
Recent Developments in Targeting the Cell Cycle in Melanoma


Simple Summary
Abstract
1. Introduction
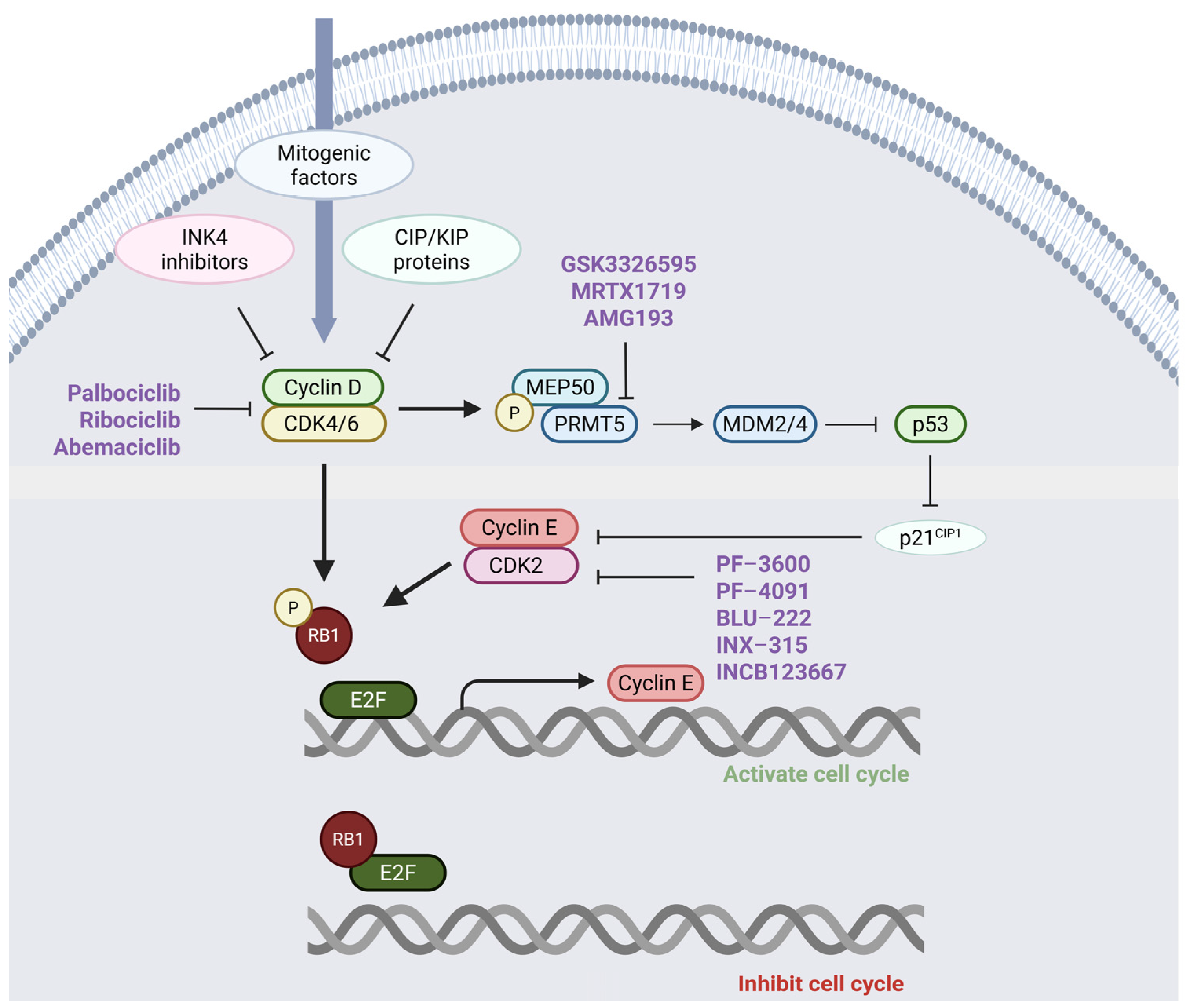
2. The Role of the Cyclin–CDK-RB Axis in Regulating Cell Proliferation
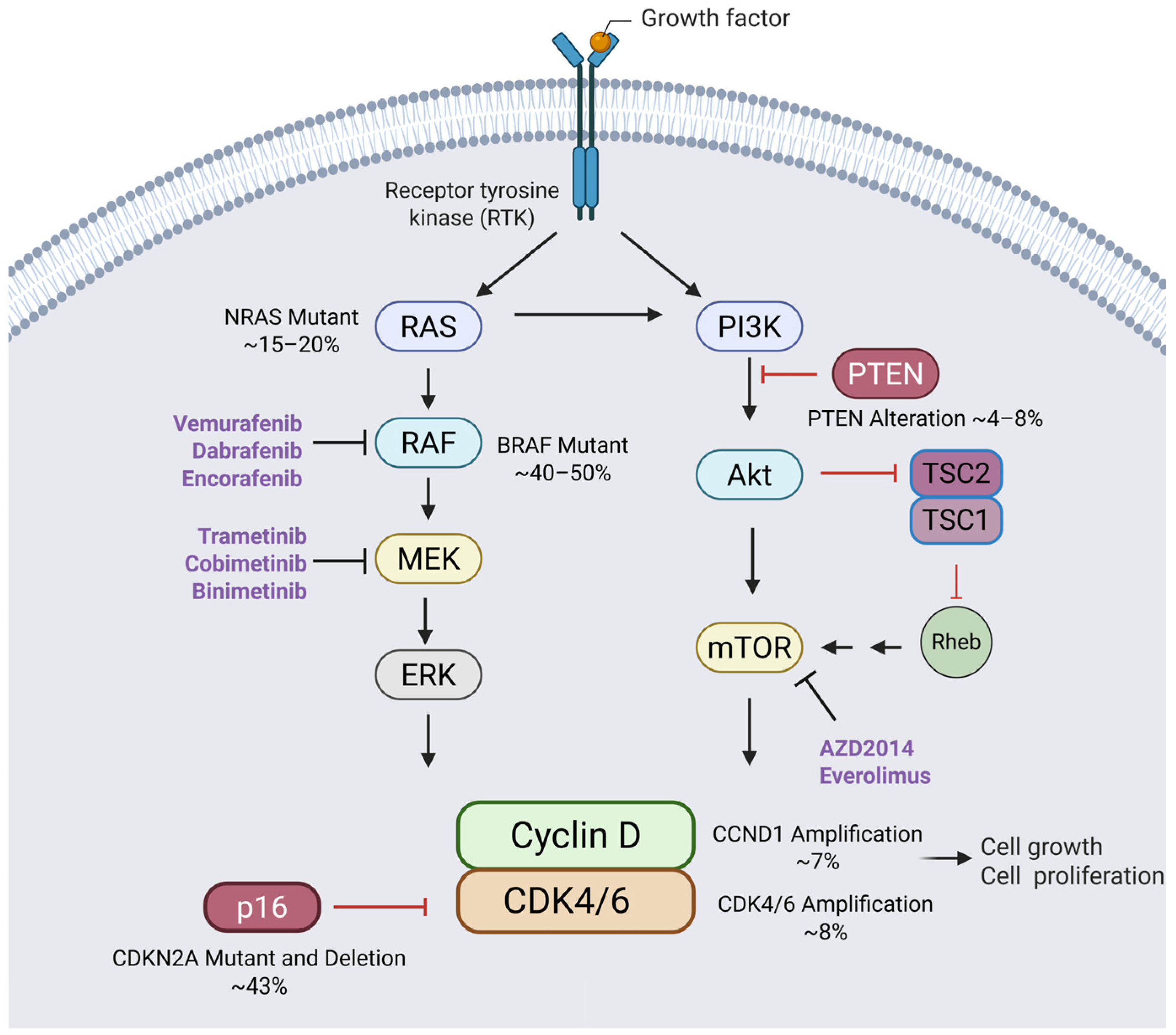
3. Alterations to the Cyclin–CDK-RB Axis in Melanoma
4. Preclinical and Clinical Studies in Melanoma Utilizing CDK4/6 Inhibitors
5. Redundancies in CDK Pathways: Implications for Cancer Therapy Resistance
6. Relative Levels of CDK4 and CDK6 Impact Efficacy of CDK4/6is in Melanoma
7. Combining CDK2 and CDK4/6 Inhibitors for Synergistic Anti-Cancer Therapy
8. Combining PI3K/Akt/mTOR and CDK4/6 Inhibitors for Enhanced Cancer Therapy
9. Immunotherapy in Combination with CDK4/6 Inhibitors
10. Other Potential Combination Treatments with CDK4/6is
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Curti, B.D.; Faries, M.B. Recent Advances in the Treatment of Melanoma. N. Engl. J. Med. 2021, 384, 2229–2240. [Google Scholar] [PubMed]
- Young, R.J.; Waldeck, K.; Martin, C.; Foo, J.H.; Cameron, D.P.; Kirby, L.; Do, H.; Mitchell, C.; Cullinane, C.; Liu, W.; et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment. Cell Melanoma Res. 2014, 27, 590–600. [Google Scholar] [PubMed]
- Morrison, L.; Loibl, S.; Turner, N.C. The CDK4/6 inhibitor revolution—A game-changing era for breast cancer treatment. Nat. Rev. Clin. Oncol. 2024, 21, 89–105. [Google Scholar]
- Lv, S.; Yang, J.; Lin, J.; Huang, X.; Zhao, H.; Zhao, C.; Yang, L. CDK4/6 inhibitors in lung cancer: Current practice and future directions. Eur. Respir. Rev. 2024, 33, 230145. [Google Scholar] [PubMed]
- Thoma, O.M.; Neurath, M.F.; Waldner, M.J. Cyclin-Dependent Kinase Inhibitors and Their Therapeutic Potential in Colorectal Cancer Treatment. Front. Pharmacol. 2021, 12, 757120. [Google Scholar]
- Olmez, I.; Zhang, Y.; Manigat, L.; Benamar, M.; Brenneman, B.; Nakano, I.; Godlewski, J.; Bronisz, A.; Lee, J.; Abbas, T.; et al. Combined c-Met/Trk Inhibition Overcomes Resistance to CDK4/6 Inhibitors in Glioblastoma. Cancer Res. 2018, 78, 4360–4369. [Google Scholar] [PubMed]
- Goel, S.; Bergholz, J.S.; Zhao, J.J. Targeting CDK4 and CDK6 in cancer. Nat. Rev. Cancer 2022, 22, 356–372. [Google Scholar] [PubMed]
- Hartwell, L.H.; Culotti, J.; Reid, B. Genetic control of the cell-division cycle in yeast. I. Detection of mutants. Proc. Natl. Acad. Sci. USA 1970, 66, 352–359. [Google Scholar]
- Hartwell, L.H.; Mortimer, R.K.; Culotti, J.; Culotti, M. Genetic Control of the Cell Division Cycle in Yeast: V. Genetic Analysis of cdc Mutants. Genetics 1973, 74, 267–286. [Google Scholar]
- Lee, M.G.; Nurse, P. Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature 1987, 327, 31–35. [Google Scholar]
- Evans, T.; Rosenthal, E.T.; Youngblom, J.; Distel, D.; Hunt, T. Cyclin: A protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 1983, 33, 389–396. [Google Scholar] [PubMed]
- Matsushime, H.; Ewen, M.E.; Strom, D.K.; Kato, J.Y.; Hanks, S.K.; Roussel, M.F.; Sherr, C.J. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell 1992, 71, 323–334. [Google Scholar]
- Matsushime, H.; Roussel, M.F.; Ashmun, R.A.; Sherr, C.J. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell 1991, 65, 701–713. [Google Scholar] [PubMed]
- Matsushime, H.; Roussel, M.F.; Sherr, C.J. Novel mammalian cyclins (CYL genes) expressed during G1. Cold Spring Harb. Symp. Quant. Biol. 1991, 56, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, M.; Harlow, E. Identification of G1 kinase activity for cdk6, a novel cyclin D partner. Mol. Cell. Biol. 1994, 14, 2077–2086. [Google Scholar] [PubMed]
