
Stromal Cells in Early Inflammation-Related Pancreatic Carcinogenesis—Biology and Its Potential Role in Therapeutic Targeting

 , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Types of Non-Hematopoietic Mesenchymal Cells in Healthy Pancreas
2.1. Tissue-Resident Fibroblasts in Pancreas—Origin and Markers
2.2. Pancreatic Stellate Cells—Overlapping and Distinct Characteristics Compared to Pancreatic Fibroblasts
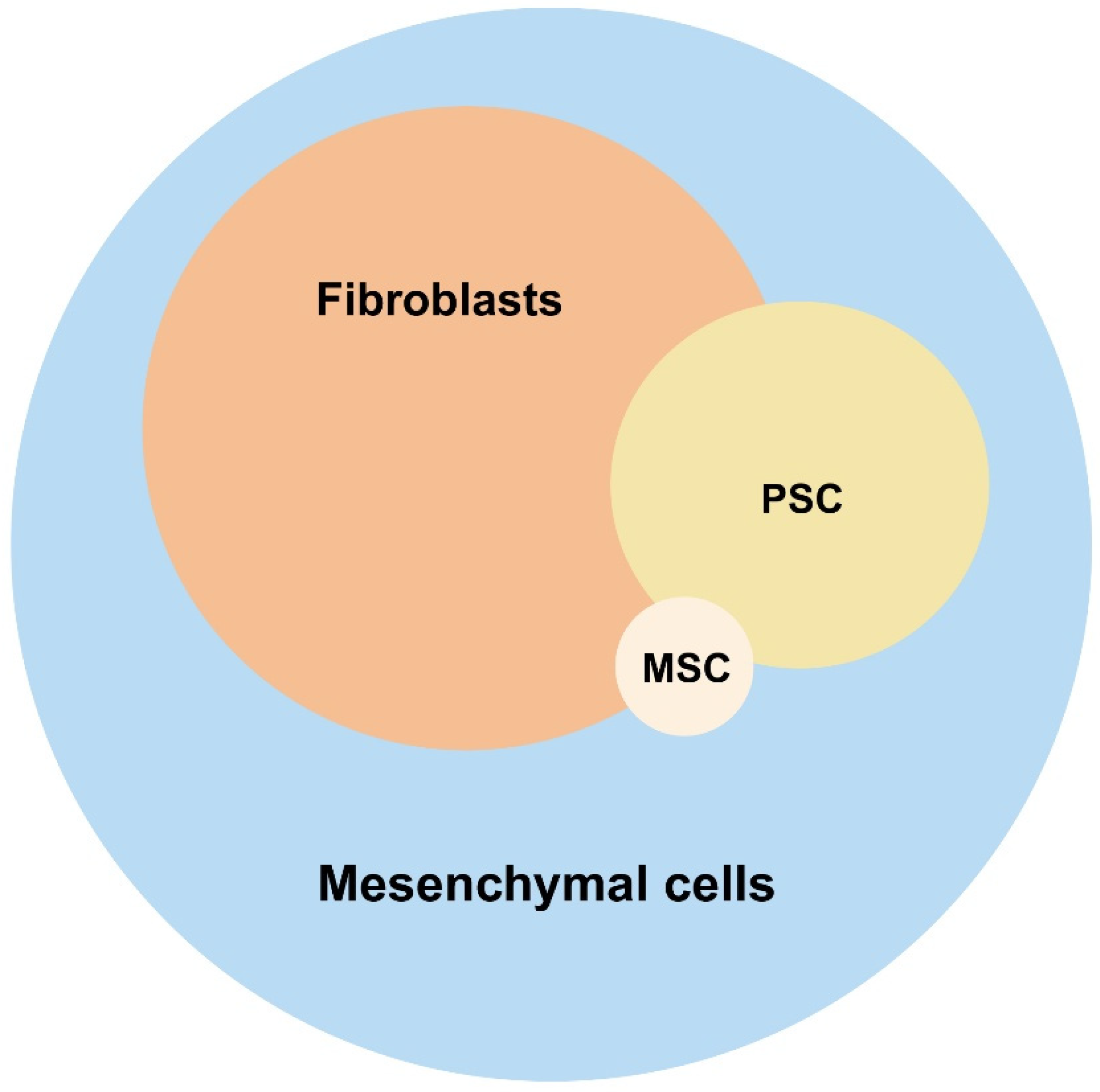
2.3. Multipotent Stroma Cells
3. Alterations and Differentiation of Stroma in Pancreatic Inflammation
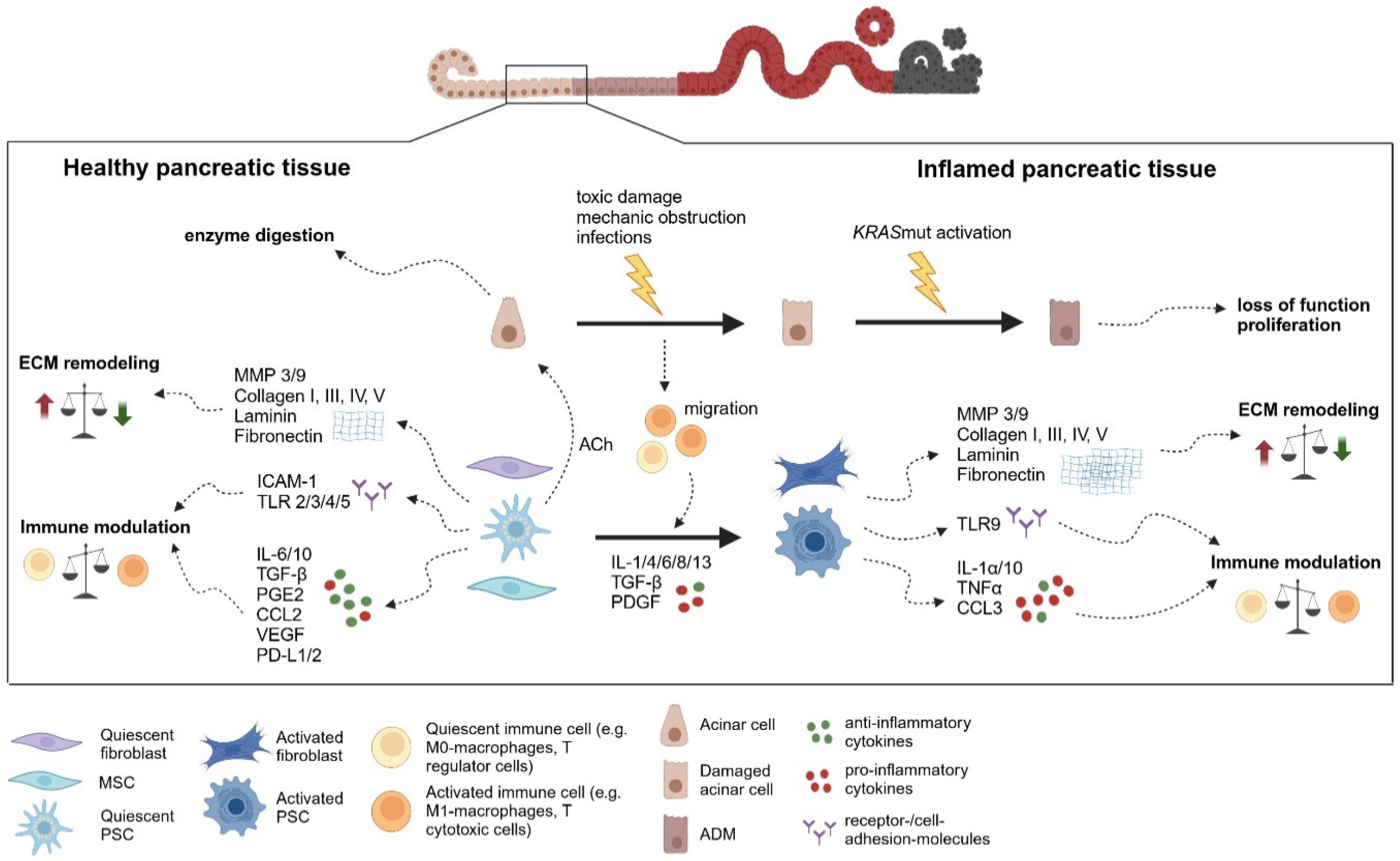
3.1. Stroma in Acute Pancreatitis
3.2. Stroma in Chronic Pancreatitis
3.3. Activation of Pancreatic Mesenchymal Cells
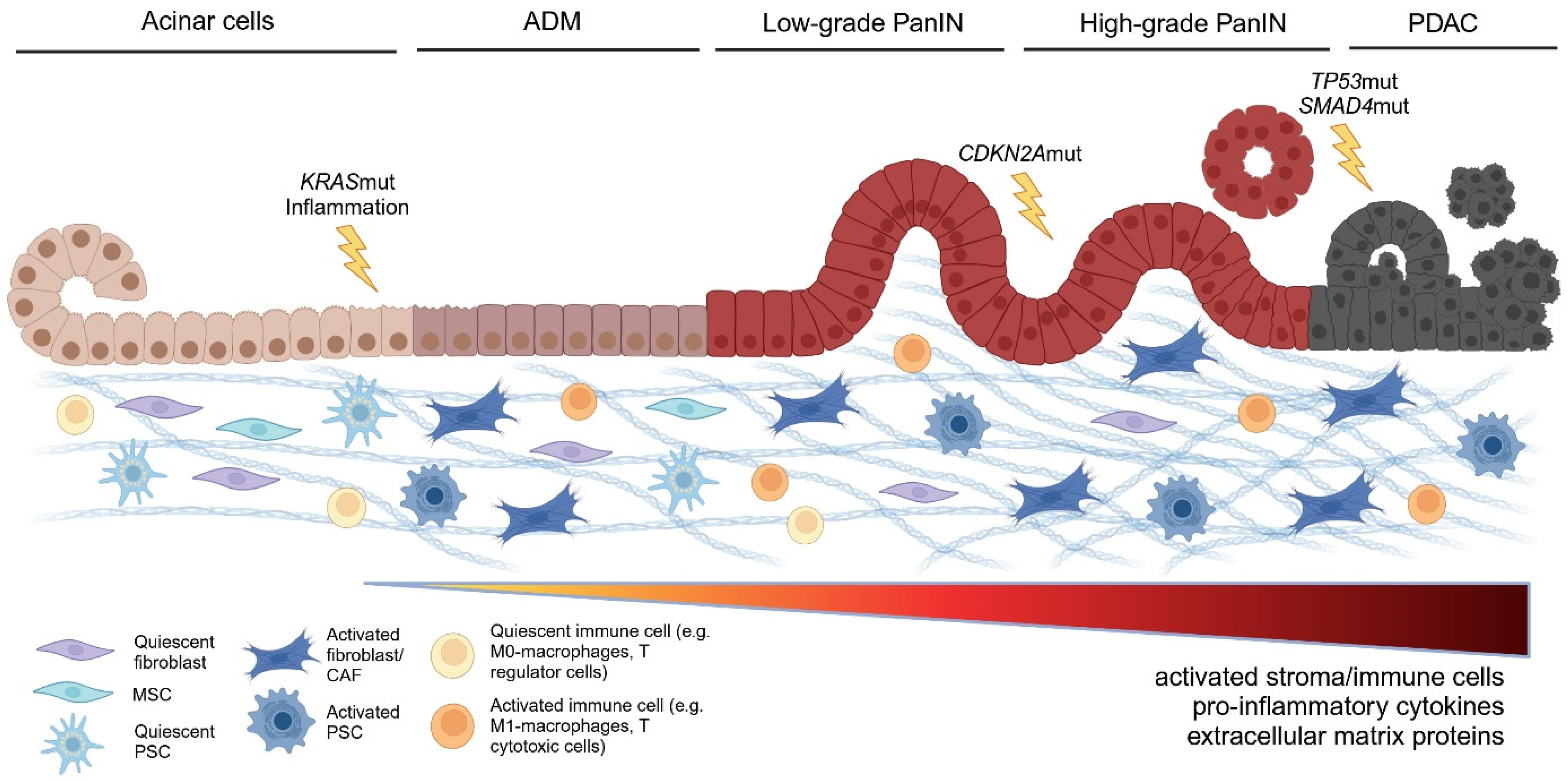
4. Role of Inflammation-Driven Stroma at Different Stages and Processes of PDAC Carcinogenesis
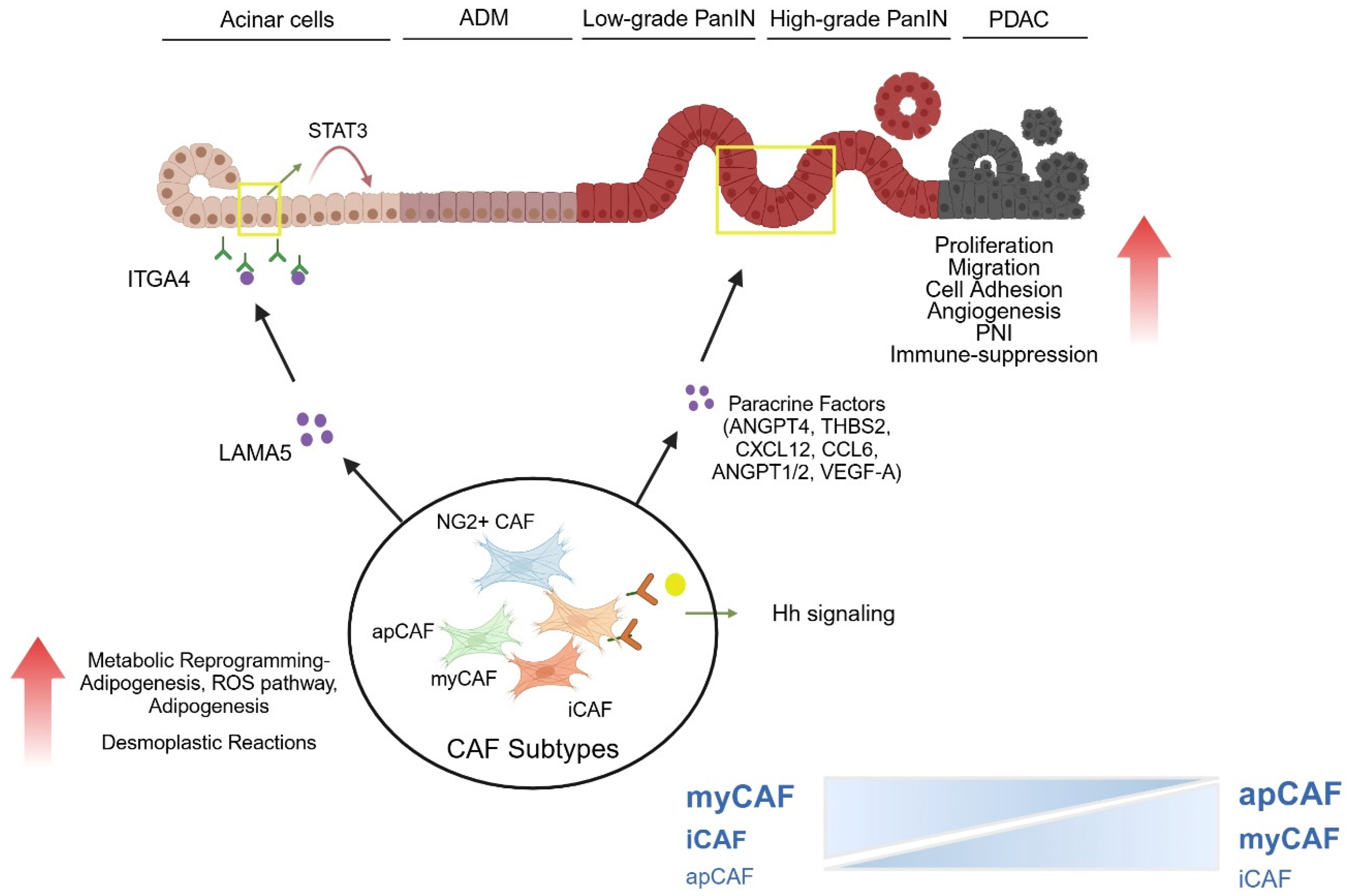
4.1. Impact of Stroma on ADM Development and ADM—PanIN Transition
4.2. Impact of Stroma on Transition from Low Grade to High Grade PanIN
4.3. Impact on Metabolism of Precursor Lesions
4.4. Angiogenesis in the Stroma During Early-Stage Pancreatic Carcinogenesis
4.5. Impact on Intrapancreatic Nerves and Precursor–Neural Interactions
4.6. Impact on Immune Cell Distribution and Precursor–Immune Cell Interactions
5. Current Translation into the Clinical Setting and Outlook
5.1. Targeting Stroma Cells to Prevent Precursor Progression
5.2. Targeting Paracrine Communication Between Stroma and Precancerous Cells
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moir, J.A.G.; Mann, J.; White, S.A. The role of pancreatic stellate cells in pancreatic cancer. Surg. Oncol. 2015, 24, 232–238. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M. Mouse models of pancreatic cancer. World J. Gastroenterol. 2012, 18, 1286. [Google Scholar] [CrossRef] [PubMed]
- Kirkegård, J.; Mortensen, F.V.; Cronin-Fenton, D. Chronic Pancreatitis and Pancreatic Cancer Risk: A Systematic Review and Meta-analysis. Am. J. Gastroenterol. 2017, 112, 1366–1372. [Google Scholar] [CrossRef]
- Manohar, M.; Verma, A.K.; Venkateshaiah, S.U.; Sanders, N.L.; Mishra, A. Pathogenic mechanisms of pancreatitis. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 10. [Google Scholar] [CrossRef]
- Garcia, P.E.; Scales, M.K.; Allen, B.L.; Pasca di Magliano, M. Pancreatic Fibroblast Heterogeneity: From Development to Cancer. Cells 2020, 9, 2464. [Google Scholar] [CrossRef]
- Han, L.; Wu, Y.; Fang, K.; Sweeney, S.; Roesner, U.K.; Parrish, M.; Patel, K.; Walter, T.; Piermattei, J.; Trimboli, A.; et al. The splanchnic mesenchyme is the tissue of origin for pancreatic fibroblasts during homeostasis and tumorigenesis. Nat. Commun. 2023, 14, 1. [Google Scholar] [CrossRef] [PubMed]
- Oñate, M.K.; Oon, C.; Bhattacharyya, S.; Low, V.; Chen, C.; Zhao, X.; Yan, Z.; Hang, Y.; Kim, S.K.; Xia, Z.; et al. Stromal KITL/SCF promotes pancreas tissue homeostasis and restrains tumor progression. bioRxiv 2024. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Desmouliere, A.; Darby, I.A.; Laverdet, B.; Bonté, F. Fibroblasts and myofibroblasts in wound healing. Clin. Cosmet. Investig. Dermatol. 2014, 7, 301–311. [Google Scholar] [CrossRef]
- Rønnov-Jessen, L.; Petersen, O.W. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab. Investig. 1993, 68, 696–707. [Google Scholar] [PubMed]
- Micallef, L.; Vedrenne, N.; Billet, F.; Coulomb, B.; Darby, I.A.; Desmoulière, A. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis Tissue Repair 2012, 5, S5. [Google Scholar] [CrossRef]
- Gabbiani, G.; Ryan, G.B.; Majne, G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia 1971, 27, 549–550. [Google Scholar] [CrossRef]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Österreicher, C.H.; Penz-Österreicher, M.; Grivennikov, S.I.; Guma, M.; Koltsova, E.K.; Datz, C.; Sasik, R.; Hardiman, G.; Karin, M.; Brenner, D.A. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc. Natl. Acad. Sci. USA 2011, 108, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.N.; Magiera, L.; Kraman, M.; Fearon, D.T. Tumoral immune suppression by macrophages expressing fibroblast activation protein-α and heme oxygenase-1. Cancer Immunol. Res. 2014, 2, 121–126. [Google Scholar] [CrossRef]
- Lau, J.; Kawahira, H.; Hebrok, M. Hedgehog signaling in pancreas development and disease. Cell. Mol. Life Sci. CMLS 2006, 63, 642–652. [Google Scholar] [CrossRef]
