1. Introduction
Cancer research over five decades has resulted in the generation of enormous amounts of information and drugs for targeted cancer therapy. Advanced and innovative cancer therapies are the following: stem cell therapy, pluripotent stem cells, and adult stem cells; immunotherapy including monoclonal antibodies, checkpoint inhibitors, CAR-T cell treatment, adoptive cell transfer, and cancer vaccines (therapeutic vaccinations and preventative vaccines); gene therapy including tailored small interfering RNAs (siRNAs) for gene silencing of antiapoptotic proteins, transcription factors, or cancer-related mutated genes and gene editing (CRISPR-Cas9); hormonal therapy; oncolytic virus therapy; molecular targeted therapy for disrupting the growth molecules responsible for inhibiting the development and metastasis of cancer; hyperthermia; and photodynamic therapy and additional treatments [
1,
2,
3,
4,
5]. However, relatively little progress has been made in the treatment of patients with solid tumors, other than extending their survival for a few months at best. Moreover, after half a century of extensive research and numerous clinical trials, even with novel designed targeted molecules, cancer remains the second-leading cause of death in the United States and, by inference, in other developed countries [
1]. Most newly developed anticancer treatments trigger tumor cell death through apoptosis, where involvement of caspases’ activity is essential. However, numerous reviews have reported mutations in apoptosis pathways that lead to cancer and favor cell resistance to apoptosis; most of these are related to caspase-dependent apoptosis pathways [
2,
3]. These include mutations involved in upstream caspase activation that leads to cancer, such as mutations involving the death receptors (DRs) [
4], various caspases [
5,
6,
7], and nucleases [
8]. Mutations or dysregulated expression of proteins of the Bcl-2 family proteins have also been widely identified in numerous cancers as well [
9,
10]. Indeed, tumor cells are often resistant to classical caspase-mediated pathways because of excessive expression of anti-apoptotic proteins, such as Bcl-2. Due to these limitations, it is becoming essential to identify and design novel, effective targeted anti-tumor agents that induce caspase-independent programmed cell death. In this study, we aimed to design a new molecule that will induce caspase-independent cell death for the treatment of solid tumors in the form of a chimeric protein.
Chimeric proteins, designed and constructed by gene fusion techniques, comprise both the cell targeting and the active moieties and belong to the “toxin delivery molecules” category of targeted cancer therapies. The first generation of these molecules, termed immunotoxins, consisted of bacterial or plant toxins as the active moiety of the chimeras that were conjugated or fused to an antibody [
11]. An example of an immunotoxin with FDA approval is moxetumomab pasudotox-tdfk (Lumoxiti™), an anti CD22 Pseudomonas exotoxin A (PE) recombinant immunotoxin, for the treatment of hairy cell leukemia [
12]. In this regard, a chimeric protein for the treatment of solid cancer cells was previously constructed by us with an analog of the decapeptide gonadotropin-releasing hormone (GnRH) serving as the targeting domain, fused to the full-length modified bacterial PE. GnRH-PE chimeric proteins had the ability to bind, internalize, and kill a large number of human-originated cancer cell lines as well as having an anti-tumor effect in vivo in a human colon xenograft mouse model [
13].
Our targeting moiety in these chimeric proteins was GnRH, which is normally synthetized by the hypothalamic neurons and secreted in a pulsatile manner [
14]. Upon reaching the anterior pituitary gland, GnRH binds to specific receptors and activates the GnRH receptor (GnRH-R). The result of its binding is the selective stimulation of the gonadotrophin cells to release luteinizing hormone and follicle-stimulating hormone, thus playing a central role in the control of human reproduction [
14]. GnRH-R belongs to the 7-transmembrane G protein coupled receptor family [
14]. There are two types of GnRH-R, type 1 and type 2. Type 1 is involved almost exclusively in the reproductive system whereas type 2 is expressed throughout the brain and not confined to the pituitary [
14]. However, in humans, the N-terminus of the predicted protein GnRHR2 contains a frameshift and premature stop codon. In humans, GnRHR2 transcription occurs, but whether the gene produces a functional protein is currently unresolved [
14]. It has become increasingly clear that GnRH-R1s are overexpressed in cancer tissues, either related (prostate, breast, endometrial, and ovarian cancers) or unrelated (colon, melanoma, glioblastoma, lung, pancreatic cancers, and more) to the reproductive system [
15]. We were the first to report that a wide variety of solid tumors, not connected to the reproduction system, i.e., non-hormonal originated cancers, overexpress the specific GnRH-R [
16]. In the last few years, the use of GnRH analogs has been proposed for treatment of various hormone-dependent cancers, via inhibition of estrogen and androgen synthesis, and today are used as an androgen-deprivation treatment of prostate cancer [
17].
The overexpression pattern of GnRH-R makes it an attractive target to deliver therapeutic agents exclusively to tumor cells. Although GnRH-R is expressed in the pituitary gland, it has been reported that GnRH-targeted proteins were not able to escape through the barriers that separate the pituitary gland from the systemic circulation so had no effect on normal pituitary function [
18]. Therefore, the GnRH-GnRH-R system can be considered as an efficient system to target a variety of solid cancer cells. Indeed, in recent years we have developed new chimeric proteins introducing human apoptosis-inducing proteins of the Bcl2 family as well as a specific apoptotic nuclease, instead of the bacterial toxins, to reduce immunogenicity and off-target problems. These chimeric proteins, GnRH-Bik, GnRH-Bak, GnRH-Bax, and GnRH-DFF40, efficiently and specifically inhibited the cell growth of human cancer cell lines [
19]. Recently, similar chimeric proteins were also developed by others with significant positive results [
20]. However, all these chimeric proteins induce the caspase-dependent apoptotic death of cancer cells both in vitro and in vivo in cancer models in mice. In the current study, we developed and tested new GnRH-based chimeric proteins that induce caspase-independent apoptotic death for treatment of solid cancers.
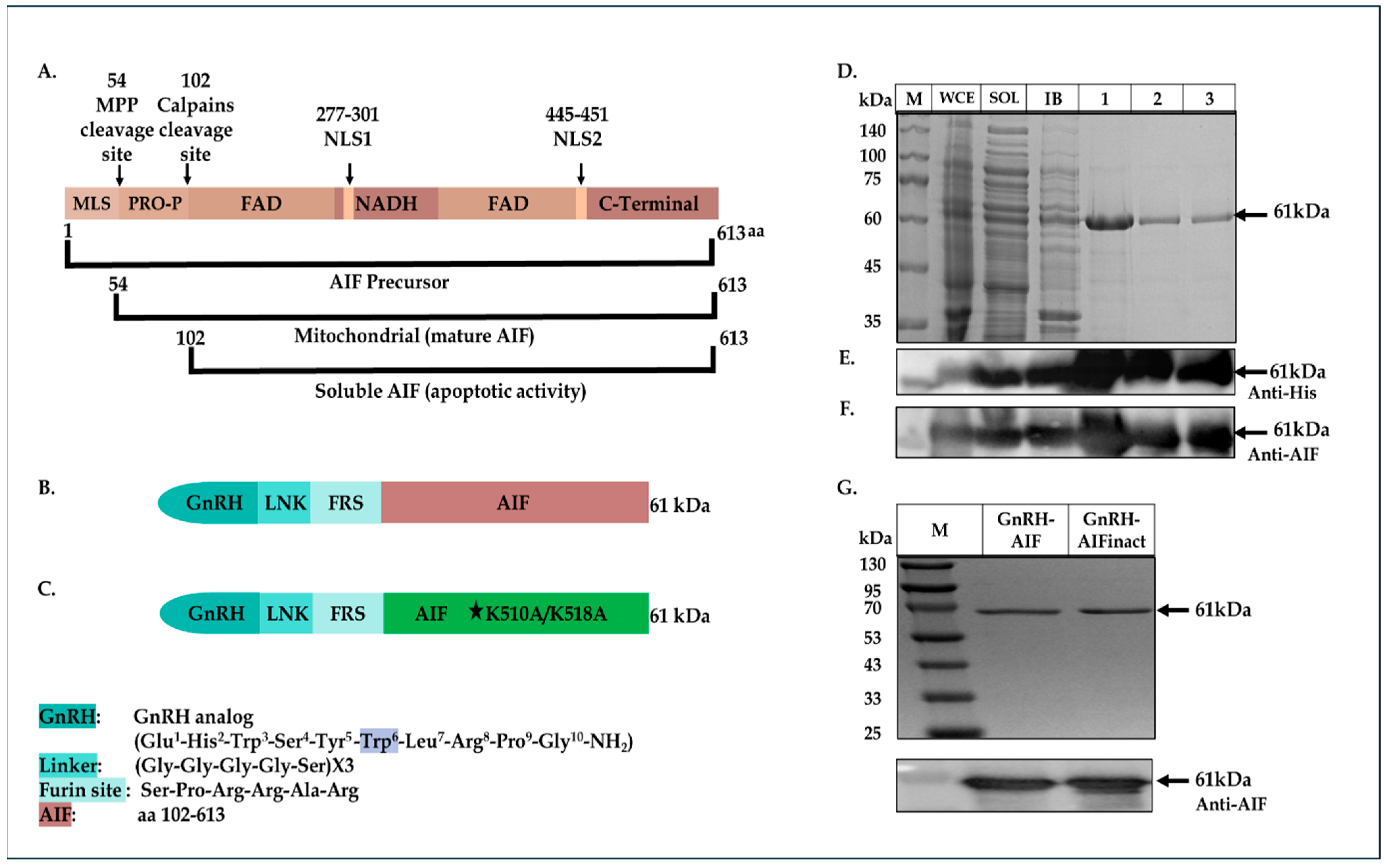
Searching for human proteins known to induce caspase-independent apoptotic death, we examined the apoptosis-inducing factor (AIF) protein, as a new “killing moiety” for our targeted chimeric proteins. AIF is a flavoprotein that is synthesized in the cytosol and imported into the mitochondrial Inner Membrane (IMS). The AIF precursor carries a mitochondrial localization sequence (MLS) in its N-terminal pre-sequence. Under normal conditions, the 54-residue MLS is first proteolytically removed after translocation into the mitochondrial intermembrane space to form the mature form (AIF-mit) where it has a NADH-dependent oxidoreductase activity [
21,
22]. During apoptosis, it is further proteolytically processed at amino-acid position 101, leading to the generation of the soluble form (AIF-sol). AIF-sol is released to the cytoplasm in response to specific death signals, and translocated to the nucleus, where it induces DNA fragmentation leading to apoptosis [
21]. AIF has a crucial role in the caspase-independent apoptotic pathway [
21] and as such was chosen as our killing moiety for the chimeric proteins, termed GnRH-AIF.