- Diehl, J.A.; Zindy, F.; Sherr, C.J. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997, 11, 957–972. [Google Scholar] [PubMed]
- Lavoie, J.N.; L’Allemain, G.; Brunet, A.; Muller, R.; Pouyssegur, J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 1996, 271, 20608–20616. [Google Scholar]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar]
- Sherr, C.J. Mammalian G1 cyclins. Cell 1993, 73, 1059–1065. [Google Scholar]
- Weinberg, R.A. The retinoblastoma protein and cell cycle control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 2022, 29, 946–960. [Google Scholar] [CrossRef]
- Blagosklonny, M.V.; Pardee, A.B. The restriction point of the cell cycle. Cell Cycle 2002, 1, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [PubMed]
- Goldstein, A.M.; Chan, M.; Harland, M.; Hayward, N.K.; Demenais, F.; Bishop, D.T.; Azizi, E.; Bergman, W.; Bianchi-Scarra, G.; Bruno, W.; et al. Features associated with germline CDKN2A mutations: A GenoMEL study of melanoma-prone families from three continents. J. Med. Genet. 2007, 44, 99–106. [Google Scholar] [CrossRef]
- Kreuger, I.Z.M.; Slieker, R.C.; van Groningen, T.; van Doorn, R. Therapeutic Strategies for Targeting CDKN2A Loss in Melanoma. J. Investig. Dermatol. 2023, 143, 18–25.e11. [Google Scholar] [PubMed]
- Zuo, L.; Weger, J.; Yang, Q.; Goldstein, A.M.; Tucker, M.A.; Walker, G.J.; Hayward, N.; Dracopoli, N.C. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat. Genet. 1996, 12, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Freedberg, D.E.; Rigas, S.H.; Russak, J.; Gai, W.; Kaplow, M.; Osman, I.; Turner, F.; Randerson-Moor, J.A.; Houghton, A.; Busam, K.; et al. Frequent p16-independent inactivation of p14ARF in human melanoma. J. Natl. Cancer Inst. 2008, 100, 784–795. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Nissan, M.H.; Pratilas, C.A.; Jones, A.M.; Ramirez, R.; Won, H.; Liu, C.; Tiwari, S.; Kong, L.; Hanrahan, A.J.; Yao, Z.; et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014, 74, 2340–2350. [Google Scholar]
- Sullivan, R.J.; Flaherty, K. MAP kinase signaling and inhibition in melanoma. Oncogene 2013, 32, 2373–2379. [Google Scholar] [PubMed]
- Kiuru, M.; Busam, K.J. The NF1 gene in tumor syndromes and melanoma. Lab. Investig. 2017, 97, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Yajima, I.; Kumasaka, M.Y.; Thang, N.D.; Goto, Y.; Takeda, K.; Yamanoshita, O.; Iida, M.; Ohgami, N.; Tamura, H.; Kawamoto, Y.; et al. RAS/RAF/MEK/ERK and PI3K/PTEN/AKT Signaling in Malignant Melanoma Progression and Therapy. Dermatol. Res. Pract. 2012, 2012, 354191. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef]
- Muise-Helmericks, R.C.; Grimes, H.L.; Bellacosa, A.; Malstrom, S.E.; Tsichlis, P.N.; Rosen, N. Cyclin D expression is controlled post-transcriptionally via a phosphatidylinositol 3-kinase/Akt-dependent pathway. J. Biol. Chem. 1998, 273, 29864–29872. [Google Scholar] [CrossRef]
- Schmidt, M.; Fernandez de Mattos, S.; van der Horst, A.; Klompmaker, R.; Kops, G.J.; Lam, E.W.; Burgering, B.M.; Medema, R.H. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell. Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef] [PubMed]
- Braal, C.L.; Jongbloed, E.M.; Wilting, S.M.; Mathijssen, R.H.J.; Koolen, S.L.W.; Jager, A. Inhibiting CDK4/6 in Breast Cancer with Palbociclib, Ribociclib, and Abemaciclib: Similarities and Differences. Drugs 2021, 81, 317–331. [Google Scholar] [PubMed]
- Kong, Y.; Sheng, X.; Wu, X.; Yan, J.; Ma, M.; Yu, J.; Si, L.; Chi, Z.; Cui, C.; Dai, J.; et al. Frequent Genetic Aberrations in the CDK4 Pathway in Acral Melanoma Indicate the Potential for CDK4/6 Inhibitors in Targeted Therapy. Clin. Cancer Res. 2017, 23, 6946–6957. [Google Scholar] [CrossRef]
