
Understanding and Targeting Metabolic Vulnerabilities in Acute Myeloid Leukemia: An Updated Comprehensive Review



Simple Summary
Abstract
1. Introduction
2. Metabolic Characteristics of Normal Hematopoietic Cells and Leukemic Cells
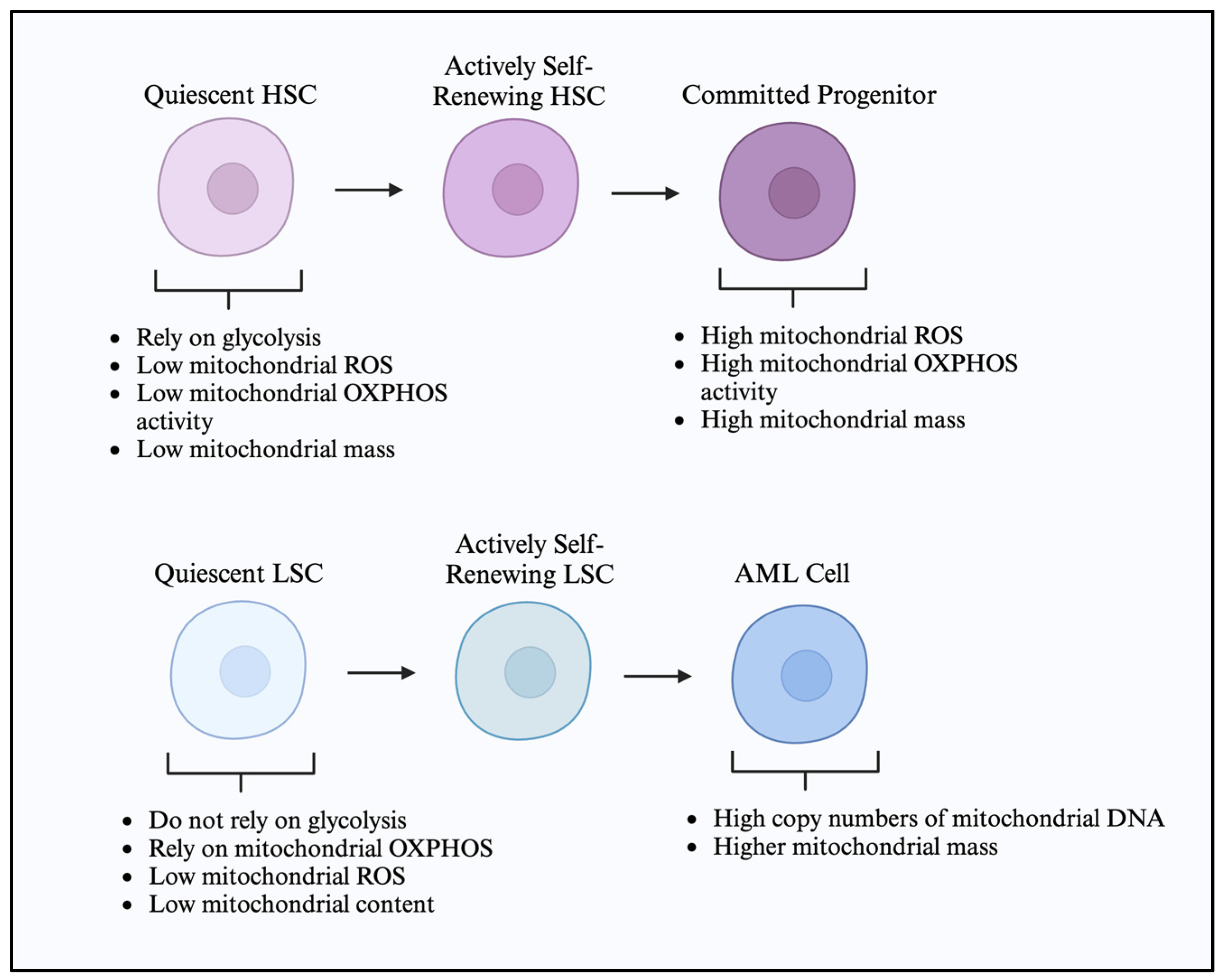
2.1. Hematopoietic Stem Cell Metabolic Characteristics
2.2. Leukemic Cells and Leukemic Stem Cell Metabolic Characteristics
3. Altered Metabolic Pathways in AML
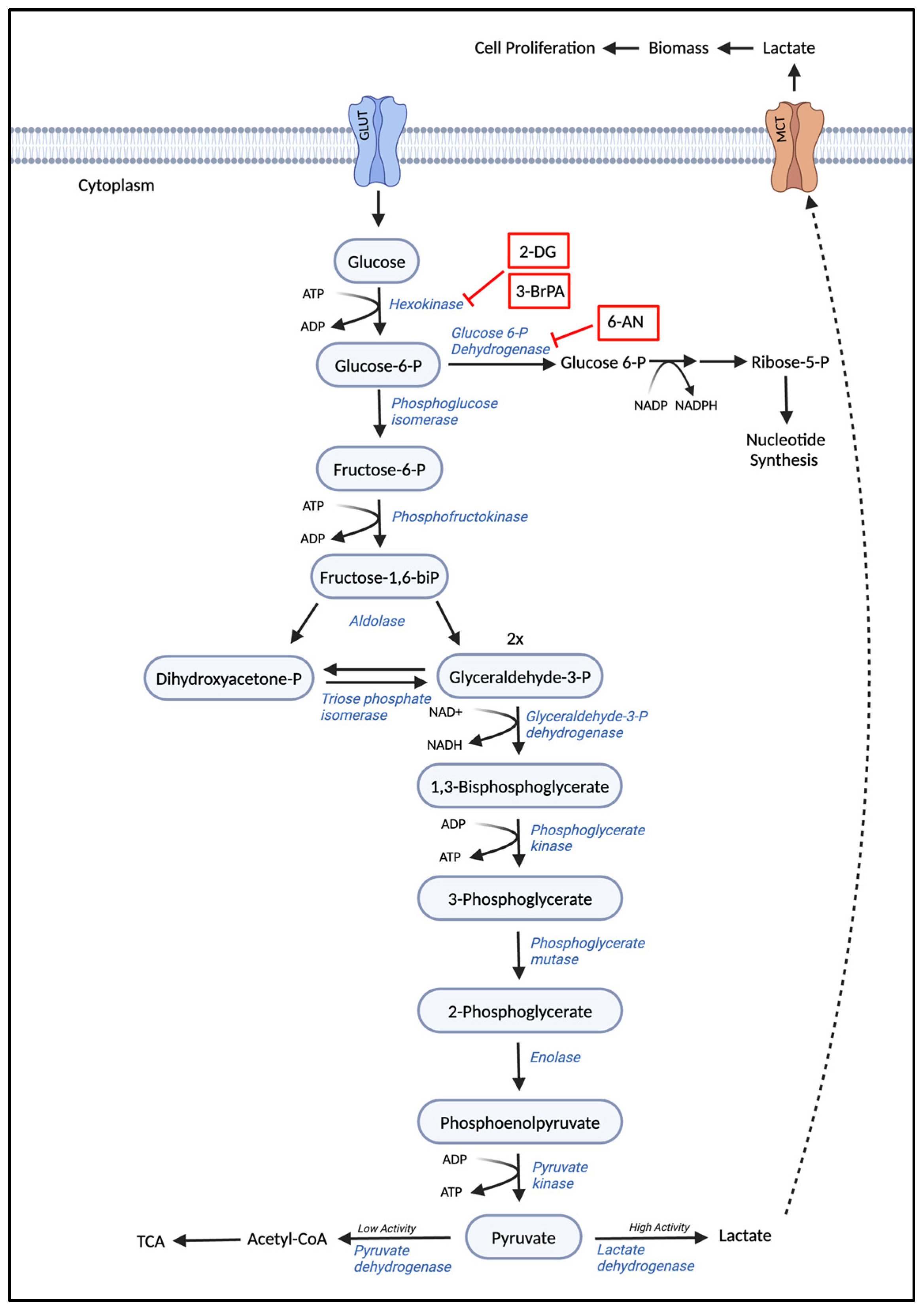
3.1. Glycolysis
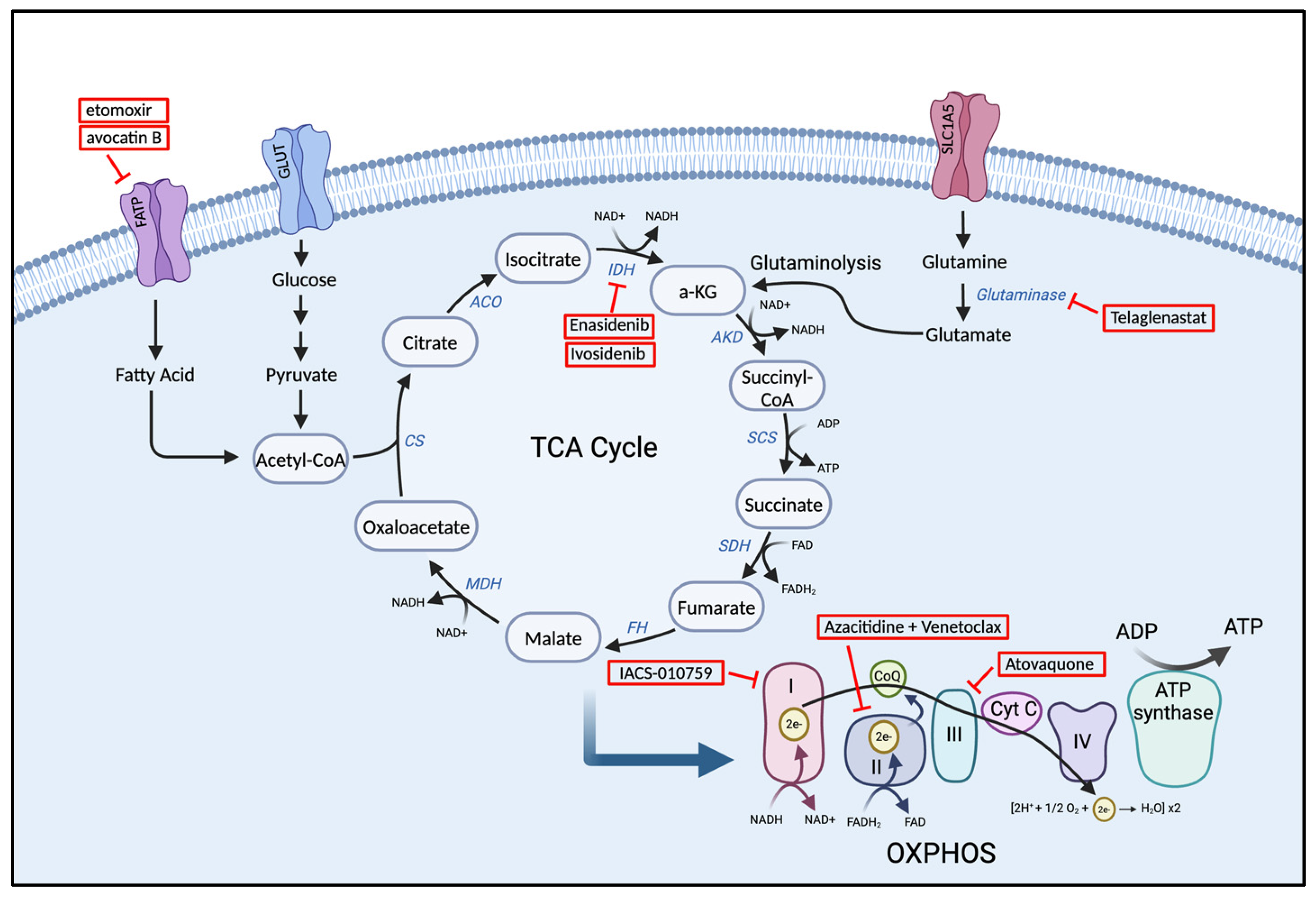
3.2. The Tricarboxylic Acid (TCA) Cycle
3.3. Amino Acid Metabolism
3.3.1. Glutamine
3.3.2. Asparagine
3.3.3. Tryptophan
3.4. Lipid Metabolism
3.4.1. Fatty Acid Biosynthesis and Oxidation
3.4.2. Lipid Steroids
3.4.3. Sphingolipids
4. Current and Emergent Metabolic Therapeutic Strategies
4.1. Agents Targeting Carbohydrate, Amino Acid Metabolism, and Fatty Acid Metabolism
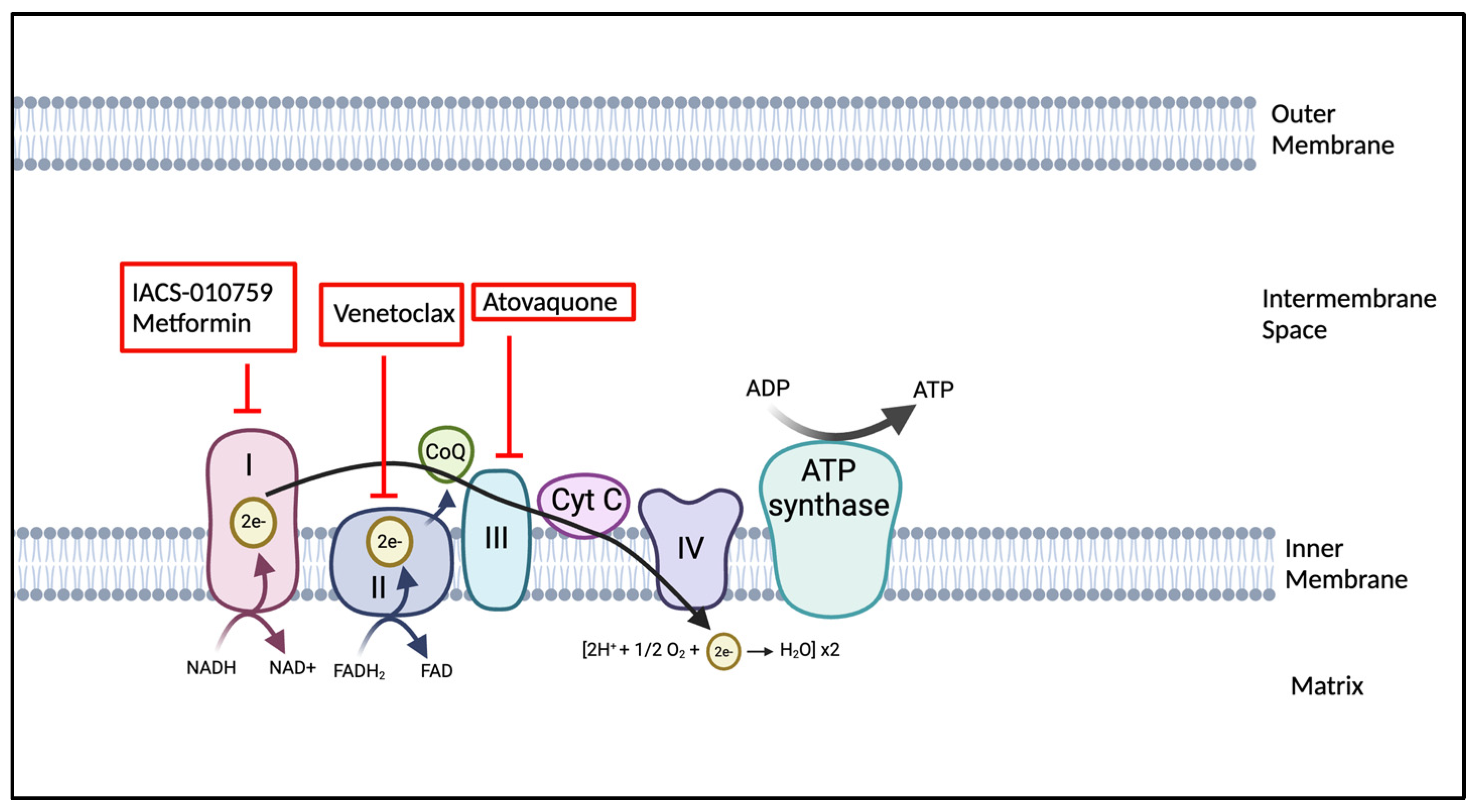
4.2. OXPHOS Inhibitors: Mechanisms and Clinical Implications
4.3. Agents Targeting Genomic Aberrations Associated with Metabolic Pathways
5. Challenges in Targeting Metabolism in AML and Clinical Translation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| HSCs | Hematopoietic stem cells |
| OXPHOS | Oxidative phosphorylation |
| LSCs | Leukemic stem cells |
| MRD | Minimal residual disease |
| Ara-C | Cytarabine |
| HSCT | Hematopoietic stem cell transplantation |
| FLT3 | FMS-related tyrosine kinase 3 gene |
| ITD | Internal tandem duplication |
| TKD | Tyrosine kinase domain |
| IDH1/2 | Isocitrate dehydrogenase mutations 1/2 |
| BM | Bone marrow |
| PDK | Pyruvate dehydrogenase kinase |
| ROS | Reactive oxygen species |
| MMP | Mitochondrial membrane potential |
| mtDNA | Mitochondrial DNA |
| FIS1 | Mitochondrial fission protein 1 |
| PDH | Pyruvate dehydrogenase |
| PPP | Pentose phosphate pathway |
| LDH | Lactate dehydrogenase |
| PKM2 | Pyruvate kinase M2 |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| 2-DG | 2-Deoxy-D-glucose |
| TCA cycle | Tricarboxylic acid cycle |
| OAA | Oxaloacetate |
| α-KG | α-ketoglutarate |
| BCAT1 | BCAA transaminase 1 |
| 2-HG | 2-hydroxyglutarate |
| SLC1A5 | Solute carrier family 1, member 5 |
| GLS | Glutaminase |
| GDH | Glutamate dehydrogenase |
| ASS1 | Argininosuccinate synthetase-1 |
| CAT | Cationic amino acid transporter |
| ASNS | Asparagine synthase |
| ALL | Acute lymphoblastic leukemia |
| IDO | Indoleamine-2,3-dioxygenase |
| 1MT | 1-methyl tryptophan |
| FASN | Fatty acid synthase |
| FAO | Fatty acid oxidation |
| FA metabolism | FA metabolism |
| PHD3 | Prolyl Hydroxylase 3 |
| ACC2 | Acetyl CoA carboxylase 2 |
| CPT1a | Carnitine palmitoyl transferase 1a |
| CPT2 | Carnitine transporter |
| LS | Lipid steroids |
| HMGCR | HMG-CoA reductase |
| SPT | Serine palmitoyltransferase |
| AC | Acid ceramidase |
| CerS | Ceramide synthase |
| S1P | Sphingosine-1-phosphate |
| SPHK | Sphingosine kinase |
| 3BrPA | 3-bromopyruvate |
| ADR | Adriamycin |
| CPT1A | Carnitine palmitoyl transferase 1A |
| AQ | Atovoquone |
| PJP | Pneumocystis jiroveci pneumonia |
| pAML | Pediatric Acute Myeloid Leukemia |
References
- Gilliland, D.G.; Jordan, C.T.; Felix, C.A. The molecular basis of leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2004, 1, 80–97. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.; Dohner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Laribi, K.; Sobh, M.; Ghez, D.; Baugier de Materre, A. Impact of age, functional status, and comorbidities on quality of life and outcomes in elderly patients with AML: Review. Ann. Hematol. 2021, 100, 1359–1376. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Cortes, J.E. Mutations in AML: Prognostic and therapeutic implications. Hematol. Am. Soc. Hematol. Educ. Program 2016, 2016, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Singh, M.; Singh, H.; Kumar, L.; Sharma, A.; Bakhshi, S.; Raina, V.; Thulkar, S. Infections in acute myeloid leukemia: An analysis of 382 febrile episodes. Med. Oncol. 2010, 27, 1037–1045. [Google Scholar] [CrossRef]
- Song, X.; Peng, Y.; Wang, X.; Chen, Y.; Jin, L.; Yang, T.; Qian, M.; Ni, W.; Tong, X.; Lan, J. Incidence, Survival, and Risk Factors for Adults with Acute Myeloid Leukemia Not Otherwise Specified and Acute Myeloid Leukemia with Recurrent Genetic Abnormalities: Analysis of the Surveillance, Epidemiology, and End Results (SEER) Database, 2001–2013. Acta Haematol. 2018, 139, 115–127. [Google Scholar] [CrossRef]
- Mrozek, K.; Bloomfield, C.D. Clinical significance of the most common chromosome translocations in adult acute myeloid leukemia. J. Natl. Cancer Inst. Monogr. 2008, 39, 52–57. [Google Scholar] [CrossRef]
- Costa, A.F.O.; Menezes, D.L.; Pinheiro, L.H.S.; Sandes, A.F.; Nunes, M.A.P.; Lyra Junior, D.P.; Schimieguel, D.M. Role of new Immunophenotypic Markers on Prognostic and Overall Survival of Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis. Sci. Rep. 2017, 7, 4138. [Google Scholar] [CrossRef]
- Estey, E.H. Acute myeloid leukemia: 2019 update on risk-stratification and management. Am. J. Hematol. 2018, 93, 1267–1291. [Google Scholar] [CrossRef] [PubMed]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Bene, M.C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef]