We constructed, expressed, and highly purified GnRH-AIF chimeric proteins. We demonstrated the ability of the chimera to enter and specifically and very efficiently kill human solid cancer cells overexpressing GnRH-R. The GnRH-AIF chimeric proteins enter target cells by binding to the GnRH-receptor, which is overexpressed on the cell surface of solid tumors cells and undergoes internalization following this binding (receptor-mediated internalization). Most importantly, death induced by GnRH-AIF chimeric protein is caspase-independent. Finally, we demonstrated the high anti-tumor efficacity of the chimera, in a colon cancer organoid model. Our study shows the potential of a GnRH-AIF chimeric protein as a novel approach to treat solid cancers that overexpress GnRH-R, via a caspase-independent apoptotic pathway.
2. Materials and Methods
2.1. Construction of the Plasmids Encoding GnRH-AIF Chimeric Proteins
A plasmid encoding the CD40-Smurf2 sequence [
23,
24] was cut with a BmtI and AgeI restriction enzyme removing the CD40 coding sequence and resulting in the vector fragment. The insert consists of the GnRH coding sequence, with tryptophan replacing glycine as the sixth amino acid, flanked by BmtI and AgeI (3′ end) restriction sites. GnRH-Smurf2 sequence was cut with EcoRI and Sac1 restriction enzymes, removing the Smurf2 coding sequence and resulting in the vector fragment. The human AIF protein sequence (amino acids 101-649) was generated and amplified by PCR using an AIFM1 clone as a template (clone number: BC139738, GE Healthcare Dharmacon Inc., Lafayette, CO, USA) with the following synthetic oligonucleotide primers:
5′-GAGGAATTCTCTCCACGGCGGGCACG TGGGCTGACACCAGAACAGAAAC-3′ (sense) and 5′-CACGAGCTCTCAGTCTTCATGAAT GTTGAATAGTTTGGC-3′ (antisense). The product fragment (containing also a Furin site before the AIF sequence, see
Figure 1B below), cut with EcoRI and Sac1 restriction enzymes, was ligated into the vector fragment using Quick Ligation Kit (New England Biolabs, Ipswich, MA, USA), 3′ to the GnRH coding sequence, thus producing the plasmid encoding the GnRH-AIF chimeric protein.
As a negative control, a plasmid containing a known deactivating mutation (alanine substitutions of lysine at the positions 510/518) [
25] in the DNA binding site of AIF was generated using PCR steps and the Gibson Assembly protocol (#E2611S, New England Biolabs). In addition, GnRH-AIFct, which encodes amino acids 353-613 of AIF, corresponding to its C-terminal domain and GnRH-AIFnt corresponding to the N-terminal region of AIF (amino acids 101–352), were generated using the same protocol. For the construction of the GnRH-AIF variants, two PCR reactions were conducted using the primers detailed in
Table 1. The amplified products were verified using 1% agarose gel. The DpnI enzyme was added to the two products, and both products were used for the Gibson reaction. The coding sequences of the chimeric proteins GnRH-AIFinact, GnRH-AIFct, or GnRH-AIFnt were confirmed by sequencing analysis.
2.2. Construction of the Plasmids Encoding GnRH-Caspase-3 Chimeric Proteins
For comparison studies, we designed GnRH-Caspase-3 chimeric proteins with caspase 3, a pro-apoptotic protein of human origin, as an active moiety. This protein causes cell death via caspase-dependent apoptosis pathways.
The Smurf2 coding sequence was removed from the plasmid encoding the GnRH-Smurf2 sequence, as described above. The caspase 3 sequence was generated and amplified by PCR using the plasmid encoding the IL2-Caspase-3 [
26] (lacking the pre-sequence) with the following synthetic oligonucleotide primers covering the whole coding sequence: 5′ CGGAATTCAACAGCTATAAAATGGATTA 3′ (sense) and 5′CCGCTCGAGTTAGTGGTA GAAGTACAGTTCTTTGG 3′ (antisense). The caspase 3 fragment, cut with EcoRI and Xho1 restriction enzymes, was ligated into the vector fragment containing the GnRH coding sequence using Quick Ligation Kit, thus producing the plasmid encoding the GnRH-Caspase-3 chimeric protein. The coding sequences of the chimeric proteins GnRH-Caspas-3 was confirmed by sequencing analysis.
2.3. Expression of GnRH-AIF and GnRH-Caspase-3 Chimeric Proteins
Following plasmid transformation into Rosetta strain Escherichia coli, the bacterial cultures were grown at 37 °C until an OD600 of 0.7–0.8. Expression was induced by adding 2 mM isopropyl-1-thio-D-galactopyranoside (IPTG, #16242352, New England Biolabs, Ipswich, MA, USA), and growth continued for additional 3 h. The cells were then centrifuged for 20 min at 4000× g, and the pellet was stored at −80 °C for 30 min. The frozen cells were thawed and suspended in lysis buffer (20 mM Tris-HCl, pH 8.0; 0.05 M NaCl; 0.2 mg/mL lysozyme; and 1 mM phenylsulfonyl fluoride) for 30 min at room temperature, followed by sonication. A sample was taken from the lysed cells and marked as whole cell extract (WCE). The rest of the cells were then centrifuged at 13,000× g for 30 min. The supernatant (marked as soluble fraction; SOL) was removed, and the pellet (containing inclusion bodies; IB) was suspended in denaturation buffer (6 M urea, 20 mM Tris-HCl, pH 8.8, 0.05 M NaCl, and 1 mM phenylsulfonyl fluoride) and continuously stirred for 90 min. The solution was cleared by centrifugation at 13,000× g for 60 min. The supernatant, containing the denatured proteins, was collected and marked as IB.
2.4. Purification of GnRH-AIF and GnRH-Caspase-3 Chimeric Proteins
The denatured chimeric proteins in 6 M urea were filtered, and a final concentration of 10 mM imidazole was added. The protein solution was loaded onto His-Trap columns (GE Healthcare, Sweden), which was pre-washed in the loading buffer (6 M urea, 20 mM Tris-HCl, pH 8.8, 0.05M NaCl, and 10 mM imidazole). To elute the desired protein, the column was washed with an increasing gradient of imidazole until a maximum concentration of 500 mM. Fractions containing the chimeric protein were pooled and slowly diluted 1:80 (vol/vol) in refolding buffer (20 mM Tris-HCl, pH 8.8, 2 mM β-mercaptoethanol (βME), 1 mM EDTA, 0.05 M NaCl, and 1 mM DTT) and then incubated at 4 °C for 72 h. The refolded chimeric protein solution was then subjected to ion exchange chromatography using a Q-Sepharose column (GE Healthcare, Upsala, Sweden) and using a SP-Sepharose column for GnRH-AIFnt (the chimeric protein is diluted in refolding buffer pH 6.5). Protein was eluted with linear gradient of 0.05–1 M NaCl in refolding buffer. Fractions containing the chimeric protein were pooled, and the protein preparation was dialyzed against phosphate buffered saline (PBS), aliquoted, and kept at −20 °C. All purification procedures were conducted using the FPLC system ÄKTA (GE Healthcare, Sweden).
2.5. Characterization of the GnRH-AIF Chimeric Proteins
2.5.1. Protein Concentration
Protein concentration was measured according to the Bradford method, using a Bradford reagent (#5000006, Bio-Rad Laboratories, Inc., Berkeley, CA, USA) and a standard curve of BSA. Protein concentration was determined at a wavelength of 595 nm.
2.5.2. Separation of Chimeric Proteins by Gel Electrophoresis
Samples from the various subcellular fractions (WCE, SOL, and IB) were separated on 12% SDS-PAGE gels and stained with Coomassie blue for visualization of the proteins.