- Nassar, K.W.; Hintzsche, J.D.; Bagby, S.M.; Espinoza, V.; Langouët-Astrié, C.; Amato, C.M.; Chimed, T.S.; Fujita, M.; Robinson, W.; Tan, A.C.; et al. Targeting CDK4/6 Represents a Therapeutic Vulnerability in Acquired BRAF/MEK Inhibitor–Resistant Melanoma. Mol. Cancer Ther. 2021, 20, 2049–2060. [Google Scholar] [CrossRef]
- Yoshida, A.; Lee, E.K.; Diehl, J.A. Induction of Therapeutic Senescence in Vemurafenib-Resistant Melanoma by Extended Inhibition of CDK4/6. Cancer Res. 2016, 76, 2990–3002. [Google Scholar] [CrossRef]
- Guo, L.; Qi, J.; Wang, H.; Jiang, X.; Liu, Y. Getting under the skin: The role of CDK4/6 in melanomas. Eur. J. Med. Chem. 2020, 204, 112531. [Google Scholar]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Dai, J.; Cao, Y.; Bai, X.; Sheng, X.; Chi, Z.; Cui, C.; Kong, Y.; Zhang, Y.; Wu, L.; et al. Palbociclib in advanced acral melanoma with genetic aberrations in the cyclin-dependent kinase 4 pathway. Eur. J. Cancer 2021, 148, 297–306. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, M.H.; Jegede, O.; Dickson, M.A.; DeMichele, A.M.; Piekarz, R.; Gray, R.J.; Wang, V.; McShane, L.M.; Rubinstein, L.V.; Patton, D.R.; et al. Phase II Study of Palbociclib in Patients with Tumors with CDK4 or CDK6 Amplification: Results from the NCI-MATCH ECOG-ACRIN Trial (EAY131) Subprotocol Z1C. Clin. Cancer Res. 2025, 31, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Bechter, O.; Wolter, P.; Lebbe, C.; Elez, E.; Miller, W.H.; Long, G.V.; Omlin, A.G.; Siena, S.; Calvo, E.; et al. A phase Ib study evaluating the recommended phase II dose, safety, tolerability, and efficacy of mivavotinib in combination with nivolumab in advanced solid tumors. J. Clin. Oncol. 2017, 35, 9518. [Google Scholar] [CrossRef]
- Schuler, M.; Zimmer, L.; Kim, K.B.; Sosman, J.A.; Ascierto, P.A.; Postow, M.A.; De Vos, F.; van Herpen, C.M.L.; Carlino, M.S.; Johnson, D.B.; et al. Phase Ib/II Trial of Ribociclib in Combination with Binimetinib in Patients with NRAS-mutant Melanoma. Clin. Cancer Res. 2022, 28, 3002–3010. [Google Scholar] [CrossRef] [PubMed]
- Louveau, B.; Resche-Rigon, M.; Lesimple, T.; Da Meda, L.; Pracht, M.; Baroudjian, B.; Delyon, J.; Amini-Adle, M.; Dutriaux, C.; Reger de Moura, C.; et al. Phase I-II Open-Label Multicenter Study of Palbociclib + Vemurafenib in BRAF (V600MUT) Metastatic Melanoma Patients: Uncovering CHEK2 as a Major Response Mechanism. Clin. Cancer Res. 2021, 27, 3876–3883. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Amaria, R.N.; Lawrence, D.P.; Brennan, J.; Leister, C.; Singh, R.; Legos, J.; Thurm, H.; Yan, L.; Flaherty, K.T.; et al. Abstract PR06: Phase 1b dose-escalation study of trametinib (MEKi) plus palbociclib (CDK4/6i) in patients with advanced solid tumors. Mol. Cancer Ther. 2015, 14, PR06. [Google Scholar] [CrossRef]
- Dummer, R.; Sandhu, S.K.; Miller, W.H.; Butler, M.O.; Blank, C.U.; Muñoz-Couselo, E.; Burris, H.A., III; Postow, M.A.; Chmielowski, B.; Middleton, M.R.; et al. A phase II, multicenter study of encorafenib/binimetinib followed by a rational triple-combination after progression in patients with advanced BRAF V600-mutated melanoma (LOGIC2). J. Clin. Oncol. 2020, 38, 10022. [Google Scholar] [CrossRef]
- Malumbres, M.; Sotillo, R.; Santamaria, D.; Galan, J.; Cerezo, A.; Ortega, S.; Dubus, P.; Barbacid, M. Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 2004, 118, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Tetsu, O.; McCormick, F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 2003, 3, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Csergeová, L.; Krbušek, D.; Janoštiak, R. CIP/KIP and INK4 families as hostages of oncogenic signaling. Cell Div. 2024, 19, 11. [Google Scholar] [PubMed]