- Hui, C.; Angers, S. Gli Proteins in Development and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef]
- Garcia, P.E.; Adoumie, M.; Kim, E.C.; Zhang, Y.; Scales, M.K.; El-Tawil, Y.S.; Shaikh, A.Z.; Wen, H.-J.; Bednar, F.; Allen, B.L.; et al. Differential Contribution of Pancreatic Fibroblast Subsets to the Pancreatic Cancer Stroma. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 581–599. [Google Scholar] [CrossRef]
- Larsen, B.M.; Hrycaj, S.M.; Newman, M.; Li, Y.; Wellik, D.M. Mesenchymal Hox6 function is required for pancreatic endocrine cell differentiation. Development 2015, 142, 3859–3868. [Google Scholar] [CrossRef] [PubMed]
- Asahina, K.; Tsai, S.Y.; Li, P.; Ishii, M.; Maxson, R.E.; Sucov, H.M.; Tsukamoto, H. Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology 2009, 49, 998–1011. [Google Scholar] [CrossRef] [PubMed]
- Apte, M.V.; Haber, P.S.; Applegate, T.L.; Norton, I.D.; McCaughan, G.W.; Korsten, M.A.; Pirola, R.C.; Wilson, J.S. Periacinar stellate shaped cells in rat pancreas: Identification, isolation, and culture. Gut 1998, 43, 128–133. [Google Scholar] [CrossRef]
- Cassiman, D.; Barlow, A.; Vander Borght, S.; Libbrecht, L.; Pachnis, V. Hepatic stellate cells do not derive from the neural crest. J. Hepatol. 2006, 44, 1098–1104. [Google Scholar] [CrossRef]
- Chan, K.; Fu, Y.; Wu, T.; Hsu, P.; Lee, W. Hepatic stellate cells promote the differentiation of embryonic stem cell-derived definitive endodermal cells into hepatic progenitor cells. Hepatol. Res. 2013, 43, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Niki, T.; Pekny, M.; Hellemans, K.; De Bleser, P.; Van Den Berg, K.; Vaeyens, F.; Quartier, E.; Schuit, F.; Geerts, A. Class Vi Intermediate Filament Protein Nestin Is Induced During Activation of Rat Hepatic Stellate Cells. Hepatology 1999, 29, 520–527. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schneider, E.; Groß, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grünert, A.; Adler, G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998, 115, 421–432. [Google Scholar] [CrossRef]
- Masamune, A.; Watanabe, T.; Kikuta, K.; Shimosegawa, T. Roles of Pancreatic Stellate Cells in Pancreatic Inflammation and Fibrosis. Clin. Gastroenterol. Hepatol. 2009, 7, S48–S54. [Google Scholar] [CrossRef]
- Walther, T.C.; Farese, R.V. Lipid Droplets and Cellular Lipid Metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef]
- Sunami, Y.; Rebelo, A.; Kleeff, J. Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers 2017, 10, 3. [Google Scholar] [CrossRef]
- Molenaar, M.R.; Haaker, M.W.; Vaandrager, A.B.; Houweling, M.; Helms, J.B. Lipidomic profiling of rat hepatic stellate cells during activation reveals a two-stage process accompanied by increased levels of lysosomal lipids. J. Biol. Chem. 2023, 299, 103042. [Google Scholar] [CrossRef]
- Birtolo, C.; Pham, H.; Morvaridi, S.; Chheda, C.; Go, V.L.W.; Ptasznik, A.; Edderkaoui, M.; Weisman, M.H.; Noss, E.; Brenner, M.B.; et al. Cadherin-11 Is a Cell Surface Marker Up-Regulated in Activated Pancreatic Stellate Cells and Is Involved in Pancreatic Cancer Cell Migration. Am. J. Pathol. 2017, 187, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Haber, P.S.; Keogh, G.W.; Apte, M.V.; Moran, C.S.; Stewart, N.L.; Crawford, D.H.G.; Pirola, R.C.; McCaughan, G.W.; Ramm, G.A.; Wilson, J.S. Activation of Pancreatic Stellate Cells in Human and Experimental Pancreatic Fibrosis. Am. J. Pathol. 1999, 155, 1087–1095. [Google Scholar] [CrossRef]
- Bansod, S.; Saifi, M.A.; Godugu, C. Inhibition of discoidin domain receptors by imatinib prevented pancreatic fibrosis demonstrated in experimental chronic pancreatitis model. Sci. Rep. 2021, 11, 12894. [Google Scholar] [CrossRef] [PubMed]
- DeLeon-Pennell, K.Y.; Barker, T.H.; Lindsey, M.L. Fibroblasts: The arbiters of extracellular matrix remodeling. Matrix Biol. 2020, 91–92, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Phillips, P.A.; Yang, L.; Shulkes, A.; Vonlaufen, A.; Poljak, A.; Bustamante, S.; Warren, A.; Xu, Z.; Guilhaus, M.; Pirola, R.; et al. Pancreatic stellate cells produce acetylcholine and may play a role in pancreatic exocrine secretion. Proc. Natl. Acad. Sci. USA 2010, 107, 17397–17402. [Google Scholar] [CrossRef]