- Fernandez, H.F. New trends in the standard of care for initial therapy of acute myeloid leukemia. Hematol. Am. Soc. Hematol. Educ. Program 2010, 2010, 56–61. [Google Scholar] [CrossRef]
- Magina, K.N.; Pregartner, G.; Zebisch, A.; Wolfler, A.; Neumeister, P.; Greinix, H.T.; Berghold, A.; Sill, H. Cytarabine dose in the consolidation treatment of AML: A systematic review and meta-analysis. Blood 2017, 130, 946–948. [Google Scholar] [CrossRef]
- Murphy, T.; Yee, K.W.L. Cytarabine and daunorubicin for the treatment of acute myeloid leukemia. Expert. Opin. Pharmacother. 2017, 18, 1765–1780. [Google Scholar] [CrossRef]
- Jaramillo, S.; Benner, A.; Krauter, J.; Martin, H.; Kindler, T.; Bentz, M.; Salih, H.R.; Held, G.; Kohne, C.H.; Gotze, K.; et al. Condensed versus standard schedule of high-dose cytarabine consolidation therapy with pegfilgrastim growth factor support in acute myeloid leukemia. Blood Cancer J. 2017, 7, e564. [Google Scholar] [CrossRef] [PubMed]
- Shimoni, A.; Hardan, I.; Shem-Tov, N.; Yeshurun, M.; Yerushalmi, R.; Avigdor, A.; Ben-Bassat, I.; Nagler, A. Allogeneic hematopoietic stem-cell transplantation in AML and MDS using myeloablative versus reduced-intensity conditioning: The role of dose intensity. Leukemia 2006, 20, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Jiang, P.Y.Z.; Sun, H.; Zhang, X.; Jiang, Z.; Li, Y.; Song, Y. Advances in targeted therapy for acute myeloid leukemia. Biomark. Res. 2020, 8, 17. [Google Scholar] [CrossRef]
- Cucchi, D.G.J.; Polak, T.B.; Ossenkoppele, G.J.; Uyl-De Groot, C.A.; Cloos, J.; Zweegman, S.; Janssen, J. Two decades of targeted therapies in acute myeloid leukemia. Leukemia 2021, 35, 651–660. [Google Scholar] [CrossRef]
- Stone, R.M.; Mandrekar, S.J.; Sanford, B.L.; Laumann, K.; Geyer, S.; Bloomfield, C.D.; Thiede, C.; Prior, T.W.; Dohner, K.; Marcucci, G.; et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017, 377, 454–464. [Google Scholar] [CrossRef]
- Perl, A.E.; Martinelli, G.; Cortes, J.E.; Neubauer, A.; Berman, E.; Paolini, S.; Montesinos, P.; Baer, M.R.; Larson, R.A.; Ustun, C.; et al. Gilteritinib or Chemotherapy for Relapsed or Refractory FLT3-Mutated AML. N. Engl. J. Med. 2019, 381, 1728–1740. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D. Ivosidenib in IDH1-Mutated Acute Myeloid Leukemia. N. Engl. J. Med. 2018, 379, 1186. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Cortes, J.E.; Dombret, H.; Merchant, A.; Tauchi, T.; DiRienzo, C.G.; Sleight, B.; Zhang, X.; Leip, E.P.; Shaik, N.; Bell, T.; et al. Glasdegib plus intensive/nonintensive chemotherapy in untreated acute myeloid leukemia: BRIGHT AML 1019 Phase III trials. Future Oncol. 2019, 15, 3531–3545. [Google Scholar] [CrossRef]
- Savona, M.R.; Pollyea, D.A.; Stock, W.; Oehler, V.G.; Schroeder, M.A.; Lancet, J.; McCloskey, J.; Kantarjian, H.M.; Ma, W.W.; Shaik, M.N.; et al. Phase Ib Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination with Standard Chemotherapy in Patients with AML or High-Risk MDS. Clin. Cancer Res. 2018, 24, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab Ozogamicin Versus Best Supportive Care in Older Patients With Newly Diagnosed Acute Myeloid Leukemia Unsuitable for Intensive Chemotherapy: Results of the Randomized Phase III EORTC-GIMEMA AML-19 Trial. J. Clin. Oncol. 2016, 34, 972–979. [Google Scholar] [CrossRef]
- Jen, E.Y.; Ko, C.W.; Lee, J.E.; Del Valle, P.L.; Aydanian, A.; Jewell, C.; Norsworthy, K.J.; Przepiorka, D.; Nie, L.; Liu, J.; et al. FDA Approval: Gemtuzumab Ozogamicin for the Treatment of Adults with Newly Diagnosed CD33-Positive Acute Myeloid Leukemia. Clin. Cancer Res. 2018, 24, 3242–3246. [Google Scholar] [CrossRef]
- Recher, C. The beginning of a new therapeutic era in acute myeloid leukemia. EJHaem 2021, 2, 823–833. [Google Scholar] [CrossRef]
- Burnett, A.; Wetzler, M.; Lowenberg, B. Therapeutic advances in acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 487–494. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef]
- Wei, A.H.; Strickland, S.A., Jr.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined with Low-Dose Cytarabine for Previously Untreated Patients with Acute Myeloid Leukemia: Results from a Phase Ib/II Study. J. Clin. Oncol. 2019, 37, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Larrue, C.; Mouche, S.; Lin, S.; Simonetta, F.; Scheidegger, N.K.; Poulain, L.; Birsen, R.; Sarry, J.E.; Stegmaier, K.; Tamburini, J. Mitochondrial fusion is a therapeutic vulnerability of acute myeloid leukemia. Leukemia 2023, 37, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Bosc, C.; Saland, E.; Bousard, A.; Gadaud, N.; Sabatier, M.; Cognet, G.; Farge, T.; Boet, E.; Gotanegre, M.; Aroua, N.; et al. Mitochondrial inhibitors circumvent adaptive resistance to venetoclax and cytarabine combination therapy in acute myeloid leukemia. Nat. Cancer 2021, 2, 1204–1223. [Google Scholar] [CrossRef] [PubMed]
- Stuani, L.; Sabatier, M.; Saland, E.; Cognet, G.; Poupin, N.; Bosc, C.; Castelli, F.A.; Gales, L.; Turtoi, E.; Montersino, C.; et al. Mitochondrial metabolism supports resistance to IDH mutant inhibitors in acute myeloid leukemia. J. Exp. Med. 2021, 218, e20200924. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef]
- Castro, I.; Sampaio-Marques, B.; Ludovico, P. Targeting Metabolic Reprogramming in Acute Myeloid Leukemia. Cells 2019, 8, 967. [Google Scholar] [CrossRef]