- AbuHammad, S.; Cullinane, C.; Martin, C.; Bacolas, Z.; Ward, T.; Chen, H.; Slater, A.; Ardley, K.; Kirby, L.; Chan, K.T.; et al. Regulation of PRMT5-MDM4 axis is critical in the response to CDK4/6 inhibitors in melanoma. Proc. Natl. Acad. Sci. USA 2019, 116, 17990–18000. [Google Scholar] [CrossRef] [PubMed]
- Gembarska, A.; Luciani, F.; Fedele, C.; Russell, E.A.; Dewaele, M.; Villar, S.; Zwolinska, A.; Haupt, S.; de Lange, J.; Yip, D.; et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat. Med. 2012, 18, 1239–1247. [Google Scholar] [CrossRef]
- Pack, L.R.; Daigh, L.H.; Chung, M.; Meyer, T. Clinical CDK4/6 inhibitors induce selective and immediate dissociation of p21 from cyclin D-CDK4 to inhibit CDK2. Nat. Commun. 2021, 12, 3356. [Google Scholar] [CrossRef]
- Wu, X.; Yang, X.; Xiong, Y.; Li, R.; Ito, T.; Ahmed, T.A.; Karoulia, Z.; Adamopoulos, C.; Wang, H.; Wang, L.; et al. Distinct CDK6 complexes determine tumor cell response to CDK4/6 inhibitors and degraders. Nat. Cancer 2021, 2, 429–443. [Google Scholar] [CrossRef]
- Hallett, S.T.; Pastok, M.W.; Morgan, R.M.L.; Wittner, A.; Blundell, K.; Felletar, I.; Wedge, S.R.; Prodromou, C.; Noble, M.E.M.; Pearl, L.H.; et al. Differential Regulation of G1 CDK Complexes by the Hsp90-Cdc37 Chaperone System. Cell Rep. 2017, 21, 1386–1398. [Google Scholar] [CrossRef]
- Brand, M.; Jiang, B.; Bauer, S.; Donovan, K.A.; Liang, Y.; Wang, E.S.; Nowak, R.P.; Yuan, J.C.; Zhang, T.; Kwiatkowski, N.; et al. Homolog-Selective Degradation as a Strategy to Probe the Function of CDK6 in AML. Cell Chem. Biol. 2019, 26, 300–306.e309. [Google Scholar] [CrossRef]
- De Dominici, M.; Porazzi, P.; Xiao, Y.; Chao, A.; Tang, H.Y.; Kumar, G.; Fortina, P.; Spinelli, O.; Rambaldi, A.; Peterson, L.F.; et al. Selective inhibition of Ph-positive ALL cell growth through kinase-dependent and -independent effects by CDK6-specific PROTACs. Blood 2020, 135, 1560–1573. [Google Scholar] [CrossRef]
- Jiang, B.; Wang, E.S.; Donovan, K.A.; Liang, Y.; Fischer, E.S.; Zhang, T.; Gray, N.S. Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew. Chem. Int. Ed. Engl. 2019, 58, 6321–6326. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Bendjennat, M.; Kour, S.; King, H.M.; Kizhake, S.; Zahid, M.; Natarajan, A. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg. Med. Chem. Lett. 2019, 29, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Yang, Z.; Gao, H.; Yang, H.; Zhu, S.; An, Z.; Wang, J.; Li, Q.; Chandarlapaty, S.; Deng, H.; et al. Potent and Preferential Degradation of CDK6 via Proteolysis Targeting Chimera Degraders. J. Med. Chem. 2019, 62, 7575–7582. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Nicolay, B.N.; Chick, J.M.; Gao, X.; Geng, Y.; Ren, H.; Gao, H.; Yang, G.; Williams, J.A.; Suski, J.M.; et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 2017, 546, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Golomb, L.; Meyerson, M. Functional Genomic Analysis of CDK4 and CDK6 Gene Dependency across Human Cancer Cell Lines. Cancer Res. 2022, 82, 2171–2184. [Google Scholar] [CrossRef]
- Arora, M.; Moser, J.; Hoffman, T.E.; Watts, L.P.; Min, M.; Musteanu, M.; Rong, Y.; Ill, C.R.; Nangia, V.; Schneider, J.; et al. Rapid adaptation to CDK2 inhibition exposes intrinsic cell-cycle plasticity. Cell 2023, 186, 2628–2643.e2621. [Google Scholar] [CrossRef] [PubMed]