- Omary, M.B.; Lugea, A.; Lowe, A.W.; Pandol, S.J. The pancreatic stellate cell: A star on the rise in pancreatic diseases. J. Clin. Investig. 2007, 117, 50–59. [Google Scholar] [CrossRef]
- Qin, A.; Shi, K.; Tindall, R.R.; Li, J.; Cheng, B.; Li, J.; Yang, B.; Yu, Q.; Zhang, Y.; Hong, B.; et al. Characterization of Pancreatic Collagen-Expressing Fibroblasts in Mouse Acute Pancreatitis. Gastro Hep Adv. 2025, 4, 100557. [Google Scholar] [CrossRef]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.-J.; et al. StellaTUM: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Powell, D.W.; Mifflin, R.C.; Valentich, J.D.; Crowe, S.E.; Saada, J.I.; West, A.B. Myofibroblasts. I. Paracrine cells important in health and disease. Am. J. Physiol. 1999, 277, C1–C19. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Sakai, Y.; Kikuta, K.; Satoh, M.; Satoh, A.; Shimosegawa, T. Activated Rat Pancreatic Stellate Cells Express Intercellular Adhesion Molecule-1 (ICAM-1) in Vitro. Pancreas 2002, 25, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Masamune, A.; Kikuta, K.; Watanabe, T.; Satoh, K.; Satoh, A.; Shimosegawa, T. Pancreatic stellate cells express Toll-like receptors. J. Gastroenterol. 2008, 43, 352–362. [Google Scholar] [CrossRef]
- Shimizu, K.; Kobayashi, M.; Tahara, J.; Shiratori, K. Cytokines and Peroxisome Proliferator-Activated Receptor γ Ligand Regulate Phagocytosis by Pancreatic Stellate Cells. Gastroenterology 2005, 128, 2105–2118. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, L.; Wang, S.; Huang, B.; Jing, Y.; Su, J. Bone Marrow Mesenchymal Stromal Cells: Identification, Classification, and Differentiation. Front. Cell Dev. Biol. 2022, 9, 787118. [Google Scholar] [CrossRef]
- Soliman, H.; Theret, M.; Scott, W.; Hill, L.; Underhill, T.M.; Hinz, B.; Rossi, F.M.V. Multipotent stromal cells: One name, multiple identities. Cell Stem Cell 2021, 28, 1690–1707. [Google Scholar] [CrossRef]
- Katzerke, C.; Schaffrath, J.; Lützkendorf, J.; Janssen, M.; Merbach, A.-K.; Nerger, K.; Binder, M.; Baum, C.; Lauer, K.; Rohde, C.; et al. Reduced proliferation of bone marrow MSC after allogeneic stem cell transplantation is associated with clinical outcome. Blood Adv. 2023, 7, 2811–2824. [Google Scholar] [CrossRef]
- Zhang, Y.; Ravikumar, M.; Ling, L.; Nurcombe, V.; Cool, S.M. Age-Related Changes in the Inflammatory Status of Human Mesenchymal Stem Cells: Implications for Cell Therapy. Stem Cell Rep. 2021, 16, 694–707. [Google Scholar] [CrossRef]
- Li, C.; Wang, B. Mesenchymal Stem/Stromal Cells in Progressive Fibrogenic Involvement and Anti-Fibrosis Therapeutic Properties. Front. Cell Dev. Biol. 2022, 10, 902677. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liu, N.; Bao, C.; Yang, D.; Ma, G.; Yi, W.; Xiao, G.; Cao, H. Mesenchymal stem cells in fibrotic diseases—The two sides of the same coin. Acta Pharmacol. Sin. 2023, 44, 268–287. [Google Scholar] [CrossRef]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of Action of Living, Apoptotic, and Dead MSCs. Front. Immunol. 2019, 10, 1191. [Google Scholar] [CrossRef]
- Glenn, J.D. Mesenchymal stem cells: Emerging mechanisms of immunomodulation and therapy. World J. Stem Cells 2014, 6, 526. [Google Scholar] [CrossRef]
- Cooper, T.T.; Sherman, S.E.; Bell, G.I.; Ma, J.; Kuljanin, M.; Jose, S.E.; Lajoie, G.A.; Hess, D.A. Characterization of a Vimentinhigh /Nestinhigh proteome and tissue regenerative secretome generated by human pancreas-derived mesenchymal stromal cells. Stem Cells 2020, 38, 666–682. [Google Scholar] [CrossRef]
- Epshtein, A.; Sakhneny, L.; Landsman, L. Isolating and Analyzing Cells of the Pancreas Mesenchyme by Flow Cytometry. J. Vis. Exp. 2017, 119, 55344. [Google Scholar] [CrossRef]
- Gopurappilly, R.; Bhat, V.; Bhonde, R. Pancreatic tissue resident mesenchymal stromal cell (MSC)-like cells as a source of in vitro islet neogenesis. J. Cell. Biochem. 2013, 114, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Boland, L.K.; Burand, A.J.; Boyt, D.T.; Dobroski, H.; Di, L.; Liszewski, J.N.; Schrodt, M.V.; Frazer, M.K.; Santillan, D.A.; Ankrum, J.A. Nature vs. Nurture: Defining the Effects of Mesenchymal Stromal Cell Isolation and Culture Conditions on Resiliency to Palmitate Challenge. Front. Immunol. 2019, 10, 1080. [Google Scholar] [CrossRef]