2.5.3. Western Blot Analyses
Protein samples were separated on SDS-PAGE gels and then electro-transferred onto a polyvinylidene fluoride (PVDF) membrane (Bio-Rad). The membrane was then blotted with one of the following antibodies: rabbit anti-AIFM1 (1:1000; ab32516, Abcam, Cambridge, UK); mouse anti-His (1:10,000; #27-4710-01, Amersham Pharmacia Biotech, Uppsala, Sweden); mouse anti-tubulin (1:10,000; ab7291, Abcam); mouse anti-lamin (1:1000, ab8983, Abcam); or rabbit anti-SF2 (1:1000, ab133689 Abcam). The secondary antibodies used were goat anti-rabbit or goat anti-mouse HRSP-conjugated (1:10,000, Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA). Band visualization was performed using the EZ-ECL chemiluminescence detection kit (Biological Industries, Beit HaEmek, Israel).
2.6. Cells Lines
All cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). MCF-7 breast adenocarcinoma cells (ATCC-HTB-22), LNCaP prostate adenocarcinoma cells (ATCC-CRL-1740), Colo205 (ATCC-CCL-22), Colo320 (ATCC-CCL-220), SW48 (ATCC-CCL-231), and HCT115 (ATCC-CCL-225) colon adenocarcinoma cells were grown in RPMI 1640 medium (L0501, Biowest, Nuaille, France) supplemented with 10% fetal bovine serum (FBS; Merck, Darmstadt, Germany), 2 mM L-glutamine (Sartorius, Göttingen, Germany), and 100 units/mL penicillin (penicillin-streptomycin solution 100×; Biowest). A204 rhabdomyosarcoma cells (ATCC-HTB-82), T24P bladder urinary transitional cell carcinoma (ATCC-HTB-4), normal fibroblasts, (ATCC-PCS-420-013), lung A549 cells (ATCC-CCL-185), and HEK-293 renal adenocarcinoma cells (ATCC-CRL-1573) were grown in DMEM medium (01-055-1A, Sartorius) supplemented with 10% FBS, 2 mM L-glutamine, and 100 units/mL penicillin. All cells were maintained in flasks and grown in a highly humidified atmosphere of 5% CO
2 at 37 °C. Expression levels of GnRH-R on the various cell lines are summarized in
Table S1.
2.7. Cell Viability Assay
A total of 0.5 × 104 cells/100 µL/well were seeded in 96-well plates, treated with the various GnRH-AIF chimeric proteins, GnRH-Caspase-3 chimeric proteins (positive control), or PBS, and incubated for 24/48/72 h. Cell viability was determined using the Cell Titer-Blue kit (G8080, Promega, Madison, WI, USA) according to manufacturer’s instructions.
In addition, Colo205 and SW48 cells treated as described above were incubated in the IncuCyte® S3 Live-Cell Analysis System and tracked for up to 72 h. Images of each well were obtained every 2 h. The data were analyzed using IncuCyte® S3 Software 2019A (Sartorius).
2.8. Confocal Microscopy
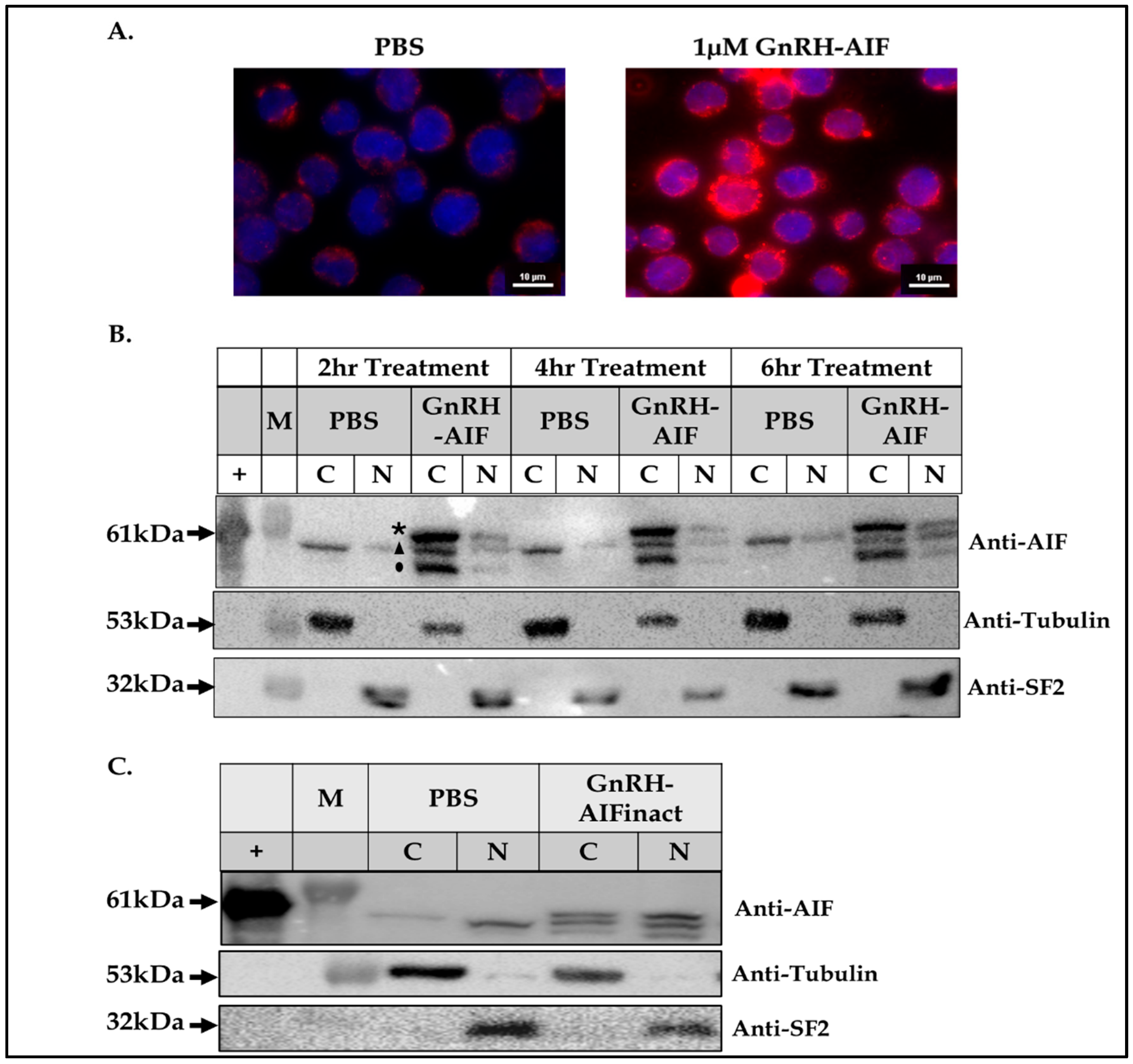
A total of 5 × 105 Colo205 cells were seeded in 3 cm plates and treated with 1 µM GnRH-AIF or PBS and incubated for 24 h. Treated cells were washed with cold PBS and then fixated with 1 mL of 4% paraformaldehyde for 10 min at room temperature. Next, cells were treated with a permeabilization solution (0.1% TritonX100 in PBS) for 10 min. To block unwanted nonspecific interactions, the cells were treated with 1% BSA and 0.3 M glycine in PBST (PBS + 0.1% Tween) for 30 min. GnRH-AIF chimeric proteins were stained with rabbit anti-AIFM1 antibodies overnight (1:500) and visualized with secondary 555-anti Rabbit antibody (1:200, AB_3095431 Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA). The cells were incubated with 1:1000 DAPI (D9542, Sigma-Aldrich, St. Louis, MO, USA) for 10 min, and then the slides were covered with mounting buffer overnight and analyzed using confocal microscope (Nikon Eclipse NI DS-Qi2, 60X, Nikon Instruments Inc., New York, NY, USA).
2.9. Internalization of GnRH-AIF Chimeric Proteins Analyzed by Western Blot
A total of 2 × 106 Colo205 cells were seeded in 6-well plates and treated with chimeric proteins or PBS for 24 h. Internalization of the chimeric proteins into target cells was examined by Western blot analysis of treated cell extracts using anti-AIFM1 antibodies (see above).
2.10. Subcellular Fractionation of Target Cells
A total of 3 × 10
6 LNCaP cells were seeded in 10 cm plates and treated with the chimeric proteins or PBS and incubated for 24 h. Subcellular fractionation to cytosolic and nuclear fractions was performed using the RIME protocol [
27]. Protein concentration was measured according to the Bradford method to ensure equal loading of nuclear and cytoplasmatic fractions.
2.11. Cell Cycle Analysis by Propidium Iodide Staining
A total of 2 × 106 Colo205 cells were seeded in 3 cm plates and treated with 1 µM GnRH-AIF or PBS and incubated for 24 h. The cells were collected, counted, and washed twice with PBS and then treated with Nicoletti buffer (50 µg/mL propidium iodide, 0.1% sodium citrate, 0.1% TritonX100), for 1 h at 4 °C in the dark. Cell cycle analysis of the cells was performed using flow cytometer analyzers (C6 Plus Flow Cytometer, Biosciences, Denver, CO, USA) and analyzed using the FCS Express7 program.