- Freeman-Cook, K.; Hoffman, R.L.; Miller, N.; Almaden, J.; Chionis, J.; Zhang, Q.; Eisele, K.; Liu, C.; Zhang, C.; Huser, N.; et al. Expanding control of the tumor cell cycle with a CDK2/4/6 inhibitor. Cancer Cell 2021, 39, 1404–1421.e1411. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Hemming, M.L.; Lundberg, M.Z.; Serrata, M.P.; Goldaracena, I.; Liu, N.; Yin, P.; Paulo, J.A.; Gygi, S.P.; George, S.; et al. Concurrent inhibition of CDK2 adds to the anti-tumour activity of CDK4/6 inhibition in GIST. Br. J. Cancer 2022, 127, 2072–2085. [Google Scholar] [CrossRef]
- Yang, L.; Wang, M.; Li, B.; Xu, S.; Lin, J. Discovery of novel CDK2 inhibitors using multistage virtual screening and in vitro melanoma cell lines. FASEB J. 2023, 37, e22889. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.; Trub, A.; Ahn, A.; Taylor, M.; Ambani, K.; Chan, K.T.; Lu, K.H.; Mahendra, C.A.; Blyth, C.; Coulson, R.; et al. INX-315, a Selective CDK2 Inhibitor, Induces Cell Cycle Arrest and Senescence in Solid Tumors. Cancer Discov. 2024, 14, 446–467. [Google Scholar] [CrossRef]
- Denz, C.R.; Grondine, M.; Fan, J.; Hsu, J.H.-R.; Jackson, A.; Robinson, J.; Guo, G.; Li, W.; Wang, Y.; Martin, M.S.; et al. Abstract ND06: First disclosure of AZD8421, a highly selective CDK2 inhibitor to address resistance to CDK4/6 inhibitors in breast and CCNE1-high cancers. Cancer Res. 2024, 84, ND06. [Google Scholar] [CrossRef]
- Juric, D.; Patel, M.R.; Duska, L.R.; Jhaveri, K.L.; Henick, B.S.; Matulonis, U.A.; Munster, P.N.; Birrer, M.J.; Moore, K.N.; Curigliano, G.; et al. BLU-222, an investigational, oral, potent, and highly selective CDK2 inhibitor (CDK2i), as monotherapy in patients (pts) with advanced solid tumors and in combination with ribociclib (RIBO) and fulvestrant (FUL) in HR+/HER2− breast cancer (BC). J. Clin. Oncol. 2024, 42, 1056. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Saleh, N.; Shattuck-Brandt, R.; Riemenschneider, K.; Slesur, L.; Chen, S.C.; Johnson, C.A.; Yang, J.; Blevins, A.; Yan, C.; et al. MDM2 antagonists overcome intrinsic resistance to CDK4/6 inhibition by inducing p21. Sci. Transl. Med. 2019, 11, eaav7171. [Google Scholar] [CrossRef] [PubMed]
- Vilgelm, A.E.; Pawlikowski, J.S.; Liu, Y.; Hawkins, O.E.; Davis, T.A.; Smith, J.; Weller, K.P.; Horton, L.W.; McClain, C.M.; Ayers, G.D.; et al. Mdm2 and aurora kinase a inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer Res. 2015, 75, 181–193. [Google Scholar] [PubMed]
- Aggarwal, P.; Vaites, L.P.; Kim, J.K.; Mellert, H.; Gurung, B.; Nakagawa, H.; Herlyn, M.; Hua, X.; Rustgi, A.K.; McMahon, S.B.; et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell 2010, 18, 329–340. [Google Scholar] [PubMed]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar]
- Romero-Pozuelo, J.; Figlia, G.; Kaya, O.; Martin-Villalba, A.; Teleman, A.A. Cdk4 and Cdk6 Couple the Cell-Cycle Machinery to Cell Growth via mTORC1. Cell Rep. 2020, 31, 107504. [Google Scholar]
- Rodriguez, M.J.; Perrone, M.C.; Riggio, M.; Palafox, M.; Salinas, V.; Elia, A.; Salgueiro, N.D.; Werbach, A.E.; Marks, M.P.; Kauffman, M.A.; et al. Targeting mTOR to overcome resistance to hormone and CDK4/6 inhibitors in ER-positive breast cancer models. Sci. Rep. 2023, 13, 2710. [Google Scholar]
- Cayo, A.; Venturini, W.; Rebolledo-Mira, D.; Moore-Carrasco, R.; Herrada, A.A.; Nova-Lamperti, E.; Valenzuela, C.; Brown, N.E. Palbociclib-Induced Cellular Senescence Is Modulated by the mTOR Complex 1 and Autophagy. Int. J. Mol. Sci. 2023, 24, 9284. [Google Scholar] [CrossRef]