- Kubik, J.; Humeniuk, E.; Adamczuk, G.; Madej-Czerwonka, B.; Korga-Plewko, A. Targeting Energy Metabolism in Cancer Treatment. Int. J. Mol. Sci. 2022, 23, 5572. [Google Scholar] [CrossRef]
- Eaves, C.J. Hematopoietic stem cells: Concepts, definitions, and the new reality. Blood 2015, 125, 2605–2613. [Google Scholar] [CrossRef]
- Kohli, L.; Passegue, E. Surviving change: The metabolic journey of hematopoietic stem cells. Trends Cell Biol. 2014, 24, 479–487. [Google Scholar] [CrossRef]
- Boulais, P.E.; Frenette, P.S. Making sense of hematopoietic stem cell niches. Blood 2015, 125, 2621–2629. [Google Scholar] [CrossRef]
- Ho, Y.H.; Mendez-Ferrer, S. Microenvironmental contributions to hematopoietic stem cell aging. Haematologica 2020, 105, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Ellis, S.L.; Nilsson, S.K. The location and cellular composition of the hemopoietic stem cell niche. Cytotherapy 2012, 14, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Domingues, M.J.; Cao, H.; Heazlewood, S.Y.; Cao, B.; Nilsson, S.K. Niche Extracellular Matrix Components and Their Influence on HSC. J. Cell Biochem. 2017, 118, 1984–1993. [Google Scholar] [CrossRef]
- Giebel, B.; Bruns, I. Self-renewal versus differentiation in hematopoietic stem and progenitor cells: A focus on asymmetric cell divisions. Curr. Stem Cell Res. Ther. 2008, 3, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Israelsen, W.J.; Lee, D.; Yu, V.W.C.; Jeanson, N.T.; Clish, C.B.; Cantley, L.C.; Vander Heiden, M.G.; Scadden, D.T. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 2014, 158, 1309–1323. [Google Scholar] [CrossRef]
- Ito, K.; Bonora, M.; Ito, K. Metabolism as master of hematopoietic stem cell fate. Int. J. Hematol. 2019, 109, 18–27. [Google Scholar] [CrossRef]
- Takubo, K. Regulation of hematopoietic stem cells by oxygen metabolism. Rinsho Ketsueki 2010, 51, 95–103. [Google Scholar]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Filippi, M.D.; Ghaffari, S. Mitochondria in the maintenance of hematopoietic stem cells: New perspectives and opportunities. Blood 2019, 133, 1943–1952. [Google Scholar] [CrossRef]
- Papa, L.; Djedaini, M.; Hoffman, R. Mitochondrial Role in Stemness and Differentiation of Hematopoietic Stem Cells. Stem Cells Int. 2019, 2019, 4067162. [Google Scholar] [CrossRef]
- Norddahl, G.L.; Pronk, C.J.; Wahlestedt, M.; Sten, G.; Nygren, J.M.; Ugale, A.; Sigvardsson, M.; Bryder, D. Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging. Cell Stem Cell 2011, 8, 499–510. [Google Scholar] [CrossRef]
- Ludin, A.; Gur-Cohen, S.; Golan, K.; Kaufmann, K.B.; Itkin, T.; Medaglia, C.; Lu, X.J.; Ledergor, G.; Kollet, O.; Lapidot, T. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid. Redox Signal. 2014, 21, 1605–1619. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Li, H.; Pazhanisamy, S.K.; Meng, A.; Wang, Y.; Zhou, D. Reactive oxygen species and hematopoietic stem cell senescence. Int. J. Hematol. 2011, 94, 24–32. [Google Scholar] [CrossRef]
- Liang, R.; Arif, T.; Kalmykova, S.; Kasianov, A.; Lin, M.; Menon, V.; Qiu, J.; Bernitz, J.M.; Moore, K.; Lin, F.; et al. Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 2020, 26, 359–376.e7. [Google Scholar] [CrossRef] [PubMed]
- Morganti, C.; Bonora, M.; Ito, K.; Ito, K. Electron transport chain complex II sustains high mitochondrial membrane potential in hematopoietic stem and progenitor cells. Stem Cell Res. 2019, 40, 101573. [Google Scholar] [CrossRef]
- Karigane, D.; Takubo, K. Metabolic regulation of hematopoietic and leukemic stem/progenitor cells under homeostatic and stress conditions. Int. J. Hematol. 2017, 106, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Panina, S.B.; Pei, J.; Kirienko, N.V. Mitochondrial metabolism as a target for acute myeloid leukemia treatment. Cancer Metab. 2021, 9, 17. [Google Scholar] [CrossRef]
- Saito, K.; Zhang, Q.; Yang, H.; Yamatani, K.; Ai, T.; Ruvolo, V.; Baran, N.; Cai, T.; Ma, H.; Jacamo, R.; et al. Exogenous mitochondrial transfer and endogenous mitochondrial fission facilitate AML resistance to OxPhos inhibition. Blood Adv. 2021, 5, 4233–4255. [Google Scholar] [CrossRef]
- Moschoi, R.; Imbert, V.; Nebout, M.; Chiche, J.; Mary, D.; Prebet, T.; Saland, E.; Castellano, R.; Pouyet, L.; Collette, Y.; et al. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 2016, 128, 253–264. [Google Scholar] [CrossRef]
- Stratmann, S.; Vesterlund, M.; Umer, H.M.; Eshtad, S.; Skaftason, A.; Herlin, M.K.; Sundstrom, C.; Eriksson, A.; Hoglund, M.; Palle, J.; et al. Proteogenomic analysis of acute myeloid leukemia associates relapsed disease with reprogrammed energy metabolism both in adults and children. Leukemia 2023, 37, 550–559. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Jung, N.; Dai, B.; Gentles, A.J.; Majeti, R.; Feinberg, A.P. An LSC epigenetic signature is largely mutation independent and implicates the HOXA cluster in AML pathogenesis. Nat. Commun. 2015, 6, 8489. [Google Scholar] [CrossRef]
- Rothenberg-Thurley, M.; Amler, S.; Goerlich, D.; Kohnke, T.; Konstandin, N.P.; Schneider, S.; Sauerland, M.C.; Herold, T.; Hubmann, M.; Ksienzyk, B.; et al. Persistence of pre-leukemic clones during first remission and risk of relapse in acute myeloid leukemia. Leukemia 2018, 32, 1598–1608. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Mangialardi, G.; Vacca, A. Stephen Paget and the ‘seed and soil’ theory of metastatic dissemination. Clin. Exp. Med. 2006, 6, 145–149. [Google Scholar] [CrossRef]