- Thirlwell, K.L.; Colligan, D.; Mountford, J.C.; Samuel, K.; Bailey, L.; Cuesta-Gomez, N.; Hewit, K.D.; Kelly, C.J.; West, C.C.; McGowan, N.W.A.; et al. Pancreas-derived mesenchymal stromal cells share immune response-modulating and angiogenic potential with bone marrow mesenchymal stromal cells and can be grown to therapeutic scale under Good Manufacturing Practice conditions. Cytotherapy 2020, 22, 762–771. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, J.; Liu, B.; Shao, C.; Shi, Y. Reciprocal regulation of mesenchymal stem cells and immune responses. Cell Stem Cell 2022, 29, 1515–1530. [Google Scholar] [CrossRef]
- Cao, H.; Chu, Y.; Zhu, H.; Sun, J.; Pu, Y.; Gao, Z.; Yang, C.; Peng, S.; Dou, Z.; Hua, J. Characterization of immortalized mesenchymal stem cells derived from foetal porcine pancreas. Cell Prolif. 2011, 44, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Seeberger, K.L.; Eshpeter, A.; Rajotte, R.V.; Korbutt, G.S. Epithelial cells within the human pancreas do not coexpress mesenchymal antigens: Epithelial-mesenchymal transition is an artifact of cell culture. Lab. Investig. 2009, 89, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Habtezion, A.; Gukovskaya, A.S.; Pandol, S.J. Acute Pancreatitis: A Multifaceted Set of Organelle and Cellular Interactions. Gastroenterology 2019, 156, 1941–1950. [Google Scholar] [CrossRef]
- Thierens, N.D.E.; Verdonk, R.C.; Löhr, J.M.; van Santvoort, H.C.; Bouwense, S.A.; van Hooft, J.E. Chronic pancreatitis. Lancet 2025, 404, 2605–2618. [Google Scholar] [CrossRef]
- Kong, X.; Sun, T.; Kong, F.; Du, Y.; Li, Z. Chronic Pancreatitis and Pancreatic Cancer. Gastrointest. Tumors 2014, 1, 123–134. [Google Scholar] [CrossRef]
- Liot, S.; Balas, J.; Aubert, A.; Prigent, L.; Mercier-Gouy, P.; Verrier, B.; Bertolino, P.; Hennino, A.; Valcourt, U.; Lambert, E. Stroma Involvement in Pancreatic Ductal Adenocarcinoma: An Overview Focusing on Extracellular Matrix Proteins. Front. Immunol. 2021, 12, 612271. [Google Scholar] [CrossRef]
- Hingorani, S.R. Epithelial and stromal co-evolution and complicity in pancreatic cancer. Nat. Rev. Cancer 2023, 23, 57–77. [Google Scholar] [CrossRef] [PubMed]
- Habtezion, A. Inflammation in acute and chronic pancreatitis. Curr. Opin. Gastroenterol. 2015, 31, 395–399. [Google Scholar] [CrossRef]
- Bachem, M.G.; Zhou, Z.; Zhou, S.; Siech, M. Role of stellate cells in pancreatic fibrogenesis associated with acute and chronic pancreatitis. J. Gastroenterol. Hepatol. 2006, 21 (Suppl. S3), S92–S96. [Google Scholar] [CrossRef]
- Murtaugh, L.C.; Keefe, M.D. Regeneration and repair of the exocrine pancreas. Annu. Rev. Physiol. 2015, 77, 229–249. [Google Scholar] [CrossRef]
- Munir, F.; Jamshed, M.B.; Shahid, N.; Muhammad, S.A.; Ghanem, N.B.; Qiyu, Z. Current status of diagnosis and Mesenchymal stem cells therapy for acute pancreatitis. Physiol. Rep. 2019, 7, e14170. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Whitcomb, D.C.; Shimosegawa, T.; Esposito, I.; Lerch, M.M.; Gress, T.; Mayerle, J.; Drewes, A.M.; Rebours, V.; Akisik, F.; et al. Chronic pancreatitis. Nat. Rev. Dis. Prim. 2017, 3, 17060. [Google Scholar] [CrossRef]
- Kirkegård, J.; Cronin-Fenton, D.; Heide-Jørgensen, U.; Mortensen, F.V. Acute Pancreatitis and Pancreatic Cancer Risk: A Nationwide Matched-Cohort Study in Denmark. Gastroenterology 2018, 154, 1729–1736. [Google Scholar] [CrossRef]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat. Commun. 2015, 6, 7158. [Google Scholar] [CrossRef] [PubMed]
- Hah, N.; Sherman, M.H.; Yu, R.T.; Downes, M.; Evans, R.M. Targeting Transcriptional and Epigenetic Reprogramming in Stromal Cells in Fibrosis and Cancer. Cold Spring Harb. Symp. Quant. Biol. 2015, 80, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Zambirinis, C.P.; Levie, E.; Nguy, S.; Avanzi, A.; Barilla, R.; Xu, Y.; Seifert, L.; Daley, D.; Greco, S.H.; Deutsch, M.; et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J. Exp. Med. 2015, 212, 2077–2094. [Google Scholar] [CrossRef]
- Koliaraki, V.; Prados, A.; Armaka, M.; Kollias, G. The mesenchymal context in inflammation, immunity and cancer. Nat. Immunol. 2020, 21, 974–982. [Google Scholar] [CrossRef]
- Caligiuri, G.; Tuveson, D.A. Activated fibroblasts in cancer: Perspectives and challenges. Cancer Cell 2023, 41, 434–449. [Google Scholar] [CrossRef]
- Öhlund, D.; Elyada, E.; Tuveson, D. Fibroblast heterogeneity in the cancer wound. J. Exp. Med. 2014, 211, 1503–1523. [Google Scholar] [CrossRef]