- Liu, X.; Li, W.; Yi, L.; Wang, J.; Liu, W.; Cheng, H.; Ren, S. CDK4/6 inhibitors dephosphorylate RNF26 to stabilize TSC1 and increase the sensitivity of ccRCC to mTOR inhibitors. Br. J. Cancer 2024, 131, 444–456. [Google Scholar]
- Michaloglou, C.; Crafter, C.; Siersbaek, R.; Delpuech, O.; Curwen, J.O.; Carnevalli, L.S.; Staniszewska, A.D.; Polanska, U.M.; Cheraghchi-Bashi, A.; Lawson, M.; et al. Combined Inhibition of mTOR and CDK4/6 Is Required for Optimal Blockade of E2F Function and Long-term Growth Inhibition in Estrogen Receptor-positive Breast Cancer. Mol. Cancer Ther. 2018, 17, 908–920. [Google Scholar]
- Yang, Z.; Feng, J.; Jing, J.; Huang, Y.; Ye, W.W.; Lei, L.; Wang, X.J.; Cao, W.M. Resistance to anti-HER2 therapy associated with the TSC2 nonsynonymous variant c.4349 C > G (p.Pro1450Arg) is reversed by CDK4/6 inhibitor in HER2-positive breast cancer. NPJ Breast Cancer 2023, 9, 36. [Google Scholar] [PubMed]
- Goel, S.; Wang, Q.; Watt, A.C.; Tolaney, S.M.; Dillon, D.A.; Li, W.; Ramm, S.; Palmer, A.C.; Yuzugullu, H.; Varadan, V.; et al. Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer Cell 2016, 29, 255–269. [Google Scholar] [PubMed]
- Teh, J.L.F.; Cheng, P.F.; Purwin, T.J.; Nikbakht, N.; Patel, P.; Chervoneva, I.; Ertel, A.; Fortina, P.M.; Kleiber, I.; HooKim, K.; et al. In Vivo E2F Reporting Reveals Efficacious Schedules of MEK1/2-CDK4/6 Targeting and mTOR-S6 Resistance Mechanisms. Cancer Discov. 2018, 8, 568–581. [Google Scholar] [PubMed]
- Yoshida, A.; Bu, Y.; Qie, S.; Wrangle, J.; Camp, E.R.; Hazard, E.S.; Hardiman, G.; de Leeuw, R.; Knudsen, K.E.; Diehl, J.A. SLC36A1-mTORC1 signaling drives acquired resistance to CDK4/6 inhibitors. Sci. Adv. 2019, 5, eaax6352. [Google Scholar] [PubMed]
- Romano, G.; Chen, P.L.; Song, P.; McQuade, J.L.; Liang, R.J.; Liu, M.; Roh, W.; Duose, D.Y.; Carapeto, F.C.L.; Li, J.; et al. A Preexisting Rare PIK3CA(E545K) Subpopulation Confers Clinical Resistance to MEK plus CDK4/6 Inhibition in NRAS Melanoma and Is Dependent on S6K1 Signaling. Cancer Discov. 2018, 8, 556–567. [Google Scholar]
- Sullivan, R.J. Dual MAPK/CDK Targeting in Melanoma: New Approaches, New Challenges. Cancer Discov. 2018, 8, 532–533. [Google Scholar]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar]
- Wolchok, J.D.; Chiarion-Sileni, V.; Rutkowski, P.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Queirolo, P.; Dummer, R.; Butler, M.O.; Hill, A.G.; et al. Final, 10-Year Outcomes with Nivolumab plus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2025, 392, 11–22. [Google Scholar]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar]
- Lelliott, E.J.; Kong, I.Y.; Zethoven, M.; Ramsbottom, K.M.; Martelotto, L.G.; Meyran, D.; Zhu, J.J.; Costacurta, M.; Kirby, L.; Sandow, J.J.; et al. CDK4/6 Inhibition Promotes Antitumor Immunity through the Induction of T-cell Memory. Cancer Discov. 2021, 11, 2582–2601. [Google Scholar] [CrossRef] [PubMed]
- Lelliott, E.J.; Mangiola, S.; Ramsbottom, K.M.; Zethoven, M.; Lim, L.; Lau, P.K.H.; Oliver, A.J.; Martelotto, L.G.; Kirby, L.; Martin, C.; et al. Combined BRAF, MEK, and CDK4/6 Inhibition Depletes Intratumoral Immune-Potentiating Myeloid Populations in Melanoma. Cancer Immunol. Res. 2021, 9, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Lelliott, E.J.; Sheppard, K.E.; McArthur, G.A. Harnessing the immunotherapeutic potential of CDK4/6 inhibitors in melanoma: Is timing everything? NPJ Precis. Oncol. 