- Gorre, M.E.; Mohammed, M.; Ellwood, K.; Hsu, N.; Paquette, R.; Rao, P.N.; Sawyers, C.L. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001, 293, 876–880. [Google Scholar] [CrossRef]
- Heidel, F.; Solem, F.K.; Breitenbuecher, F.; Lipka, D.B.; Kasper, S.; Thiede, M.H.; Brandts, C.; Serve, H.; Roesel, J.; Giles, F.; et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood 2006, 107, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.H.; Winter, P.S.; Xie, A.; Roth, C.; Martz, C.A.; Stein, E.M.; Anderson, G.R.; Tingley, J.P.; Wood, K.C. Targeting MCL-1/BCL-XL Forestalls the Acquisition of Resistance to ABT-199 in Acute Myeloid Leukemia. Sci. Rep. 2016, 6, 27696. [Google Scholar] [CrossRef]
- Boyd, A.L.; Aslostovar, L.; Reid, J.; Ye, W.; Tanasijevic, B.; Porras, D.P.; Shapovalova, Z.; Almakadi, M.; Foley, R.; Leber, B.; et al. Identification of Chemotherapy-Induced Leukemic-Regenerating Cells Reveals a Transient Vulnerability of Human AML Recurrence. Cancer Cell 2018, 34, 483–498.e5. [Google Scholar] [CrossRef]
- Skrtic, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef]
- Sriskanthadevan, S.; Jeyaraju, D.V.; Chung, T.E.; Prabha, S.; Xu, W.; Skrtic, M.; Jhas, B.; Hurren, R.; Gronda, M.; Wang, X.; et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 2015, 125, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Marlein, C.R.; Zaitseva, L.; Piddock, R.E.; Robinson, S.D.; Edwards, D.R.; Shafat, M.S.; Zhou, Z.; Lawes, M.; Bowles, K.M.; Rushworth, S.A. NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 2017, 130, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Raffel, S.; Klimmeck, D.; Falcone, M.; Demir, A.; Pouya, A.; Zeisberger, P.; Lutz, C.; Tinelli, M.; Bischel, O.; Bullinger, L.; et al. Quantitative proteomics reveals specific metabolic features of acute myeloid leukemia stem cells. Blood 2020, 136, 1507–1519. [Google Scholar] [CrossRef]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 2018, 23, 86–100.e6. [Google Scholar] [CrossRef]
- Tian, Y.; Huang, Z.; Wang, Z.; Yin, C.; Zhou, L.; Zhang, L.; Huang, K.; Zhou, H.; Jiang, X.; Li, J.; et al. Identification of novel molecular markers for prognosis estimation of acute myeloid leukemia: Over-expression of PDCD7, FIS1 and Ang2 may indicate poor prognosis in pretreatment patients with acute myeloid leukemia. PLoS ONE 2014, 9, e84150. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.H.; Mullokandov, M.; Wu, Y.; Voisin, V.; Gronda, M.; Hurren, R.; Wang, X.; MacLean, N.; Jeyaraju, D.V.; Jitkova, Y.; et al. Mitochondrial carrier homolog 2 is necessary for AML survival. Blood 2020, 136, 81–92. [Google Scholar] [CrossRef]
- Bahat, A.; Goldman, A.; Zaltsman, Y.; Khan, D.H.; Halperin, C.; Amzallag, E.; Krupalnik, V.; Mullokandov, M.; Silberman, A.; Erez, A.; et al. MTCH2-mediated mitochondrial fusion drives exit from naive pluripotency in embryonic stem cells. Nat. Commun. 2018, 9, 5132. [Google Scholar] [CrossRef]
- Maryanovich, M.; Zaltsman, Y.; Ruggiero, A.; Goldman, A.; Shachnai, L.; Zaidman, S.L.; Porat, Z.; Golan, K.; Lapidot, T.; Gross, A. An MTCH2 pathway repressing mitochondria metabolism regulates haematopoietic stem cell fate. Nat. Commun. 2015, 6, 7901. [Google Scholar] [CrossRef]
- Singh, R.P.; Jeyaraju, D.V.; Voisin, V.; Hurren, R.; Xu, C.; Hawley, J.R.; Barghout, S.H.; Khan, D.H.; Gronda, M.; Wang, X.; et al. Disrupting Mitochondrial Copper Distribution Inhibits Leukemic Stem Cell Self-Renewal. Cell Stem Cell 2020, 26, 926–937.e10. [Google Scholar] [CrossRef]
- Herault, O.; Hope, K.J.; Deneault, E.; Mayotte, N.; Chagraoui, J.; Wilhelm, B.T.; Cellot, S.; Sauvageau, M.; Andrade-Navarro, M.A.; Hebert, J.; et al. A role for GPx3 in activity of normal and leukemia stem cells. J. Exp. Med. 2012, 209, 895–901. [Google Scholar] [CrossRef]
- Adane, B.; Ye, H.; Khan, N.; Pei, S.; Minhajuddin, M.; Stevens, B.M.; Jones, C.L.; D’Alessandro, A.; Reisz, J.A.; Zaberezhnyy, V.; et al. The Hematopoietic Oxidase NOX2 Regulates Self-Renewal of Leukemic Stem Cells. Cell Rep. 2019, 27, 238–254.e6. [Google Scholar] [CrossRef]
- Hanekamp, D.; Cloos, J.; Schuurhuis, G.J. Leukemic stem cells: Identification and clinical application. Int. J. Hematol. 2017, 105, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Locasale, J.W.; Swanson, K.D.; Sharfi, H.; Heffron, G.J.; Amador-Noguez, D.; Christofk, H.R.; Wagner, G.; Rabinowitz, J.D.; Asara, J.M.; et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 2010, 329, 1492–1499. [Google Scholar] [CrossRef]
- Goodwin, M.L.; Gladden, L.B.; Nijsten, M.W.; Jones, K.B. Lactate and cancer: Revisiting the warburg effect in an era of lactate shuttling. Front. Nutr. 2014, 1, 27. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef]
- Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Jiang, J.K.; Boxer, M.B.; Hong, B.S.; Tempel, W.; Dimov, S.; Shen, M.; Jha, A.; et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 2012, 8, 839–847. [Google Scholar] [CrossRef]
- Locasale, J.W.; Vander Heiden, M.G.; Cantley, L.C. Rewiring of glycolysis in cancer cell metabolism. Cell Cycle 2010, 9, 4253. [Google Scholar] [CrossRef]