- Lavie, D.; Ben-Shmuel, A.; Erez, N.; Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 2022, 3, 793–807. [Google Scholar] [CrossRef]
- Hu, Z.-Y.; Ding, D.; Song, Y.; Deng, Y.-F.; Zhang, C.-M.; Yu, T. Molecular mechanism of pancreatic ductal adenocarcinoma: The heterogeneity of cancer-associated fibroblasts and key signaling pathways. World J. Clin. Oncol. 2025, 16, 97007. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M.; et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef]
- Brichkina, A.; Polo, P.; Sharma, S.D.; Visestamkul, N.; Lauth, M. A Quick Guide to CAF Subtypes in Pancreatic Cancer. Cancers 2023, 15, 2614. [Google Scholar] [CrossRef]
- Peiffer, R.; Boumahd, Y.; Gullo, C.; Crake, R.; Letellier, E.; Bellahcène, A.; Peulen, O. Cancer-Associated Fibroblast Diversity Shapes Tumor Metabolism in Pancreatic Cancer. Cancers 2022, 15, 61. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.-C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of Carcinoma-Associated Fibroblasts and Fibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell 2015, 28, 831–833. [Google Scholar] [CrossRef]
- Sunami, Y.; Häußler, J.; Kleeff, J. Cellular Heterogeneity of Pancreatic Stellate Cells, Mesenchymal Stem Cells, and Cancer-Associated Fibroblasts in Pancreatic Cancer. Cancers 2020, 12, 3770. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef]
- de La, O.J.-P.; Emerson, L.L.; Goodman, J.L.; Froebe, S.C.; Illum, B.E.; Curtis, A.B.; Murtaugh, L.C. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc. Natl. Acad. Sci. USA 2008, 105, 18907–18912. [Google Scholar] [CrossRef]
- Kopp, J.L.; von Figura, G.; Mayes, E.; Liu, F.-F.; Dubois, C.L.; Morris, J.P.; Pan, F.C.; Akiyama, H.; Wright, C.V.E.; Jensen, K.; et al. Identification of Sox9-Dependent Acinar-to-Ductal Reprogramming as the Principal Mechanism for Initiation of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2012, 22, 737–750. [Google Scholar] [CrossRef]
- Helms, E.J.; Berry, M.W.; Chaw, R.C.; DuFort, C.C.; Sun, D.; Onate, M.K.; Oon, C.; Bhattacharyya, S.; Sanford-Crane, H.; Horton, W.; et al. Mesenchymal Lineage Heterogeneity Underlies Nonredundant Functions of Pancreatic Cancer–Associated Fibroblasts. Cancer Discov. 2022, 12, 484–501. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, R.; Siddiqui, J.A.; Ganguly, K.; Thompson, C.M.; Cannon, A.; Aithal, A.; Perumal, N.; Maurya, S.K.; Li, X.; Cox, J.L.; et al. Muc4 loss mitigates epidermal growth factor receptor activity essential for PDAC tumorigenesis. Oncogene 2023, 42, 759–770. [Google Scholar] [CrossRef]
- Nimmakayala, R.K.; Ogunleye, A.O.; Parte, S.; Krishna Kumar, N.; Raut, P.; Varadharaj, V.; Perumal, N.K.; Nallasamy, P.; Rauth, S.; Cox, J.L.; et al. PAF1 cooperates with YAP1 in metaplastic ducts to promote pancreatic cancer. Cell Death Dis. 2022, 13, 839. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Tsou, L.; Wang, H.; Gaiser, S.; Chang, D.Z.; Daniluk, J.; Bi, Y.; Grote, T.; Longnecker, D.S.; Logsdon, C.D. Ras Activity Levels Control the Development of Pancreatic Diseases. Gastroenterology 2009, 137, 1072–1082.e6. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Döppler, H.; Necela, B.; Krishna, M.; Crawford, H.C.; Raimondo, M.; Storz, P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-κB and MMPs. J. Cell Biol. 2013, 202, 563–577. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Contino, G.; Deshpande, V.; Tzatsos, A.; Conrad, C.; Benes, C.H.; Levy, D.E.; Settleman, J.; Engelman, J.A.; Bardeesy, N. STAT3 Plays a Critical Role in KRAS -Induced Pancreatic Tumorigenesis. Cancer Res. 2011, 71, 5020–5029. [Google Scholar] [CrossRef] [PubMed]
- Means, A.L.; Logsdon, C.D. Acinar Ductal Metaplasia: Yap Fills a Gap. Gastroenterology 2016, 151, 393–395. [Google Scholar] [CrossRef]
- Velez-Delgado, A.; Donahue, K.L.; Brown, K.L.; Du, W.; Irizarry-Negron, V.; Menjivar, R.E.; Lasse Opsahl, E.L.; Steele, N.G.; The, S.; Lazarus, J.; et al. Extrinsic KRAS Signaling Shapes the Pancreatic Microenvironment Through Fibroblast Reprogramming. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 1673–1699. [Google Scholar] [CrossRef]