2022, 6, 26. [Google Scholar] [CrossRef]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Wekking, D.; Leoni, V.P.; Lambertini, M.; Dessi, M.; Pretta, A.; Cadoni, A.; Atzori, L.; Scartozzi, M.; Solinas, C. CDK4/6 inhibition in hormone receptor-positive/HER2-negative breast cancer: Biological and clinical aspects. Cytokine Growth Factor. Rev. 2024, 75, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Ding, D.; Yan, Y.; Li, H.; Wang, B.; Ma, L.; Ye, Z.; Ma, T.; Wu, Q.; Rodrigues, D.N.; et al. Phosphorylated RB Promotes Cancer Immunity by Inhibiting NF-kappaB Activation and PD-L1 Expression. Mol. Cell 2019, 73, 22–35.e26. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yan, J.; Guo, Q.; Chi, Z.; Tang, B.; Zheng, B.; Yu, J.; Yin, T.; Cheng, Z.; Wu, X.; et al. Genetic Aberrations in the CDK4 Pathway Are Associated with Innate Resistance to PD-1 Blockade in Chinese Patients with Non-Cutaneous Melanoma. Clin. Cancer Res. 2019, 25, 6511–6523. [Google Scholar] [CrossRef] [PubMed]
- Teh, J.L.F.; Erkes, D.A.; Cheng, P.F.; Tiago, M.; Wilski, N.A.; Field, C.O.; Chervoneva, I.; Levesque, M.P.; Xu, X.; Dummer, R.; et al. Activation of CD8+ T Cells Contributes to Antitumor Effects of CDK4/6 Inhibitors plus MEK Inhibitors. Cancer Immunol. Res. 2020, 8, 1114–1121. [Google Scholar] [CrossRef]
- Lau, P.K.H.; Cullinane, C.; Jackson, S.; Walker, R.; Smith, L.K.; Slater, A.; Kirby, L.; Patel, R.P.; von Scheidt, B.; Slaney, C.Y.; et al. Enhancing Adoptive Cell Transfer with Combination BRAF-MEK and CDK4/6 Inhibitors in Melanoma. Cancers 2021, 13, 6342. [Google Scholar] [CrossRef]
- Randic, T.; Magni, S.; Philippidou, D.; Margue, C.; Grzyb, K.; Preis, J.R.; Wroblewska, J.P.; Nazarov, P.V.; Mittelbronn, M.; Frauenknecht, K.B.M.; et al. Single-cell transcriptomics of NRAS-mutated melanoma transitioning to drug resistance reveals P2RX7 as an indicator of early drug response. Cell Rep. 2023, 42, 112696. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, A.; De Marchi, E.; Ferracin, M.; Orioli, E.; Zanoni, M.; Bassi, C.; Tesei, A.; Capece, M.; Dika, E.; Negrini, M.; et al. P2X7 promotes metastatic spreading and triggers release of miRNA-containing exosomes and microvesicles from melanoma cells. Cell Death Dis. 2021, 12, 1088. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Clinical Trial Identifier | Study Name | Phase/Recruitment Status | Last Update Posted | Therapies |
|---|---|---|---|---|
| CDK4/6 inhibition alone | ||||
| NCT02308020 | A Study of Abemaciclib (LY2835219) in Participants with Breast Cancer, Non-small Cell Lung Cancer, or Melanoma That Has Spread to the Brain | Phase 2; Completed | 19 December 2020 | CDK4/6i (abemaciclib) |
| NCT01037790 | Phase II Trial of the Cyclin-Dependent Kinase Inhibitor PD 0332991 in Patients with Cancer | Phase 2; Completed | 11 March 2021 | CDK4/6i (palbociclib) |
| NCT05063058 | Biomarker-driven Therapy for Melanoma (TREAT20plus) | Phase N/A; Completed | 30 September 2021 | CDK4/6i (palbociclib) |
| NCT01394016 | A Phase 1 Study of LY2835219 In Participants With Advanced Cancer | Phase 1; Completed | 19 September 2024 | CDK4/6i (abemaciclib) |
| NCT03310879 | Study of the CDK4/6 Inhibitor Abemaciclib in Solid Tumors Harboring Genetic Alterations in Genes Encoding D-type Cyclins or Amplification of CDK4 or CDK6 | Recruiting; Phase 2 | 24 January 2025 | CDK4/6i (abemaciclib) |
| NCT02465060 | Targeted Therapy Directed by Genetic Testing in Treating Patients with Advanced Refractory Solid Tumors, Lymphomas, or Multiple Myeloma (The MATCH Screening Trial) | Phase 2; Active, not recruiting | 09 April 2025 | CDK4/6i (palbociclib) |
| CDK4/6 inhibition + MAPK inhibition | ||||
| NCT02065063 | A Study to Investigate the Safety, Pharmacokinetics, Pharmacodynamics, and Anti-Cancer Activity of Trametinib in Combination with Palbociclib in Subjects With Solid Tumors | Phase 1; Completed | 1 June 2018 | CDK4/6i (palbociclib) and MEKi (trametinib) |