- Goncalves, M.D.; Cantley, L.C. A Glycolysis Outsider Steps into the Cancer Spotlight. Cell Metab. 2018, 28, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Mercher, T.; Schwaller, J. From hypoxia single-cell gene signatures to HIF targeting of AML leukemic stem cells. Hemasphere 2024, 8, e59. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, J.E.; Krebs, H. The evolution of metabolic cycles. Nature 1981, 291, 381–382. [Google Scholar] [CrossRef]
- Arnold, P.K.; Finley, L.W.S. Regulation and function of the mammalian tricarboxylic acid cycle. J. Biol. Chem. 2023, 299, 102838. [Google Scholar] [CrossRef]
- Boet, E.; Sarry, J.E. Targeting Metabolic Dependencies Fueling the TCA Cycle to Circumvent Therapy Resistance in Acute Myeloid Leukemia. Cancer Res. 2024, 84, 950–952. [Google Scholar] [CrossRef]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Janin, M.; Mylonas, E.; Saada, V.; Micol, J.B.; Renneville, A.; Quivoron, C.; Koscielny, S.; Scourzic, L.; Forget, S.; Pautas, C.; et al. Serum 2-hydroxyglutarate production in IDH1- and IDH2-mutated de novo acute myeloid leukemia: A study by the Acute Leukemia French Association group. J. Clin. Oncol. 2014, 32, 297–305. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Emadi, A. Exploiting AML vulnerability: Glutamine dependency. Blood 2015, 126, 1269–1270. [Google Scholar] [CrossRef] [PubMed]
- Labow, B.I.; Souba, W.W. Glutamine. World J. Surg. 2000, 24, 1503–1513. [Google Scholar] [CrossRef]
- Meister, A. Metabolism of glutamine. Physiol. Rev. 1956, 36, 103–127. [Google Scholar] [CrossRef]
- Bhutia, Y.D.; Ganapathy, V. Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta 2016, 1863, 2531–2539. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 749. [Google Scholar] [CrossRef]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Srivastava, S.; Zhang, J. Starve Cancer Cells of Glutamine: Break the Spell or Make a Hungry Monster? Cancers 2019, 11, 804. [Google Scholar] [CrossRef]
- Ni, F.; Yu, W.M.; Li, Z.; Graham, D.K.; Jin, L.; Kang, S.; Rossi, M.R.; Li, S.; Broxmeyer, H.E.; Qu, C.K. Critical role of ASCT2-mediated amino acid metabolism in promoting leukaemia development and progression. Nat. Metab. 2019, 1, 390–403. [Google Scholar] [CrossRef]
- Gregory, M.A.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.T.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. 2019, 25, 4079–4090. [Google Scholar] [CrossRef]
- Miraki-Moud, F.; Ghazaly, E.; Ariza-McNaughton, L.; Hodby, K.A.; Clear, A.; Anjos-Afonso, F.; Liapis, K.; Grantham, M.; Sohrabi, F.; Cavenagh, J.; et al. Arginine deprivation using pegylated arginine deiminase has activity against primary acute myeloid leukemia cells in vivo. Blood 2015, 125, 4060–4068. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; Egan, S.; Higginbotham-Jones, J.; Perry, T.; Beggs, A.; Odintsova, E.; Loke, J.; Pratt, G.; Pong U, K.; Lo, A.; et al. Arginine dependence of acute myeloid leukemia blast proliferation: A novel therapeutic target. Blood 2015, 125, 2386–2396. [Google Scholar] [CrossRef]
- Chen, C.L.; Hsu, S.C.; Ann, D.K.; Yen, Y.; Kung, H.J. Arginine Signaling and Cancer Metabolism. Cancers 2021, 13, 3541. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Hunt, N.H.; Ball, H.J. Indoleamine 2,3-dioxygenase 2 (IDO2) and the kynurenine pathway: Characteristics and potential roles in health and disease. Amino Acids 2013, 45, 1319–1329. [Google Scholar] [CrossRef]
- Ye, Z.; Yue, L.; Shi, J.; Shao, M.; Wu, T. Role of IDO and TDO in Cancers and Related Diseases and the Therapeutic Implications. J. Cancer 2019, 10, 2771–2782. [Google Scholar] [CrossRef]
- El Kholy, N.M.; Sallam, M.M.; Ahmed, M.B.; Sallam, R.M.; Asfour, I.A.; Hammouda, J.A.; Habib, H.Z.; Abu-Zahra, F. Expression of indoleamine 2,3-dioxygenase in acute myeloid leukemia and the effect of its inhibition on cultured leukemia blast cells. Med. Oncol. 2011, 28, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Chamuleau, M.E.; van de Loosdrecht, A.A.; Hess, C.J.; Janssen, J.J.; Zevenbergen, A.; Delwel, R.; Valk, P.J.; Lowenberg, B.; Ossenkoppele, G.J. High INDO (indoleamine 2,3-dioxygenase) mRNA level in blasts of acute myeloid leukemic patients predicts poor clinical outcome. Haematologica 2008, 93, 1894–1898. [Google Scholar] [CrossRef]
- Curti, A.; Aluigi, M.; Pandolfi, S.; Ferri, E.; Isidori, A.; Salvestrini, V.; Durelli, I.; Horenstein, A.L.; Fiore, F.; Massaia, M.; et al. Acute myeloid leukemia cells constitutively express the immunoregulatory enzyme indoleamine 2,3-dioxygenase. Leukemia 2007, 21, 353–355. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Leibundgut, M.; Ban, N. The crystal structure of a mammalian fatty acid synthase. Science 2008, 321, 1315–1322. [Google Scholar] [CrossRef]
- Tabe, Y.; Yamamoto, S.; Saitoh, K.; Sekihara, K.; Monma, N.; Ikeo, K.; Mogushi, K.; Shikami, M.; Ruvolo, V.; Ishizawa, J.; et al. Bone Marrow Adipocytes Facilitate Fatty Acid Oxidation Activating AMPK and a Transcriptional Network Supporting Survival of Acute Monocytic Leukemia Cells. Cancer Res. 2017, 77, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- German, N.J.; Yoon, H.; Yusuf, R.Z.; Murphy, J.P.; Finley, L.W.; Laurent, G.; Haas, W.; Satterstrom, F.K.; Guarnerio, J.; Zaganjor, E.; et al. PHD3 Loss in Cancer Enables Metabolic Reliance on Fatty Acid Oxidation via Deactivation of ACC2. Mol. Cell 2016, 63, 1006–1020. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef]