2.12. Annexin V Assay
A total of 2 × 106 Colo205 cells were seeded in 6-well plates treated with 1 µM GnRH-AIF or PBS and incubated overnight. Cells were harvested, washed with PBS, and resuspended in an Annexin-binding buffer at a concentration of 1 × 106 cells/mL. The samples were stained with 5 µL of Annexin V and 5 µL of 7AAD and then incubated for 15 min at RT (#62700-80, Annexin V APC Kit, Biogems, Westlake Village, CA, USA). Following incubation, cells were diluted with 400 mL of Annexin-binding buffer and analyzed by flow cytometry immediately. Unstained and single-stained cells were used for compensation and statistics. Cells stained with Annexin represent early apoptotic cells. Double-stained cells represent late apoptotic cells, and cells stained with 7AAD represent necrotic cells. Samples were analyzed with a LSR II flow cytometer (Biosciences, Denver, CO, USA).
2.13. Real-Time PCR Analysis of Apoptosis-Related mRNAs
A total of 1.0 × 10
6 Colo205 cells were seeded in 3 cm plates treated with GnRH-AIF (1 µM) or PBS and incubated for 48 h. Total RNA was extracted from cells using a commercial kit (#GZXD200, GENEzol™ TriRNA Pure Kit, Geneaid, New Taipei City, Taiwan). RNA concentrations were determined using a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA, USA). One µg of each RNA sample was reverse transcribed using a reverse transcription kit (qScript cDNA Synthesis Kit, Quantabio, Beverly, MA, USA, Cat# 95047-100-2). The resulting cDNA was diluted in DNase-free water (1:10) before quantification by real-time quantitative PCR. Individual mRNA levels were quantified using real-time PCR. Each sample contained 2 µL of cDNA, 1 µL of primers, 5 µL of SYBR
® Green, and 3 µL of H
2O in a total volume of 10 µL per sample. The primers used are listed in
Table 2. The data were analyzed using the primer express program (Applied Biosystems, Foster City, CA, USA). All data are expressed as the ratio between the expression level of the target gene mRNA and that of actin.
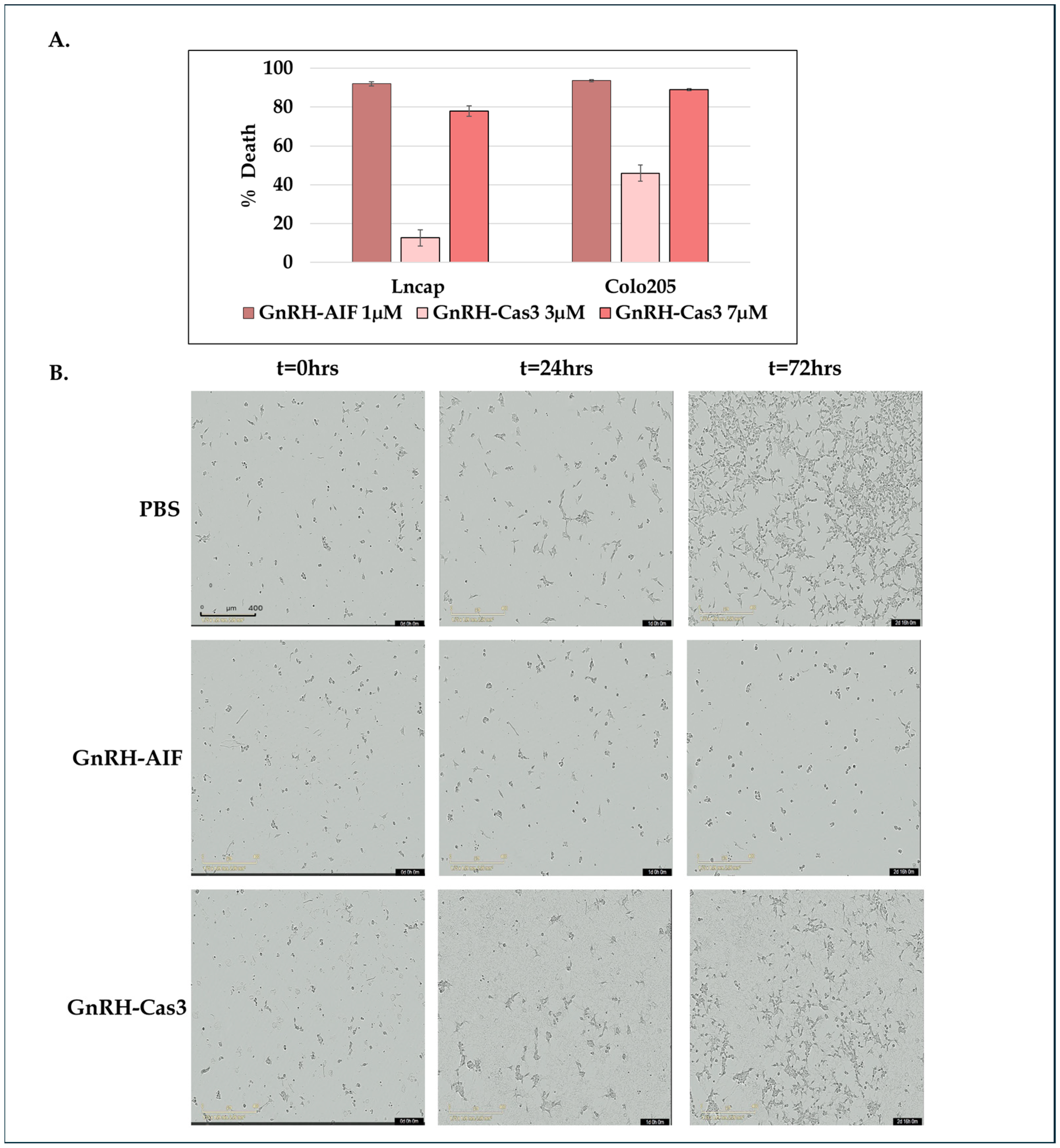
2.14. In Vitro Caspase 3 Activity Assay
A total of 0.5 × 104 cells/100 µL/well of Colo205 were seeded in 96-well plates and cultured in medium with or without 20 µM zVAD-FMK (sc-3067, Santa Cruz Biotechnology Inc., Dallas, TX, USA) for 5 h and then were treated with GnRH-Caspase-3 (7 µM) and GnRH-AIF (1 µM) and incubated for 48 h. Caspase 3 activity within the cells was assessed by a Apo-ONE® Homogeneous Caspase 3/7 Assay Kit (Promega), and caspase 3 activity was analyzed using a FLU Ostar plate reader at the following wavelengths: excitation/emission—485/520 nm. Results are shown as fold increases in caspase 3 activity in the treated cells compared to control cells and were normalized to number of cells.
2.15. Gel Retardation Assay
DNA-binding activity of GnRH-AIF chimeric proteins was determined by gel retardation assays. A total of 2.5 μg of 1 kb DNA ladder (DM015-R500, Hylabs, Rehovot, Israel) was incubated for 30 min at room temperature with GnRH-AIF proteins and then analyzed by agarose gel electrophoresis (1%) in the presence of ethidium bromide to visualize the DNA.
2.16. Nuclease Activity Assays
Assays were performed by mixing 250 ng of a double-stranded, supercoiled pET-28 plasmid or 500 ng of genomic DNA as substrate with 12 μg of GnRH-AIF chimeric proteins, GnRH-AIFinact, and GnRH-Caspase-3 (or 1 IU of DNase for 1 min and 5 min) in 20 mM Tris, having a pH 8.0 and a final volume of 10 μL, with 0.1 mM CaCl2 and 1 mM MgCl2. The samples were incubated for 20 min at 37 °C, after which they were mixed with 6× DNA loading dye (Thermo Scientific). Samples were then loaded onto 0.8% agarose gel with ethidium bromide and run for 1 h at 100 V.
2.17. DNA Laddering
A total of 1.5 × 106 Colo205 cells were seeded in 1 mL of medium and treated with 1 µM GnRH-AIF or PBS for 24 h. The cells were then collected and washed twice with PBS, and DNA was extracted using the GenElute Blood Genomic DNA Kit (NA2010-1KT, Sigma-Aldrich). A total of 10 µg of DNA of each sample was loaded on a 1.5% agarose gel.
2.18. Immunoprecipitation and Co-IP Assay
AIF was immunoprecipitated using nuclear fractions of treated and control Colo205 cell extract, 1 µg of rabbit monoclonal anti-AIF antibody (ab32516, Abcam, Cambridge, UK), and 30 µL of Protein A/G beads (sc-2003, Santa Cruz Biotechnology, Dallas, TX, USA) The antibodies were incubated under agitation with Protein A/G beads for 4 h, and then the nuclear fraction was added to each sample and incubated overnight under agitation at 4 °C. The beads were washed with PBS to remove non-specific binding. Proteins were eluted by the addition of 40 µL SDS loading buffer (X2) and incubated for 10 min at 95 °C; 20 µL of each sample was separated on a SDS-PAGE gel, transferred to a nitrocellulose membrane, and probed with anti-ENDOG (#22148-1-AP, Proteintech Group, Inc., Munich, Germany), rabbit anti-AIFM1 (1:1000; ab32516, Abcam), or anti-lamin (1:1000, ab8983, Abcam).