- Parte, S.; Kaur, A.B.; Nimmakayala, R.K.; Ogunleye, A.O.; Chirravuri, R.; Vengoji, R.; Leon, F.; Nallasamy, P.; Rauth, S.; Alsafwani, Z.W.; et al. Cancer-Associated Fibroblast Induces Acinar-to-Ductal Cell Transdifferentiation and Pancreatic Cancer Initiation Via LAMA5/ITGA4 Axis. Gastroenterology 2024, 166, 842–858.e5. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.T.F.; Mitchell, J.T.; Kiemen, A.L.; Lyman, M.; Fujikura, K.; Lee, J.W.; Coyne, E.; Shin, S.M.; Nagaraj, S.; Deshpande, A.; et al. PanIN and CAF transitions in pancreatic carcinogenesis revealed with spatial data integration. Cell Syst. 2024, 15, 753–769.e5. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Huang, H.; Wang, Z.; Parmar, K.; Du, W.; Huang, J.; Maitra, A.; Olson, E.; Verma, U.; Brekken, R.A. Cellular heterogeneity during mouse pancreatic ductal adenocarcinoma progression at single-cell resolution. JCI Insight 2019, 4, e129212. [Google Scholar] [CrossRef] [PubMed]
- Cui Zhou, D.; Jayasinghe, R.G.; Chen, S.; Herndon, J.M.; Iglesia, M.D.; Navale, P.; Wendl, M.C.; Caravan, W.; Sato, K.; Storrs, E.; et al. Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer. Nat. Genet. 2022, 54, 1390–1405. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef]
- Niu, N.; Shen, X.; Wang, Z.; Chen, Y.; Weng, Y.; Yu, F.; Tang, Y.; Lu, P.; Liu, M.; Wang, L.; et al. Tumor cell-intrinsic epigenetic dysregulation shapes cancer-associated fibroblasts heterogeneity to metabolically support pancreatic cancer. Cancer Cell 2024, 42, 869–884.e9. [Google Scholar] [CrossRef]
- Ohri, N.; Häußler, J.; Javakhishvili, N.; Vieweg, D.; Zourelidis, A.; Trojanowicz, B.; Haemmerle, M.; Esposito, I.; Glaß, M.; Sunami, Y.; et al. Gene expression dynamics in fibroblasts during early-stage murine pancreatic carcinogenesis. iScience 2024, 28, 111572. [Google Scholar] [CrossRef]
- Zińczuk, J.; Zaręba, K.; Romaniuk, W.; Kamińska, D.; Nizioł, M.; Baszun, M.; Kędra, B.; Guzińska-Ustymowicz, K.; Pryczynicz, A. Expression of Chosen Carcinoembryonic-Related Cell Adhesion Molecules in Pancreatic Intraepithelial Neoplasia (PanIN) Associated with Chronic Pancreatitis and Pancreatic Ductal Adenocarcinoma (PDAC). Int. J. Med. Sci. 2019, 16, 583–592. [Google Scholar] [CrossRef]
- Criscimanna, A.; Duan, L.-J.; Rhodes, J.A.; Fendrich, V.; Wickline, E.; Hartman, D.J.; Monga, S.P.S.; Lotze, M.T.; Gittes, G.K.; Fong, G.-H.; et al. PanIN-Specific Regulation of Wnt Signaling by HIF2α during Early Pancreatic Tumorigenesis. Cancer Res. 2013, 73, 4781–4790. [Google Scholar] [CrossRef]
- Cheng, M.-F.; Tsai, W.-C.; Hsia, K.-T.; Yang, Y.-S.; Jin, J.-S. Expression of urocortin in pancreatic ductal adenocarcinoma and pancreatic intraepithelial neoplasia. APMIS 2014, 122, 147–154. [Google Scholar] [CrossRef]
- Waldmann, J.; Fendrich, V.; Reichert, M.; Hecker, A.; Bartsch, D.K.; Padberg, W.; Holler, J.P.N. Expression of neuropeptide Y and its receptors Y1 and Y2 in pancreatic intraepithelial neoplasia and invasive pancreatic cancer in a transgenic mouse model and human samples of pancreatic cancer. J. Surg. Res. 2018, 223, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Nan, P.; Dong, X.; Bai, X.; Lu, H.; Liu, F.; Sun, Y.; Zhao, X. Tumor-stroma TGF-β1-THBS2 feedback circuit drives pancreatic ductal adenocarcinoma progression via integrin αvβ3/CD36-mediated activation of the MAPK pathway. Cancer Lett. 2022, 528, 59–75. [Google Scholar] [CrossRef]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef]
- Mathew, E.; Zhang, Y.; Holtz, A.M.; Kane, K.T.; Song, J.Y.; Allen, B.L.; Pasca di Magliano, M. Dosage-Dependent Regulation of Pancreatic Cancer Growth and Angiogenesis by Hedgehog Signaling. Cell Rep. 2014, 9, 484–494. [Google Scholar] [CrossRef]
- Hampton, R.F.; Jimenez-Gonzalez, M.; Stanley, S.A. Unravelling innervation of pancreatic islets. Diabetologia 2022, 65, 1069–1084. [Google Scholar] [CrossRef]
- Nathan, J.D.; Peng, R.Y.; Wang, Y.; McVey, D.C.; Vigna, S.R.; Liddle, R.A. Primary sensory neurons: A common final pathway for inflammation in experimental pancreatitis in rats. Am. J. Physiol. Liver Physiol. 2002, 283, G938–G946. [Google Scholar] [CrossRef]
- Saloman, J.L.; Albers, K.M.; Li, D.; Hartman, D.J.; Crawford, H.C.; Muha, E.A.; Rhim, A.D.; Davis, B.M. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 3078–3083. [Google Scholar] [CrossRef]
- Demir, I.E.; Boldis, A.; Pfitzinger, P.L.; Teller, S.; Brunner, E.; Klose, N.; Kehl, T.; Maak, M.; Lesina, M.; Laschinger, M.; et al. Investigation of Schwann Cells at Neoplastic Cell Sites Before the Onset of Cancer Invasion. JNCI J. Natl. Cancer Inst. 2014, 106, dju184. [Google Scholar] [CrossRef] [PubMed]