- Ricciardi, M.R.; Mirabilii, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.R.; Foa, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the leukemia cell metabolism by the CPT1a inhibition: Functional preclinical effects in leukemias. Blood 2015, 126, 1925–1929. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hu, J.W.; He, X.R.; Jin, W.L.; He, X.Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef] [PubMed]
- Dimitroulakos, J.; Thai, S.; Wasfy, G.H.; Hedley, D.W.; Minden, M.D.; Penn, L.Z. Lovastatin induces a pronounced differentiation response in acute myeloid leukemias. Leuk. Lymphoma 2000, 40, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Tan, M.M.; Wong, W.W.; Dimitroulakos, J.; Minden, M.D.; Penn, L.Z. Blocking protein geranylgeranylation is essential for lovastatin-induced apoptosis of human acute myeloid leukemia cells. Leukemia 2001, 15, 1398–1407. [Google Scholar] [CrossRef]
- Wong, W.W.; Dimitroulakos, J.; Minden, M.D.; Penn, L.Z. HMG-CoA reductase inhibitors and the malignant cell: The statin family of drugs as triggers of tumor-specific apoptosis. Leukemia 2002, 16, 508–519. [Google Scholar] [CrossRef]
- Cafforio, P.; Dammacco, F.; Gernone, A.; Silvestris, F. Statins activate the mitochondrial pathway of apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis 2005, 26, 883–891. [Google Scholar] [CrossRef]
- Advani, A.S.; Li, H.; Michaelis, L.C.; Medeiros, B.C.; Liedtke, M.; List, A.F.; O’Dwyer, K.; Othus, M.; Erba, H.P.; Appelbaum, F.R. Report of the relapsed/refractory cohort of SWOG S0919: A phase 2 study of idarubicin and cytarabine in combination with pravastatin for acute myelogenous leukemia (AML). Leuk. Res. 2018, 67, 17–20. [Google Scholar] [CrossRef]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef]
- Tan, S.F.; Liu, X.; Fox, T.E.; Barth, B.M.; Sharma, A.; Turner, S.D.; Awwad, A.; Dewey, A.; Doi, K.; Spitzer, B.; et al. Acid ceramidase is upregulated in AML and represents a novel therapeutic target. Oncotarget 2016, 7, 83208–83222. [Google Scholar] [CrossRef]
- Dany, M.; Gencer, S.; Nganga, R.; Thomas, R.J.; Oleinik, N.; Baron, K.D.; Szulc, Z.M.; Ruvolo, P.; Kornblau, S.; Andreeff, M.; et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 2016, 128, 1944–1958. [Google Scholar] [CrossRef]
- Powell, J.A.; Wallington-Beddoe, C.T.; Pitson, S.M. Targeting sphingosine kinase 1 in acute myeloid leukemia: Translation to clinic. Int. J. Hematol. Oncol. 2017, 6, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Wallington-Beddoe, C.T.; Xie, V.; Tong, D.; Powell, J.A.; Lewis, A.C.; Davies, L.; Pitson, S.M.; Bradstock, K.F.; Bendall, L.J. Identification of sphingosine kinase 1 as a therapeutic target in B-lineage acute lymphoblastic leukaemia. Br. J. Haematol. 2019, 184, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Wang, J.H.; Zhao, A.H.; Xu, X.; Wang, Y.H.; Chen, T.L.; Li, J.M.; Mi, J.Q.; Zhu, Y.M.; Liu, Y.F.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef]
- Ju, H.Q.; Zhan, G.; Huang, A.; Sun, Y.; Wen, S.; Yang, J.; Lu, W.H.; Xu, R.H.; Li, J.; Li, Y.; et al. ITD mutation in FLT3 tyrosine kinase promotes Warburg effect and renders therapeutic sensitivity to glycolytic inhibition. Leukemia 2017, 31, 2143–2150. [Google Scholar] [CrossRef]
- Song, K.; Li, M.; Xu, X.; Xuan, L.I.; Huang, G.; Liu, Q. Resistance to chemotherapy is associated with altered glucose metabolism in acute myeloid leukemia. Oncol. Lett. 2016, 12, 334–342. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Verma, D.; Baran, N.; Bhagat, T.D.; Skwarska, A.; Lodi, A.; Saxena, K.; Cai, T.; Su, X.; Guerra, V.A.; et al. Glutaminase inhibition in combination with azacytidine in myelodysplastic syndromes: A phase 1b/2 clinical trial and correlative analyses. Nat. Cancer 2024, 5, 1515–1533. [Google Scholar] [CrossRef]
- Ahmed, T.; Holwerda, S.; Klepin, H.D.; Isom, S.; Ellis, L.R.; Lyerly, S.; Manuel, M.; Dralle, S.; Berenzon, D.; Powell, B.L.; et al. High dose cytarabine, mitoxantrone and l-asparaginase (HAMA) salvage for relapsed or refractory acute myeloid leukemia (AML) in the elderly. Leuk. Res. 2015, 39, 945–949. [Google Scholar] [CrossRef]
- Michelozzi, I.M.; Granata, V.; De Ponti, G.; Alberti, G.; Tomasoni, C.; Antolini, L.; Gambacorti-Passerini, C.; Gentner, B.; Dazzi, F.; Biondi, A.; et al. Acute myeloid leukaemia niche regulates response to L-asparaginase. Br. J. Haematol. 2019, 186, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Fu, H.; Jia, Z.; He, K.; Fu, L.; Wang, W. High Expression of CPT1A Predicts Adverse Outcomes: A Potential Therapeutic Target for Acute Myeloid Leukemia. EBioMedicine 2016, 14, 55–64. [Google Scholar] [CrossRef]
- Lee, E.A.; Angka, L.; Rota, S.G.; Hanlon, T.; Mitchell, A.; Hurren, R.; Wang, X.M.; Gronda, M.; Boyaci, E.; Bojko, B.; et al. Targeting Mitochondria with Avocatin B Induces Selective Leukemia Cell Death. Cancer Res. 2015, 75, 2478–2488. [Google Scholar] [CrossRef]
- Lo Presti, C.; Yamaryo-Botte, Y.; Mondet, J.; Berthier, S.; Nutiu, D.; Botte, C.; Mossuz, P. Variation in Lipid Species Profiles among Leukemic Cells Significantly Impacts Their Sensitivity to the Drug Targeting of Lipid Metabolism and the Prognosis of AML Patients. Int. J. Mol. Sci. 2023, 24, 5988. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Daver, N.; Mahendra, M.; Zhang, J.; Kamiya-Matsuoka, C.; Meric-Bernstam, F.; Kantarjian, H.M.; Ravandi, F.; Collins, M.E.; Francesco, M.E.D.; et al. Complex I inhibitor of oxidative phosphorylation in advanced solid tumors and acute myeloid leukemia: Phase I trials. Nat. Med. 2023, 29, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef]