2.19. Knockdown of Endonuclease G and Cyclophilin A (PPIA) by siRNA
esiRNA (# EHU-904001, Sigma-Aldrich) targeting human ENDOG gene transcripts was used for the knockdown of ENDOG. esiRNA (#EHU107101, Sigma-Aldrich) targeting human PPIA gene transcripts was used to knockdown PPIA. LNCaP cells were transfected with 240 µL of reduced serum medium (# 31-985-070, Opti-MEM™, Thermo Fisher Scientific), 3 µL of TransIT-X2 (#MC-MIR-6000, Mirus, Marietta, GA, USA), and 3 µL of a 14 µM siRNA stock solution (20 nM final concentration per well), which were mixed and incubated at room temperature for 30 min. In a 6-well plate, 245 µL of the mix were added in each well, and then 2 × 10
5 cells/1.5 mL were seeded in RPMI medium supplemented with 10% serum (without antibiotics). A total of 24 h later, the media was replaced with a complete growth medium, and the cells were seeded in 96-well plates for 4 h prior to treatment. Wild type and cells after knocking down were treated with various concentrations of GnRH-AIF or GnRH-Caspase-3 and incubated for a further 24 h. Cell viability was determined using the Cell Titer-Blue kit (G8080, Promega) according to manufacturer’s instructions. Validation of the knockdown was confirmed using primers of these genes (see
Table 2) and real-time PCR (see above), as well as at the protein level (
Figure S2) by Western blot analysis using anti-ENDOG antibodies (see above).
2.20. Organoid Culture
Colorectal cancer (CRC) as well as healthy, adjacent colon tissues were obtained from a colorectal carcinoma patient (male, aged 85) and a gastric carcinoma patient (female, aged 15) following surgery under the Helsinki authorization #HMO-0921-20. Biopsies were minced into 2–3 mm fragments and processed within 90 min in digestion medium (CRC—Advanced DMEM/F-12 supplemented (#12634010, Gibco™ Thermo Fisher Scientific Inc., USA) with 1% Anti-Anti, 1% HEPES (1 M solution), 1% GlutaMAX, 1 mg/mL collagenase type II, 10 µg/mL hyaluronidase, and 10.5 µM Y-27632 for 60 min at 37 °C with gentle agitation; Healthy colon—10 mL 0.01 M EDTA in PBS for 90 min at 4 °C and are then mechanically tritured to extract intestinal crypts). Digestion is stopped by the addition of Advanced DMEM/F-12 supplemented with 0.1% BSA. CRC samples are then filtered through a 100 µm mesh. Samples are centrifuged at 250× g (CRC) or 200× g (Healthy colon) for 5 min at 4 °C. Red blood cells are lysed if necessary.
The resulting pellets are resuspended in 50-100% ice-cold growth factor-reduced Matrigel (CRC—1.6 × 106 cancer cells/mL; Healthy colon—8 crypts/10 uL) and distributed in pre-heated 24-well plates (30 μL domes). Plates were inverted and incubated at 37 °C for 20 min. Then, 500 µL of growth medium was added per well. CRC growth medium included the following: Advanced DMEM/F-12 (#12634010, Gibco™ Thermo Fisher Scientific Inc., USA, 1% Anti-Anti, 1% HEPES, 1% GlutaMAX, 500 nM A83-01(SML0788 Merck), 0.02X B27 without vitamin A (17504044, Gibco™), 1.25 mM N-acetyl-L-cysteine (A9165, Sigma-Aldrich), 10 mM nicotinamide (N0630, Sigma-Aldrich), 1 µM PEG2 (2296/10 R&D Systems)), 100 µg/mL Primocin (ant-pm-2 Invivogen), 50 ng/mL recombinant human EGF (Peprotech), 100 ng/mL recombinant human Noggin (Peprotech), 500 ng/mL recombinant human R-spondin-1 (Peprotech), 0.3 µM SB202190 (S7067 Sigma-Aldrich), and 10.5 µM Y-27632 (10005583 Cayman Chemicals). Healthy colon medium included the following: IntestiCult™ Organoid Growth Medium (#06010, Stem Cell Technologies, Vancouver, BC, Canada) supplemented with primocin and 10.5 µM Y-27632.
Organoids formed typically within four days, and the medium was refreshed twice a week. Organoids were passaged by mechanical disruption of the Matrigel, washing in Advanced DMEM/F12 medium and resuspension in ice-cold GFR Matrigel.
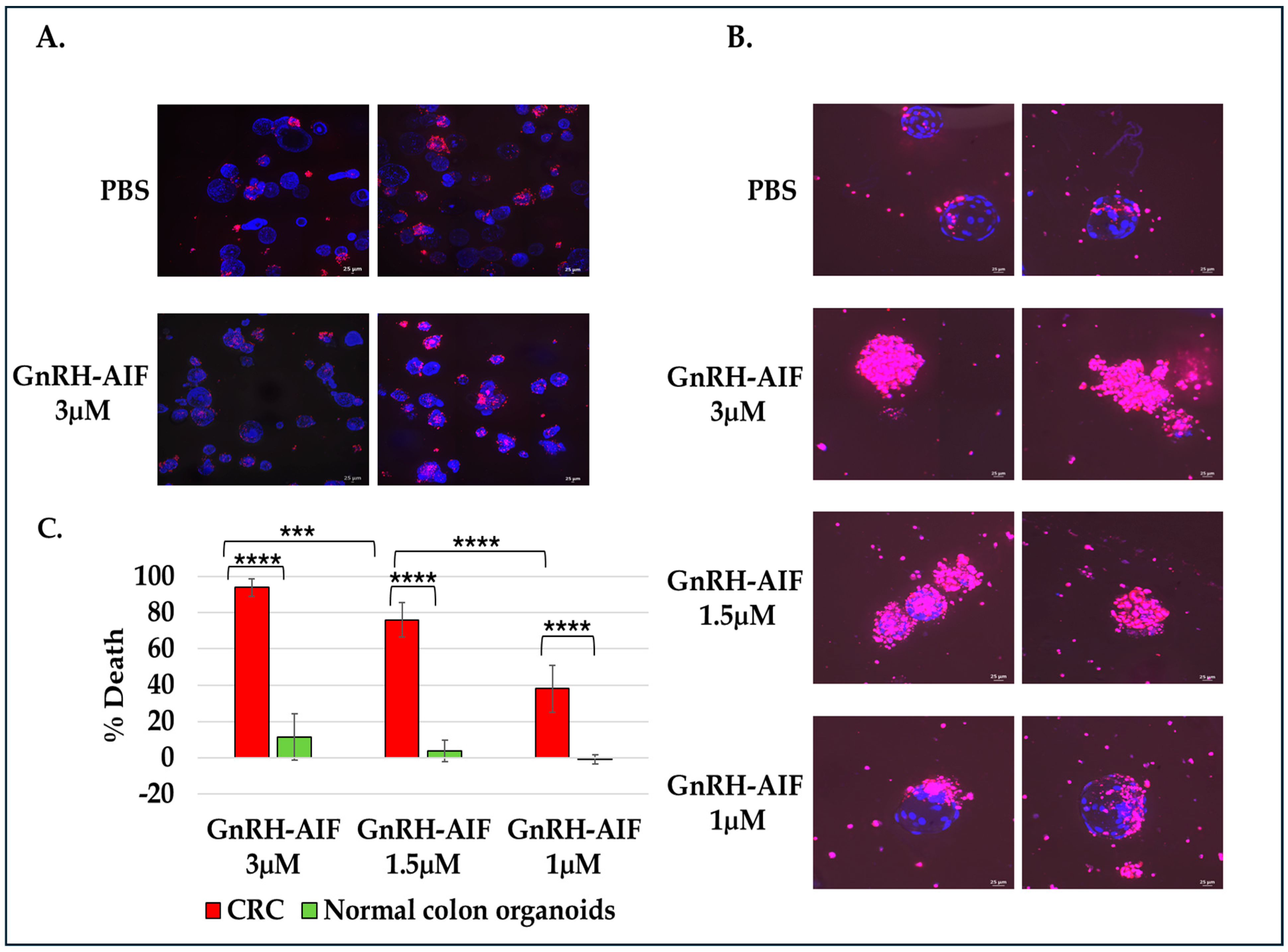
For cell viability assay, organoids were seeded on an 8-well IBIDI slide at a concentration of 5 organoids per well in 12 µL Matrigel and cultures in complete medium supplemented with various concentrations of GnRH-AIF (1, 1.5, and 3 µM) or PBS for three days. Cultures were then incubated with propidium iodide (PI) and Hoechst 33,342 according to the manufacturer’s instructions. Images were acquired on a spinning disk confocal microscope (Yokogawa W1 Spinning Disk) (4× and 20× objective), and analysis was performed using the NIS program (NIS Elements software #46002). For all experiments, three independent experiments were conducted. Areas of individual organoids were measured and exported as Excel files. Hoechst 33,342 and propidium iodide (PI) channels were separated and loaded as matched channels. Then, the total cell nuclei area (Hoechst 33342) and PI staining area were calculated for each organoid and exported to a spreadsheet. The cell death was calculated by dividing PI area per nuclei area. Results are shown as the percentage of organoid deaths calculated for treated organoids as compared to control (treated with PBS only).