- Demir, I.E.; Kujundzic, K.; Pfitzinger, P.L.; Saricaoglu, Ö.C.; Teller, S.; Kehl, T.; Reyes, C.M.; Ertl, L.S.; Miao, Z.; Schall, T.J.; et al. Early pancreatic cancer lesions suppress pain through CXCL12-mediated chemoattraction of Schwann cells. Proc. Natl. Acad. Sci. USA 2017, 114, E85–E94. [Google Scholar] [CrossRef]
- Harari, N.; Sakhneny, L.; Khalifa-Malka, L.; Busch, A.; Hertel, K.J.; Hebrok, M.; Landsman, L. Pancreatic pericytes originate from the embryonic pancreatic mesenchyme. Dev. Biol. 2019, 449, 14–20. [Google Scholar] [CrossRef]
- Natarajan, V.; Ha, S.; Delgado, A.; Jacobson, R.; Alhalhooly, L.; Choi, Y.; Kim, J. Acquired αSMA Expression in Pericytes Coincides with Aberrant Vascular Structure and Function in Pancreatic Ductal Adenocarcinoma. Cancers 2022, 14, 2448. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-Y.; Peng, S.-J.; Shen, C.-N.; Pasricha, P.J.; Tang, S.-C. PanIN-associated pericyte, glial, and islet remodeling in mice revealed by 3D pancreatic duct lesion histology. Am. J. Physiol. Liver Physiol. 2016, 311, G412–G422. [Google Scholar] [CrossRef]
- Liang, J.; Li, H.; Han, J.; Jiang, J.; Wang, J.; Li, Y.; Feng, Z.; Zhao, R.; Sun, Z.; Lv, B.; et al. Mex3a interacts with LAMA2 to promote lung adenocarcinoma metastasis via PI3K/AKT pathway. Cell Death Dis. 2020, 11, 614. [Google Scholar] [CrossRef] [PubMed]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the Immune Reaction to Pancreatic Cancer from Inception to Invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.-J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef]
- Bernard, V.; Semaan, A.; Huang, J.; San Lucas, F.A.; Mulu, F.C.; Stephens, B.M.; Guerrero, P.A.; Huang, Y.; Zhao, J.; Kamyabi, N.; et al. Single-Cell Transcriptomics of Pancreatic Cancer Precursors Demonstrates Epithelial and Microenvironmental Heterogeneity as an Early Event in Neoplastic Progression. Clin. Cancer Res. 2019, 25, 2194–2205. [Google Scholar] [CrossRef]
- Mucciolo, G.; Curcio, C.; Roux, C.; Li, W.Y.; Capello, M.; Curto, R.; Chiarle, R.; Giordano, D.; Satolli, M.A.; Lawlor, R.; et al. IL17A critically shapes the transcriptional program of fibroblasts in pancreatic cancer and switches on their protumorigenic functions. Proc. Natl. Acad. Sci. USA 2021, 118, e2020395118. [Google Scholar] [CrossRef]
- Sunami, Y.; Böker, V.; Kleeff, J. Targeting and Reprograming Cancer-Associated Fibroblasts and the Tumor Microenvironment in Pancreatic Cancer. Cancers 2021, 13, 697. [Google Scholar] [CrossRef]
- Kim, H.-G.; Tan, L.; Weisberg, E.L.; Liu, F.; Canning, P.; Choi, H.G.; Ezell, S.A.; Wu, H.; Zhao, Z.; Wang, J.; et al. Discovery of a potent and selective DDR1 receptor tyrosine kinase inhibitor. ACS Chem. Biol. 2013, 8, 2145–2150. [Google Scholar] [CrossRef]
- Berestjuk, I.; Lecacheur, M.; Carminati, A.; Diazzi, S.; Rovera, C.; Prod’homme, V.; Ohanna, M.; Popovic, A.; Mallavialle, A.; Larbret, F.; et al. Targeting Discoidin Domain Receptors DDR1 and DDR2 overcomes matrix-mediated tumor cell adaptation and tolerance to BRAF-targeted therapy in melanoma. EMBO Mol. Med. 2022, 14, e11814. [Google Scholar] [CrossRef]
- Zeng, X.-P.; Wang, L.-J.; Guo, H.-L.; He, L.; Bi, Y.-W.; Xu, Z.-L.; Li, Z.-S.; Hu, L.-H. Dasatinib ameliorates chronic pancreatitis induced by caerulein via anti-fibrotic and anti-inflammatory mechanism. Pharmacol. Res. 2019, 147, 104357. [Google Scholar] [CrossRef] [PubMed]
- El-Hamoly, T.; Hajnády, Z.; Nagy-Pénzes, M.; Bakondi, E.; Regdon, Z.; Demény, M.A.; Kovács, K.; Hegedűs, C.; Abd El-Rahman, S.S.; Szabó, É.; et al. Poly(ADP-Ribose) Polymerase 1 Promotes Inflammation and Fibrosis in a Mouse Model of Chronic Pancreatitis. Int. J. Mol. Sci. 2021, 22, 3593. [Google Scholar] [CrossRef]
- McAndrews, K.M.; Chen, Y.; Darpolor, J.K.; Zheng, X.; Yang, S.; Carstens, J.L.; Li, B.; Wang, H.; Miyake, T.; Correa de Sampaio, P.; et al. Identification of Functional Heterogeneity of Carcinoma-Associated Fibroblasts with Distinct IL6-Mediated Therapy Resistance in Pancreatic Cancer. Cancer Discov. 2022, 12, 1580–1597. [Google Scholar] [CrossRef] [PubMed]
- Chandekar, K.R.; Prashanth, A.; Vinjamuri, S.; Kumar, R. FAPI PET/CT Imaging-An Updated Review. Diagnostics 2023, 13, 2018. [Google Scholar] [CrossRef]