| NCT01781572 | A Phase Ib/II Study of LEE011 in Combination With MEK162 in Patients With NRAS Mutant Melanoma | Phases 1 and 2; Completed | 7 December 2020 | CDK4/6i (ribociclib) and MEKi (MEK162) |
| NCT02857270 | A Study of LY3214996 Administered Alone or in Combination with Other Agents in Participants With Advanced/Metastatic Cancer | Phase 1; Completed | 22 November 2022 | CDK4/6i (abemaciclib) and ERK1/2i (temuterkib) |
| NCT02159066 (LOGIC-2) | LGX818 and MEK162 in Combination with a Third Agent (BKM120, LEE011, BGJ398 or INC280) in Advanced BRAF Melanoma (LOGIC-2) | Phase 2; Completed | 5 March 2024 | CDK4/6i (ribociclib), MEKi (binimetinib), and BRAFi (encorafenib) |
| NCT04720768 (CELEBRATE) | Encorafenib, Binimetinib and Palbociclib in BRAF-mutant Metastatic Melanoma CELEBRATE (CELEBRATE) | Recruiting; Phase 1b | 5 January 2024 | CDK4/6i (palbociclib), MEKi (binimetinib), and BRAFi (encorafenib) |
| NCT03454035 | Ulixertinib/Palbociclib in Patients with Advanced Pancreatic and Other Solid Tumors | Recruiting; Phase 1 | 18 March 2025 | CDK4/6i (palbociclib) and ERK1/2i (ulixertinib) |
| NCT02645149 (MatchMel) | Molecular Profiling and Matched Targeted Therapy for Patients with Metastatic Melanoma (MatchMel) | Recruiting; Phase 2 | 07 January 2025 | CDK4/6i (ribociclib) and MEKi (trametinib) |
| NCT04594005 | CDK4/6 Tumor, Abemaciclib, Paclitaxel | Phases 1 and 2; Active, not recruiting | 18 December 2024 | CDK4/6i (abemaciclib) and paclitaxel |
| NCT01543698 | A Phase Ib/II Study of LGX818 in Combination With MEK162 in Adult Patients With BRAF Dependent Advanced Solid Tumors | Phases 1 and 2; Completed | 13 March 2024 | CDK4/6i (ribociclib), MEKi (MEK162), and BRAFi (encorafenib) |
| NCT04417621 | Study of Efficacy and Safety of LXH254 Combinations in Patients with Previously Treated Unresectable or Metastatic Melanoma | Phase 2; Active, not recruiting | 04 April 2025 | CDK4/6i (ribociclib) and RAFi (LXH254) |
| NCT02974725 | A Phase Ib Study of LXH254-centric Combinations in NSCLC or Melanoma | Phase 1; Terminated | 17 May 2024 | CDK4/6i (ribociclib) and RAFi (LXH254) |
| CDK4/6 inhibition + immunotherapy | ||||
| NCT03484923 (PLATforM) | Study of Efficacy and Safety of Novel Spartalizumab Combinations in Patients with Previously Treated Unresectable or Metastatic Melanoma (PLATforM) | Phase 2; Completed | 18 June 2024 | CDK4/6i (ribociclib) and PD-1 receptor antibody (Spartalizumab) |
| NCT02791334 (PACT) | A Study of Anti-PD-L1 Checkpoint Antibody (LY3300054) Alone and in Combination in Participants with Advanced Refractory Solid Tumors (PACT) | Phase 1; Completed | 27 September 2024 | CDK4/6i (abemaciclib) and PD-L1 antibody (LY3300054) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, C.; Nguyen, T.T.T.; Poulikakos, P.I.; Polsky, D. Recent Developments in Targeting the Cell Cycle in Melanoma. Cancers 2025, 17, 1291. https://doi.org/10.3390/cancers17081291
Hung C, Nguyen TTT, Poulikakos PI, Polsky D. Recent Developments in Targeting the Cell Cycle in Melanoma. Cancers. 2025; 17(8):1291. https://doi.org/10.3390/cancers17081291
Chicago/Turabian StyleHung, Christie, Trang T. T. Nguyen, Poulikos I. Poulikakos, and David Polsky. 2025. "Recent Developments in Targeting the Cell Cycle in Melanoma" Cancers 17, no. 8: 1291. https://doi.org/10.3390/cancers17081291
APA StyleHung, C., Nguyen, T. T. T., Poulikakos, P. I., & Polsky, D. (2025). Recent Developments in Targeting the Cell Cycle in Melanoma. Cancers, 17(8), 1291. https://doi.org/10.3390/cancers17081291