2.21. Statistical Analyses
Results of cell viability assays are the average of 3–6 independent biological experiments ± SD. The results of the Western blot analyses were evaluated by densitometry using the Image Lab Software version 5.2.1 (Bio-Rad). Statistical analysis was performed using Microsoft Excel 2017 and an unpaired, two-tailed t test was used to determine p values.
4. Discussion
Most anticancer treatments trigger tumor cell death through apoptosis, for which initiation of proteolytic action of caspases is a requirement. However, cancerous cells are known to highly regulate the apoptotic pathways, and numerous mutations in various types of cancer cells have been reported to be implicated in chemoresistance and treatment outcome [
3,
4,
5,
6,
7,
8]. Therefore, in this study we aimed to construct a new chimeric cytotoxic protein using AIF in its cleaved apoptotic form as the killing moiety, to induce caspase-independent cell death. The GnRH/GnRH-R system was used to direct the AIF protein into solid cancer cells overexpressing the GnRH-R [
19,
20,
33].
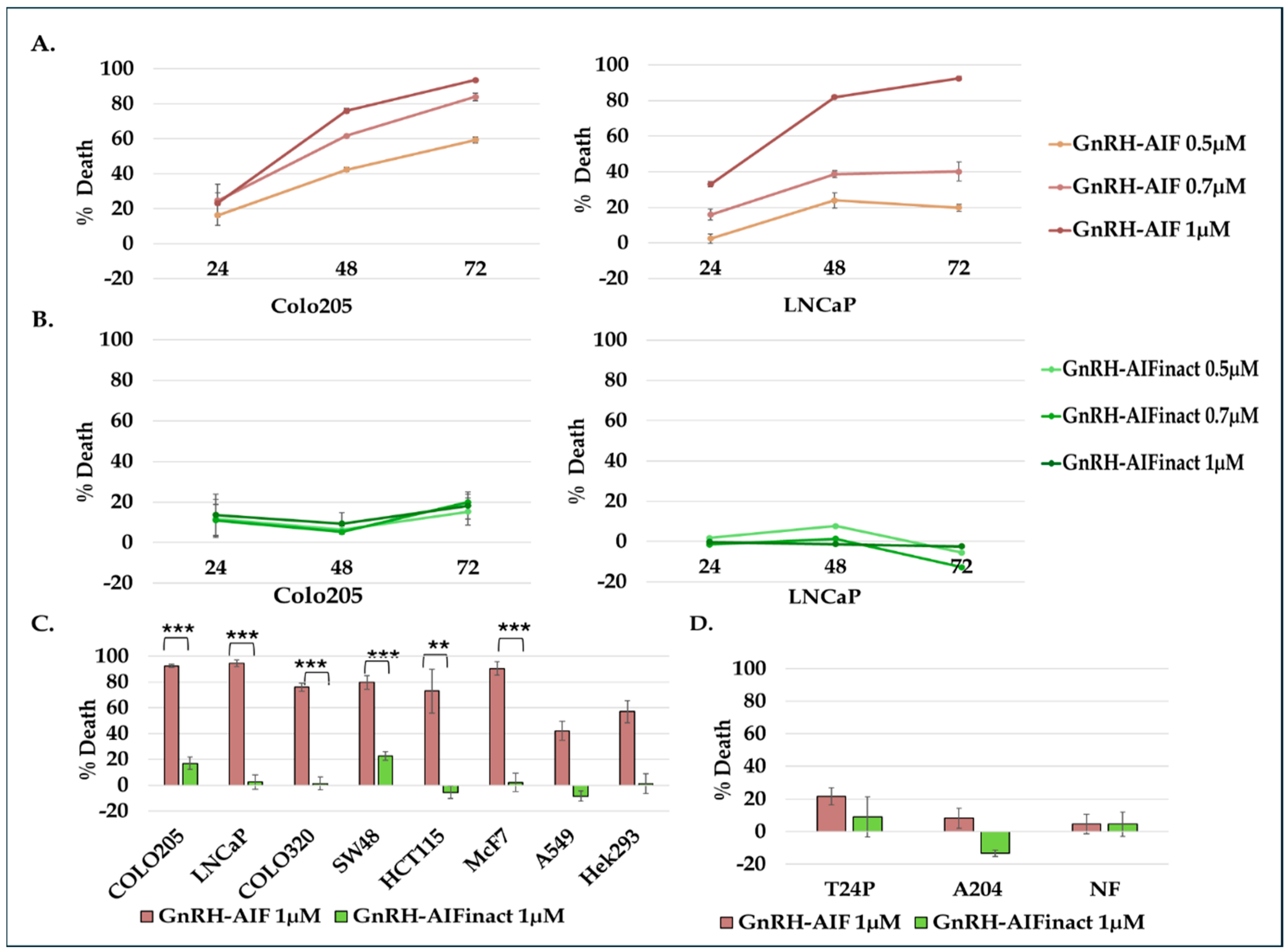
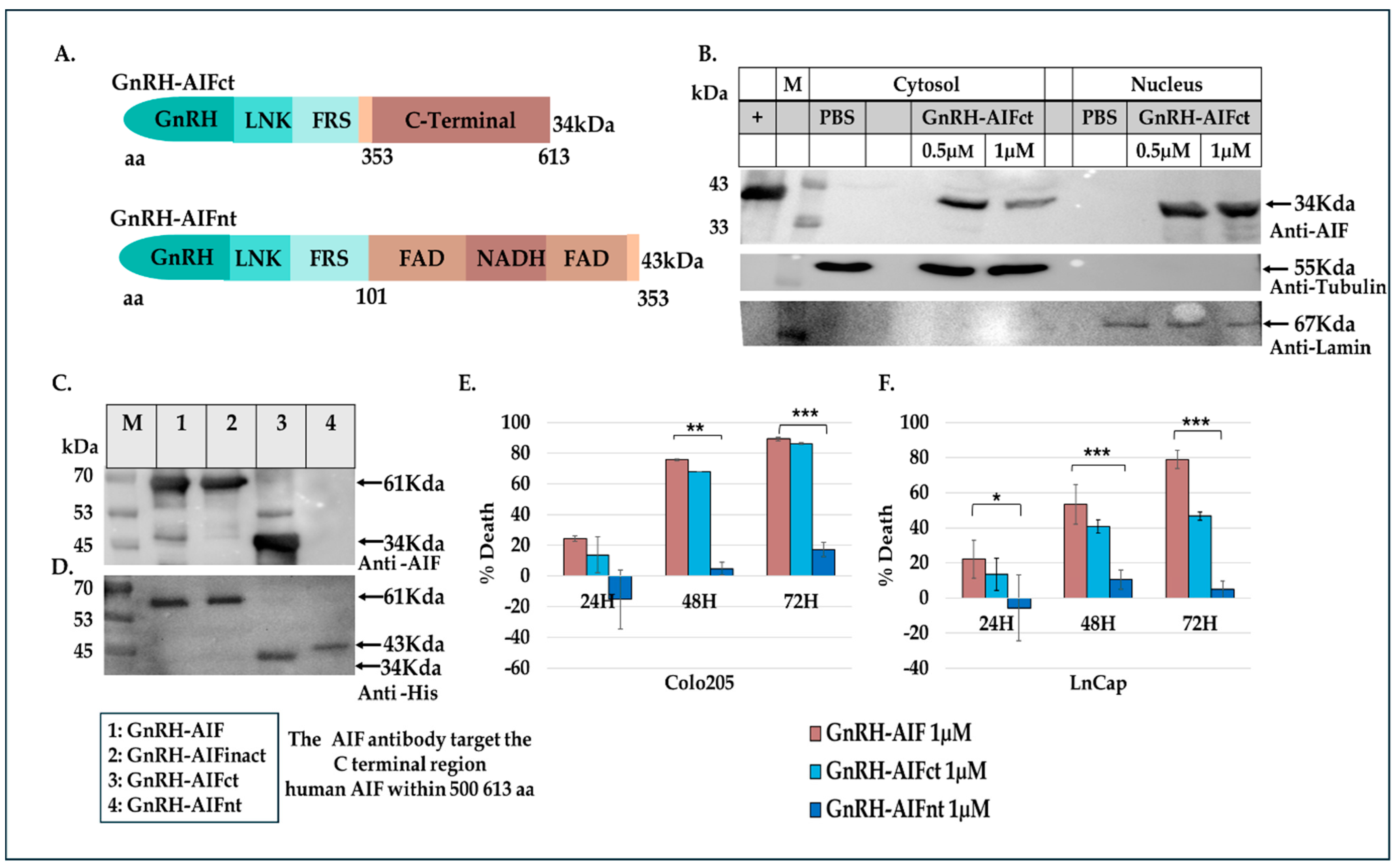
In this study, we constructed, expressed, and highly purified GnRH-AIF chimeric proteins (
Figure 1D–F). We demonstrated that GnRH-AIF chimeric proteins enter and kill specifically and very efficiently human cancer cell lines overexpressing GnRH-R (
Figure 2 and
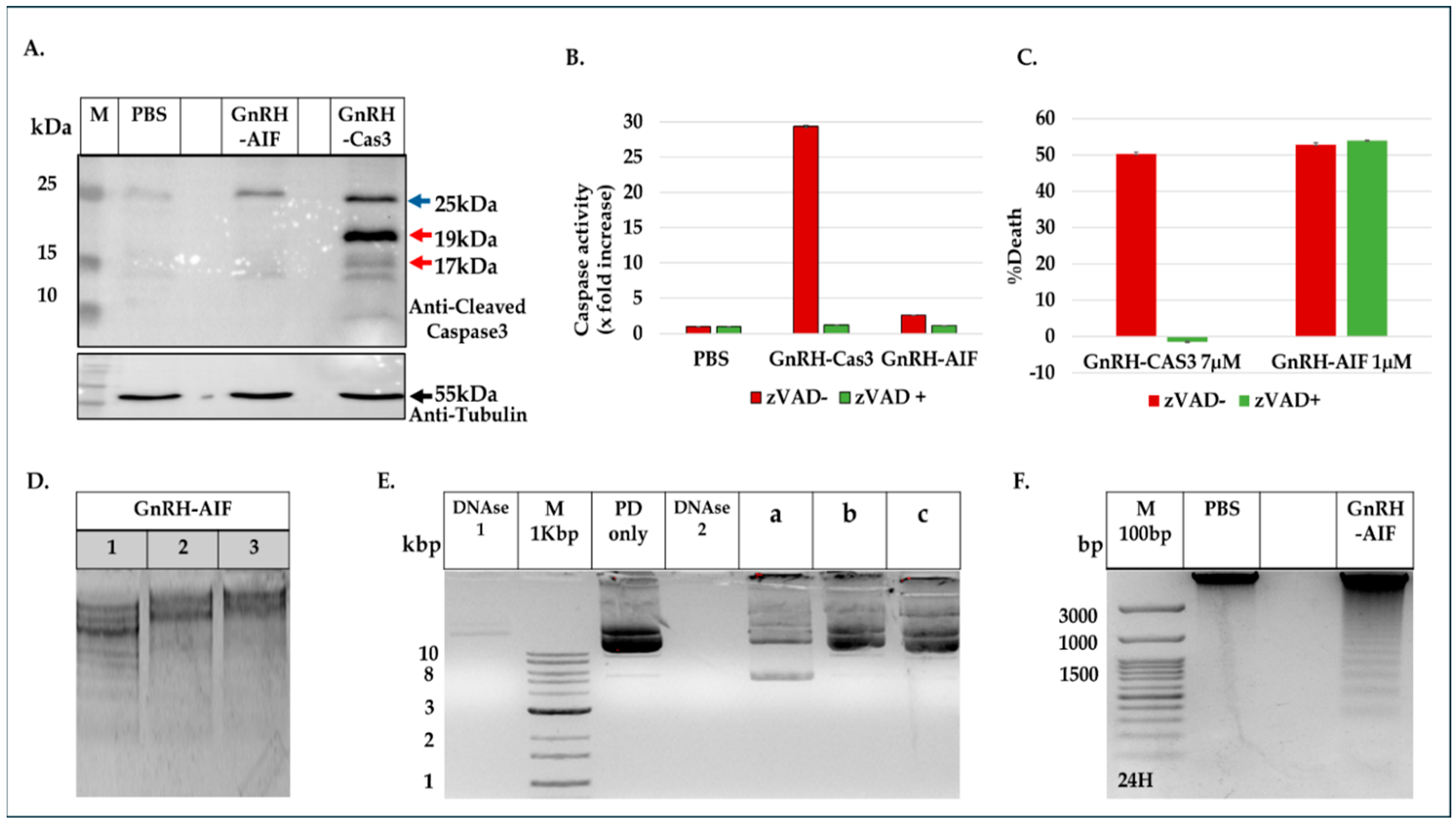
Figure 4). In addition, we successfully demonstrated that the delivery of AIF via the GnRH targeting system into cancer cells induces a specific caspase-independent apoptotic death (
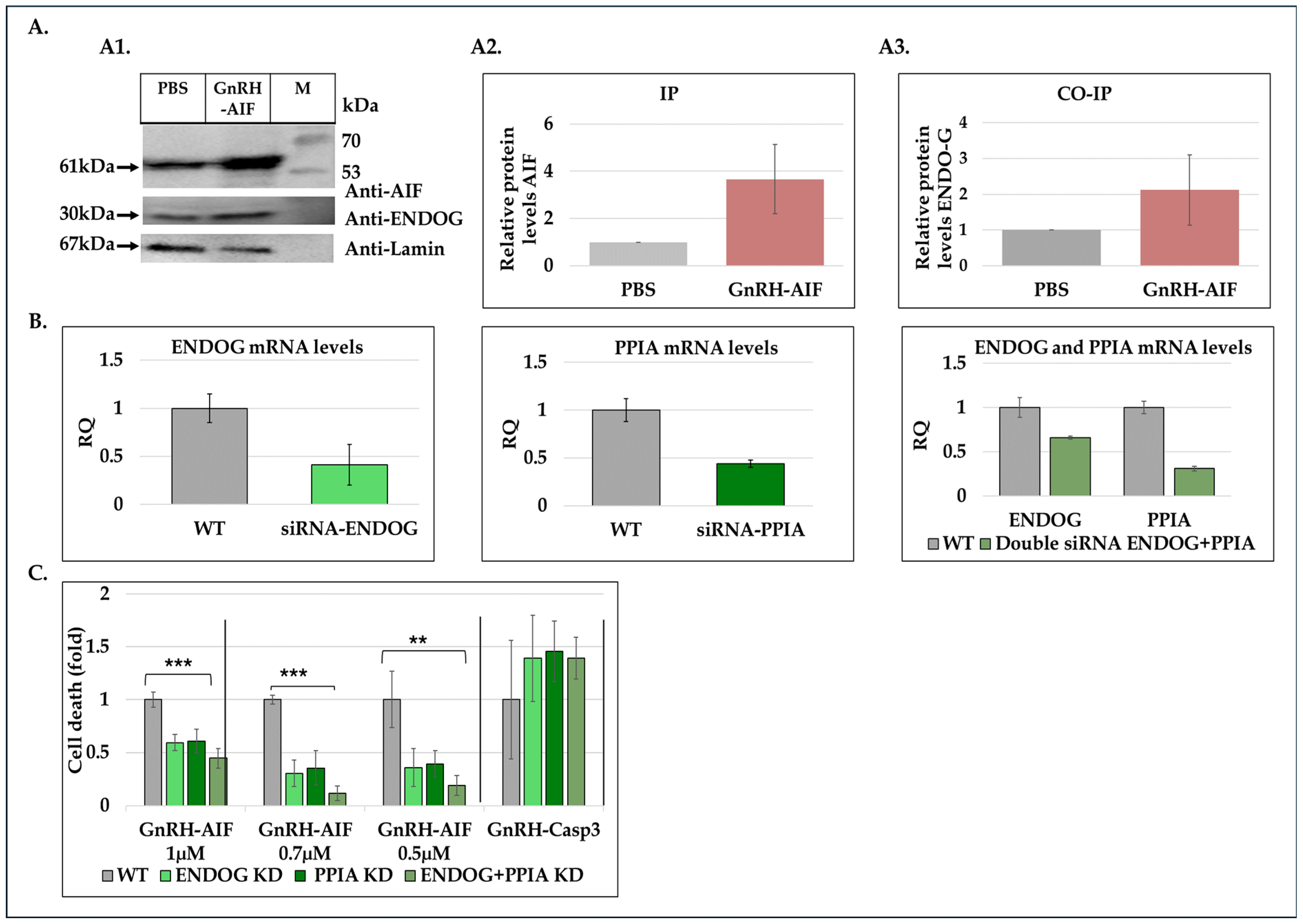
Figure 6 and
Figure 7). Furthermore, we produced a clone of GnRH-AIF with an inactive killing component [
25], thus confirming that death of the target cells was indeed caused by the killing component of the chimeric proteins (
Figure 4B–D). We also designed and produced two additional variants of the chimeric protein, the first including the amino acids 353–613 of AIF corresponding to its C-terminal domain (GnRH-AIFct) and the second variant corresponding to the N-terminal region of AIF (GnRH-AIFnt). Our results demonstrated that the pro-apoptotic activity of AIF resides in its C-terminal domain and that the first N-terminal 352 amino acids of AIF are, most probably, not required for its killing activity (
Figure 5E,F). As AIF lacks nuclease activity, it is proposed to directly interact with DNA and disrupt chromatin structure by displacing chromatin-associated proteins and by recruiting nucleases to form DNA-degrading complexes. To demonstrate that these interactions are relevant to the apoptogenic action of AIF, we knocked down relevant partners, PPIA/Endo G, two of those nuclear proteins known to be part of the DNA-degrading complexes within target cells, and tested the ability of GnRH-AIF chimeric protein-treated cells to cause cell death (