- Greifenstein, L.; Gunkel, A.; Hoehne, A.; Osterkamp, F.; Smerling, C.; Landvogt, C.; Mueller, C.; Baum, R.P. 3BP-3940, a highly potent FAP-targeting peptide for theranostics—Production, validation and first in human experience with Ga-68 and Lu-177. iScience 2023, 26, 108541. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Spektor, A.-M.; Hielscher, T.; Hoppner, J.; Glatting, F.M.; Bicu, F.; Hackert, T.; Heger, U.; Pausch, T.; Gutjahr, E.; et al. Static and Dynamic 68Ga-FAPI PET/CT for the Detection of Malignant Transformation of Intraductal Papillary Mucinous Neoplasia of the Pancreas. J. Nucl. Med. 2023, 64, 244–251. [Google Scholar] [CrossRef]
- McGahan, W.; Gough, M.; Liu, C.; Hoyte, S.; Gill, A.J.; Cavallucci, D. Fibroblast Activation Protein Is Overexpressed on Both Stromal and Epithelial Cells Before Pancreatic Ductal Adenocarcinoma: Implications for Early Diagnosis on 68Ga-FAPI-PET/CT. Gastroenterology 2024, 167, 1217–1220. [Google Scholar] [CrossRef]
- Yang, X.; Lin, Y.; Shi, Y.; Li, B.; Liu, W.; Yin, W.; Dang, Y.; Chu, Y.; Fan, J.; He, R. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar] [CrossRef]
- Deng, D.; Begum, H.; Liu, T.; Zhang, J.; Zhang, Q.; Chu, T.-Y.; Li, H.; Lemenze, A.; Hoque, M.; Soteropoulos, P.; et al. NFAT5 governs cellular plasticity-driven resistance to KRAS-targeted therapy in pancreatic cancer. J. Exp. Med. 2024, 221, e20240766. [Google Scholar] [CrossRef]
- Bhatia, M.; Ramnath, R.D.; Chevali, L.; Guglielmotti, A. Treatment with bindarit, a blocker of MCP-1 synthesis, protects mice against acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1259–G1265. [Google Scholar] [CrossRef]
- Hu, F.; Lou, N.; Jiao, J.; Guo, F.; Xiang, H.; Shang, D. Macrophages in pancreatitis: Mechanisms and therapeutic potential. Biomed. Pharmacother. 2020, 131, 110693. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Khare, T.; Bissonnette, M.; Khare, S. CXCL12-CXCR4/CXCR7 Axis in Colorectal Cancer: Therapeutic Target in Preclinical and Clinical Studies. Int. J. Mol. Sci. 2021, 22, 7371. [Google Scholar] [CrossRef]
- Roberto, M.; Arrivi, G.; Di Civita, M.A.; Barchiesi, G.; Pilozzi, E.; Marchetti, P.; Santini, D.; Mazzuca, F.; Tomao, S. The role of CXCL12 axis in pancreatic cancer: New biomarkers and potential targets. Front. Oncol. 2023, 13, 1154581. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, R.; Anam, K.; Ahmed, H. Development of Galectin-3 Targeting Drugs for Therapeutic Applications in Various Diseases. Int. J. Mol. Sci. 2023, 24, 8116. [Google Scholar] [CrossRef]
- Zhao, W.; Ajani, J.A.; Sushovan, G.; Ochi, N.; Hwang, R.; Hafley, M.; Johnson, R.L.; Bresalier, R.S.; Logsdon, C.D.; Zhang, Z.; et al. Galectin-3 Mediates Tumor Cell-Stroma Interactions by Activating Pancreatic Stellate Cells to Produce Cytokines via Integrin Signaling. Gastroenterology 2018, 154, 1524–1537.e6. [Google Scholar] [CrossRef]
- Yang, D.; Sun, X.; Moniruzzaman, R.; Wang, H.; Citu, C.; Zhao, Z.; Wistuba, I.I.; Wang, H.; Maitra, A.; Chen, Y. Genetic Deletion of Galectin-3 Inhibits Pancreatic Cancer Progression and Enhances the Efficacy of Immunotherapy. Gastroenterology 2024, 167, 298–314. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seidel, T.; Ohri, N.; Glaß, M.; Sunami, Y.; Müller, L.P.; Kleeff, J. Stromal Cells in Early Inflammation-Related Pancreatic Carcinogenesis—Biology and Its Potential Role in Therapeutic Targeting. Cancers 2025, 17, 1541. https://doi.org/10.3390/cancers17091541
Seidel T, Ohri N, Glaß M, Sunami Y, Müller LP, Kleeff J. Stromal Cells in Early Inflammation-Related Pancreatic Carcinogenesis—Biology and Its Potential Role in Therapeutic Targeting. Cancers. 2025; 17(9):1541. https://doi.org/10.3390/cancers17091541
Chicago/Turabian StyleSeidel, Tina, Nupur Ohri, Markus Glaß, Yoshiaki Sunami, Lutz P. Müller, and Jörg Kleeff. 2025. "Stromal Cells in Early Inflammation-Related Pancreatic Carcinogenesis—Biology and Its Potential Role in Therapeutic Targeting" Cancers 17, no. 9: 1541. https://doi.org/10.3390/cancers17091541
APA StyleSeidel, T., Ohri, N., Glaß, M., Sunami, Y., Müller, L. P., & Kleeff, J. (2025). Stromal Cells in Early Inflammation-Related Pancreatic Carcinogenesis—Biology and Its Potential Role in Therapeutic Targeting. Cancers, 17(9), 1541. https://doi.org/10.3390/cancers17091541