- Matre, P.; Velez, J.; Jacamo, R.; Qi, Y.; Su, X.; Cai, T.; Chan, S.M.; Lodi, A.; Sweeney, S.R.; Ma, H.; et al. Inhibiting glutaminase in acute myeloid leukemia: Metabolic dependency of selected AML subtypes. Oncotarget 2016, 7, 79722–79735. [Google Scholar] [CrossRef]
- Tsai, H.J.; Jiang, S.S.; Hung, W.C.; Borthakur, G.; Lin, S.F.; Pemmaraju, N.; Jabbour, E.; Bomalaski, J.S.; Chen, Y.P.; Hsiao, H.H.; et al. A Phase II Study of Arginine Deiminase (ADI-PEG20) in Relapsed/Refractory or Poor-Risk Acute Myeloid Leukemia Patients. Sci. Rep. 2017, 7, 11253. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Senapati, J.; Kantarjian, H.M.; Bazinet, A.; Reville, P.; Short, N.J.; Daver, N.; Borthakur, G.; Bataller, A.; Jabbour, E.; DiNardo, C.; et al. Lower intensity therapy with cladribine/low dose cytarabine/venetoclax in older patients with acute myeloid leukemia compares favorably with intensive chemotherapy among patients undergoing allogeneic stem cell transplantation. Cancer 2024, 130, 3333–3343. [Google Scholar] [CrossRef]
- Reed, G.A.; Schiller, G.J.; Kambhampati, S.; Tallman, M.S.; Douer, D.; Minden, M.D.; Yee, K.W.; Gupta, V.; Brandwein, J.; Jitkova, Y.; et al. A Phase 1 study of intravenous infusions of tigecycline in patients with acute myeloid leukemia. Cancer Med. 2016, 5, 3031–3040. [Google Scholar] [CrossRef]
- Stemer, G.; Rowe, J.M.; Ofran, Y. Efficacy and Safety Profile of Ivosidenib in the Management of Patients with Acute Myeloid Leukemia (AML): An Update on the Emerging Evidence. Blood Lymphat. Cancer 2021, 11, 41–54. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Watts, J.M.; Baer, M.R.; Yang, J.; Prebet, T.; Lee, S.; Schiller, G.J.; Dinner, S.N.; Pigneux, A.; Montesinos, P.; Wang, E.S.; et al. Olutasidenib alone or with azacitidine in IDH1-mutated acute myeloid leukaemia and myelodysplastic syndrome: Phase 1 results of a phase 1/2 trial. Lancet Haematol. 2023, 10, e46–e58. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Wang, H.; DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Watts, J.M.; Pollyea, D.A.; et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020, 4, 1894–1905. [Google Scholar] [CrossRef]
- Stevens, A.M.; Schafer, E.S.; Li, M.; Terrell, M.; Rashid, R.; Paek, H.; Bernhardt, M.B.; Weisnicht, A.; Smith, W.T.; Keogh, N.J.; et al. Repurposing Atovaquone as a Therapeutic against Acute Myeloid Leukemia (AML): Combination with Conventional Chemotherapy Is Feasible and Well Tolerated. Cancers 2023, 15, 1344. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Mao, L.; Yang, M.; Qian, P.; Lu, H.; Tong, H.; Xie, W.; Zhou, D.; Huang, X.; Wang, Y.; et al. Venetoclax plus 3 + 7 daunorubicin and cytarabine chemotherapy as first-line treatment for adults with acute myeloid leukaemia: A multicentre, single-arm, phase 2 trial. Lancet Haematol. 2022, 9, e415–e424. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Lachowiez, C.A.; Takahashi, K.; Loghavi, S.; Kadia, T.; Daver, N.; Xiao, L.; Adeoti, M.; Short, N.J.; Sasaki, K.; et al. Venetoclax combined with FLAG-IDA induction and consolidation in newly diagnosed acute myeloid leukemia. Am. J. Hematol. 2022, 97, 1035–1043. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Kim, H.; Ho, V.T.; Walker, S.R.; Bar-Natan, M.; Anahtar, M.; Liu, S.; Toniolo, P.A.; Kroll, Y.; Jones, N.; et al. Gene expression-based discovery of atovaquone as a STAT3 inhibitor and anticancer agent. Blood 2016, 128, 1845–1853. [Google Scholar] [CrossRef]
- Stevens, A.M.; Xiang, M.; Heppler, L.N.; Tosic, I.; Jiang, K.; Munoz, J.O.; Gaikwad, A.S.; Horton, T.M.; Long, X.; Narayanan, P.; et al. Atovaquone is active against AML by upregulating the integrated stress pathway and suppressing oxidative phosphorylation. Blood Adv. 2019, 3, 4215–4227. [Google Scholar] [CrossRef]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef]
- Ward, P.S.; Lu, C.; Cross, J.R.; Abdel-Wahab, O.; Levine, R.L.; Schwartz, G.K.; Thompson, C.B. The potential for isocitrate dehydrogenase mutations to produce 2-hydroxyglutarate depends on allele specificity and subcellular compartmentalization. J. Biol. Chem. 2013, 288, 3804–3815. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Araujo Cruz, M.M.; Jyotsana, N.; Sharma, A.; Yun, H.; Gorlich, K.; Wichmann, M.; Schwarzer, A.; Preller, M.; Thol, F.; et al. Mutant IDH1 promotes leukemogenesis in vivo and can be specifically targeted in human AML. Blood 2013, 122, 2877–2887. [Google Scholar] [CrossRef]
- Ogawara, Y.; Katsumoto, T.; Aikawa, Y.; Shima, Y.; Kagiyama, Y.; Soga, T.; Matsunaga, H.; Seki, T.; Araki, K.; Kitabayashi, I. IDH2 and NPM1 Mutations Cooperate to Activate Hoxa9/Meis1 and Hypoxia Pathways in Acute Myeloid Leukemia. Cancer Res. 2015, 75, 2005–2016. [Google Scholar] [CrossRef]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Kronke, J.; Bullinger, L.; Spath, D.; Kayser, S.; Zucknick, M.; Gotze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, P.; de Botton, S.; Dohner, H. Ivosidenib and Azacitidine in IDH1-Mutated AML. N. Engl. J. Med. 2022, 386, 2536–2537. [Google Scholar] [CrossRef] [PubMed]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef]