Figure 8C). Finally, we tested the effect of GnRH-AIF chimeric proteins ex vivo in a human colon cancer organoid model and demonstrated very promising anti-tumor efficacy (
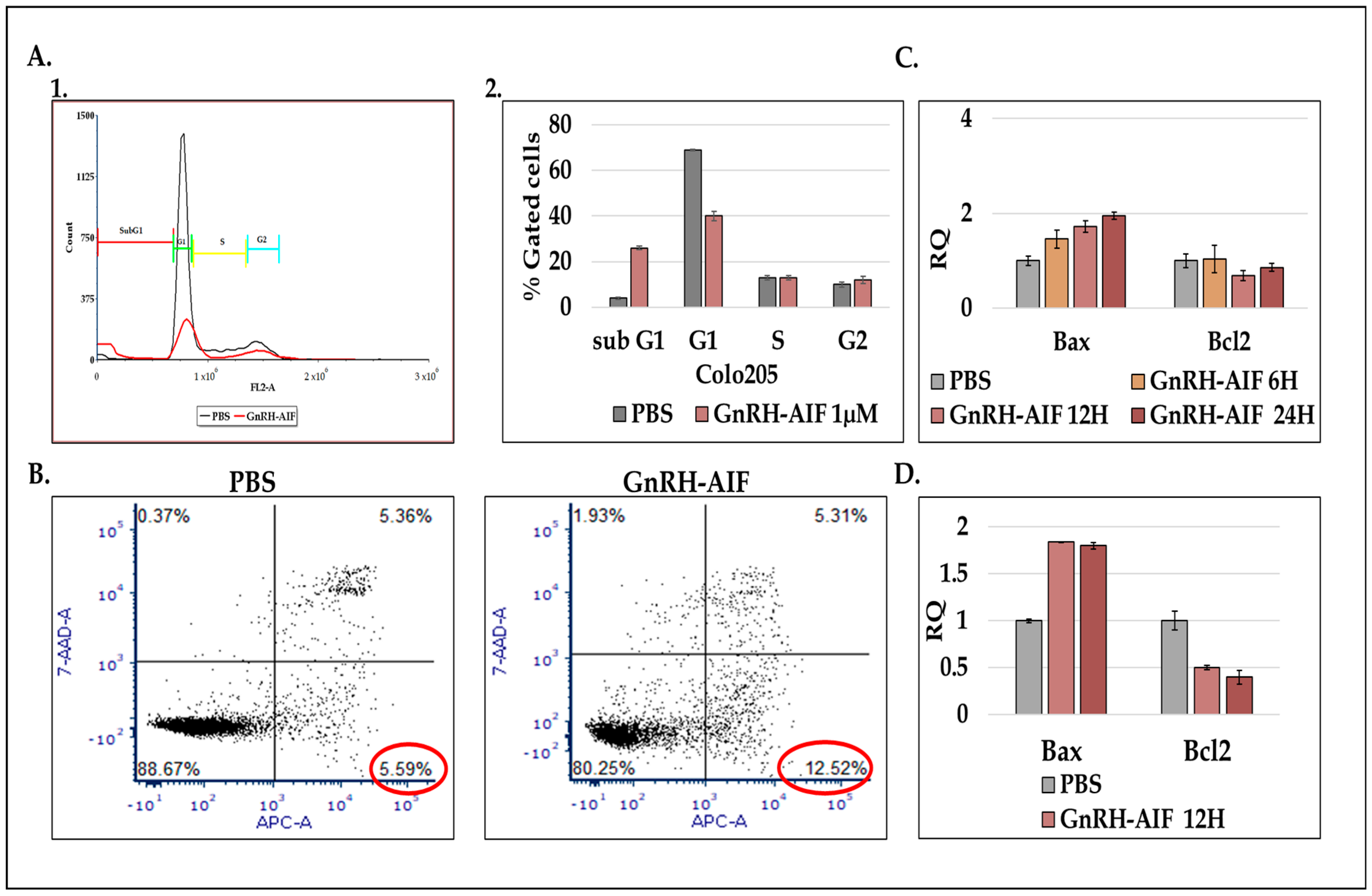
Figure 9). Our results established that caspase-independent pathways can be used for cause-specific cell death and suppress tumors as a new approach to treat cancer, in particular solid tumors.
The chimeric protein GnRH-AIF was expressed in a bacterial expression system and aggregated in the IB. Therefore, we had to develop a purification protocol including denaturation of the IB, followed by two steps of purification. In addition, a very sensitive refolding of the protein was needed to produce a highly purified and biologically active chimeric protein. GnRH-AIF chimeric proteins specifically targeted GnRH-R overexpressing cells and induced cells’ specific apoptosis in a dose- and time-dependent manner (
Figure 4 and
Figure 6). The specificity of the chimeric protein was demonstrated by distinguishing between cancer cells lines expressing and not expressing the GnRH-R (
Figure 4). In addition, we found that manipulating the AIF-killing moiety by two point mutations, forming its inactive variant [
25], resulted in no killing activity in the targeted cells (
Figure 4). Cell cycle analysis and real-time PCR for apoptotic-related proteins indicated that the GnRH-AIF chimeric proteins caused apoptotic cell death (
Figure 6). However, measuring caspase 3 activity demonstrated caspase-independent apoptotic cell death, as was our aim (
Figure 7A,B). To support our results, another chimeric protein, GnRH-Caspase-3, known to cause caspase-dependent apoptosis, was designed and purified under the same protocol as the GnRH-AIF. Western blot analysis of the cleaved caspase 3 and measurement of caspase 3 activity in vitro demonstrated that GnRH-AIF caused caspase-independent death in its initial phase, unlike GnRH-Caspase-3 which used caspase-dependent apoptotic pathways (
Figure 7A,B). In addition, we tested the ability of GnRH-AIF to induce apoptotic cell death in cancer cells under the inhibition of caspase activity (by zVAD treatment). Contrary to GnRH-Caspase-3, the cell death of GnRH-AIF-treated target cells was not affected by zVAD treatment (
Figure 7C), confirming that the GnRH-AIF chimeric proteins utilize a caspase-independent pathway to induce cell death within target cells.
Our results demonstrate that GnRH-AIF death is more rapid and effective compared to GnRH-Caspase-3 death (
Figure 3), possibly through the activation of a direct apoptotic pathway. On the other hand, GnRH-Caspase-3 may trigger cell death through a different mechanism, likely involving the activation of caspase 3, which might be relatively slower or less effective. Tumor cells lacking a functional caspase-dependent pathway can resist conventional chemotherapeutic agents or radiation, which often work by triggering caspase-dependent apoptosis pathways [
