |
| Pralsetinib | RET | Thyroid carcinoma | ≥12 | USA | Active, not recruiting | NCT04759911 |
| Pralsetinib | RET | DTC | ≥12 | USA | Recruiting | NCT05668962 |
| Pralsetinib | RET | Agnostic | ≥12 | International | Active, not recruiting | NCT03157128 |
| Oncogene-specific kinase inhibitors | NTRK, RET, ALK, BRAFV600E | DTC | ≥0 | USA, Australia | Recruiting | NCT05024929 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanderniet, J.A.; Fuentes-Bolanos, N.A.; Cho, Y.H.; Tucker, K.M.; Anazodo, A.; Bauer, A.J.; Benitez-Aguirre, P.Z. Paediatric Thyroid Carcinoma: The Genetic Revolution and Its Implications for Therapy and Outcomes. Cancers 2025, 17, 1549. https://doi.org/10.3390/cancers17091549
Vanderniet JA, Fuentes-Bolanos NA, Cho YH, Tucker KM, Anazodo A, Bauer AJ, Benitez-Aguirre PZ. Paediatric Thyroid Carcinoma: The Genetic Revolution and Its Implications for Therapy and Outcomes. Cancers. 2025; 17(9):1549. https://doi.org/10.3390/cancers17091549
Chicago/Turabian StyleVanderniet, Joel A., Noemi A. Fuentes-Bolanos, Yoon Hi Cho, Katherine M. Tucker, Antoinette Anazodo, Andrew J. Bauer, and Paul Z. Benitez-Aguirre. 2025. "Paediatric Thyroid Carcinoma: The Genetic Revolution and Its Implications for Therapy and Outcomes" Cancers 17, no. 9: 1549. https://doi.org/10.3390/cancers17091549
APA StyleVanderniet, J. A., Fuentes-Bolanos, N. A., Cho, Y. H., Tucker, K. M., Anazodo, A., Bauer, A. J., & Benitez-Aguirre, P. Z. (2025). Paediatric Thyroid Carcinoma: The Genetic Revolution and Its Implications for Therapy and Outcomes. Cancers, 17(9), 1549. https://doi.org/10.3390/cancers17091549