- de Botton, S.; Montesinos, P.; Schuh, A.C.; Papayannidis, C.; Vyas, P.; Wei, A.H.; Ommen, H.; Semochkin, S.; Kim, H.J.; Larson, R.A.; et al. Enasidenib vs. conventional care in older patients with late-stage mutant-IDH2 relapsed/refractory AML: A randomized phase 3 trial. Blood 2023, 141, 156–167. [Google Scholar] [CrossRef]
- Zeng, Z.; Wang, R.Y.; Qiu, Y.H.; Mak, D.H.; Coombes, K.; Yoo, S.Y.; Zhang, Q.; Jessen, K.; Liu, Y.; Rommel, C.; et al. MLN0128, a novel mTOR kinase inhibitor, disrupts survival signaling and triggers apoptosis in AML and AML stem/ progenitor cells. Oncotarget 2016, 7, 55083–55097. [Google Scholar] [CrossRef]
- Huang, A.; Ju, H.Q.; Liu, K.; Zhan, G.; Liu, D.; Wen, S.; Garcia-Manero, G.; Huang, P.; Hu, Y. Metabolic alterations and drug sensitivity of tyrosine kinase inhibitor resistant leukemia cells with a FLT3/ITD mutation. Cancer Lett. 2016, 377, 149–157. [Google Scholar] [CrossRef]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Abdel-Wahab, O.; Nahas, M.K.; Wang, K.; Rampal, R.K.; Intlekofer, A.M.; Patel, J.; Krivstov, A.; Frampton, G.M.; Young, L.E.; et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood 2016, 127, 3004–3014. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Inguva, A.; Jordan, C.T. Targeting Energy Metabolism in Cancer Stem Cells: Progress and Challenges in Leukemia and Solid Tumors. Cell Stem Cell 2021, 28, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Konopleva, M. Resistance to energy metabolism—Targeted therapy of AML cells residual in the bone marrow microenvironment. Cancer Drug Resist. 2023, 6, 138–150. [Google Scholar] [CrossRef]
- Phillips, C.L.; Lane, A.; Gerbing, R.B.; Alonzo, T.A.; Wilkey, A.; Radloff, G.; Lange, B.; Gamazon, E.R.; Dolan, M.E.; Davies, S.M. Genomic Variants of Cytarabine Sensitivity Associated with Treatment-Related Mortality in Pediatric AML: A Report from the Children’s Oncology Group. Clin. Cancer Res. 2020, 26, 2891–2897. [Google Scholar] [CrossRef]
- Ellinghaus, P.; Heisler, I.; Unterschemmann, K.; Haerter, M.; Beck, H.; Greschat, S.; Ehrmann, A.; Summer, H.; Flamme, I.; Oehme, F.; et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2013, 2, 611–624. [Google Scholar] [CrossRef]
- Perez-Herrero, E.; Fernandez-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B., Jr.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Dohner, H.; Campbell, P.J. Genomic Classification in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 375, 900–901. [Google Scholar] [CrossRef] [PubMed]
- Meshinchi, S.; Arceci, R.J. Prognostic factors and risk-based therapy in pediatric acute myeloid leukemia. Oncologist 2007, 12, 341–355. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Metabolic Target/Effect | Mechanism of Action | Stage of Development/NCT Number | Ref. |
|---|---|---|---|---|
| 2-Deoxy-D-glucose (2-DG) | Glycolysis | Inhibition of hexokinase 2 | Pre-clinical (in vitro and in vivo) | [144] |
| 3-bromopyruvate (3BrPA) | Glycolysis | Inhibition of hexokinase 2 | Pre-clinical (in vitro and in vivo) | [145] |
| IACS-010759 | OXPHOS | Inhibition ETC1 activity | Phase I (NCT02882321) | [152] |
| Telaglenastat (CB-839) | Glutaminolysis | Inhibited GLS1 and glutathione | Phase I (NCT02071927) | [153,154] |
| BCT-100 | Arginine metabolism | Arginine depletion | Phase I/II (NCT03455140) | [116] |
| ADI-PEG 20 | Arginine metabolism | Arginine depletion | Phase II trial (NCT01910012) | [155] |
| Venetoclax | OXPHOS | BCL-2 inhibition | Phase I/II/III (NCT02993523, NCT04801797, NCT05177731, NCT05048615,NCT03586609) | [156,157] |
| LCL204 | Lipid metabolism | Acid ceramide inhibition | Pre-clinical (in vitro and in vivo) | [139] |
| Tigecyclin | OXPHOS | Mitochondrial translation inhibition | Phase I (NCT01332786) | [158] |
| Ivosidenib (AG-120) | Genetic mutations | Inhibitor of mutant IDH1 | Phase I/II/III (NCT02074839, NCT03173248) | [22,159,160] |
| Olutasidenib (FT-2102) | Genetic mutations | Inhibitor of mutant IDH1 | Phase I/II (NCT02719574) | [161] |
| Enasidenib (AG-221) | Genetic mutations | Inhibitor of mutant IDH2 | Phase I/II/III (NCT02577406) | [162] |
| Atovaquone | OXPHOS | Upregulating the ISR pathway and downregulating oxidative phosphorylation | Pre-clinical (in vitro and in vivo) Feasibility Trial (NCT03568994) | [163] |
| Avocatin B, Etomoxir, and ST-1326 | Lipid metabolsim | Altering fatty acid oxidation | Pre-clinical (in vitro and in vivo) | [71,128,150] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Addanki, S.; Kim, L.; Stevens, A. Understanding and Targeting Metabolic Vulnerabilities in Acute Myeloid Leukemia: An Updated Comprehensive Review. Cancers 2025, 17, 1355. https://doi.org/10.3390/cancers17081355
Addanki S, Kim L, Stevens A. Understanding and Targeting Metabolic Vulnerabilities in Acute Myeloid Leukemia: An Updated Comprehensive Review. Cancers. 2025; 17(8):1355. https://doi.org/10.3390/cancers17081355
Chicago/Turabian StyleAddanki, Sridevi, Lana Kim, and Alexandra Stevens. 2025. "Understanding and Targeting Metabolic Vulnerabilities in Acute Myeloid Leukemia: An Updated Comprehensive Review" Cancers 17, no. 8: 1355. https://doi.org/10.3390/cancers17081355
APA StyleAddanki, S., Kim, L., & Stevens, A. (2025). Understanding and Targeting Metabolic Vulnerabilities in Acute Myeloid Leukemia: An Updated Comprehensive Review. Cancers, 17(8), 1355. https://doi.org/10.3390/cancers17081355