40]. Research is ongoing to develop treatments that can bypass or target alternative cell death pathways [
41]. Therefore, our study illustrates the promising potential of GnRH-AIF chimeric protein treatment to overcome the problems encountered with defects in caspase-dependent apoptotic pathways and moreover to be effective for a large number of solid cancers overexpressing GnRH-R.
In order to get a deep understanding of AIF’s mechanism of action while inducing cell death, we investigated how AIF, lacking nuclease activity itself, contributes to DNA degradation [
36]. Previous research demonstrated the implication of several proteins, such as PPIA and ENDOG, in the apoptotic role of AIF [
35,
38,
42]. Immunoprecipitation and co-immunoprecipitation experiments using nuclear extracts from treated and untreated cells demonstrated the physical interaction between GnRH-AIF and Endonuclease G (
Figure 8A). By utilizing siRNAs targeting ENDOG and PPIA proteins, we also demonstrated that their knockdown significantly reduced cell death induced by the GnRH-AIF chimeric protein (
Figure 8C). One possible explanation is that both ENDOG and PPIA recruitment to the AIF facilitates DNA degradation. Silencing ENDOG and PPIA genes could abrogate these interactions, thereby inhibiting the AIF downstream effects, leading to decreased cell death. This interpretation implies a dependency of GnRH-AIF-mediated cell death on the presence and activity of ENDOG and PPIA. Further investigations are needed to fully understand the precise mechanism by which AIF forms a complex with ENDOG and PPIA and probably other undescribed proteins to perform its apoptotic role. In addition, it is critical to understand the role of each protein within the complex known as the degradome complex [
35,
38,
42].
We also tested the nuclease activity of AIF in vitro, as it was recently reported that AIF has the ability to act as a nuclease in certain conditions and without the assistance of any proteins [
42]. However, our results showed that AIF, in the form of a chimeric protein, only has the ability to bind; however, it does not bind to cleaved genomic DNA or plasmid DNA in vitro (
Figure 7E). This article [
42] contradicts our current findings, which point out the involvement of ENDOG and PPIA in the mechanism by which the GnRH-AIF chimeric protein induces apoptosis in target cells. Further investigation is needed to clarify the detailed molecular mechanism of AIF protein.
The AIF protein is composed of three structural domains: the FAD-binding domain, the NADH-binding domain (oxidoreductase part of AIF), and the C-terminal domain (apoptotic function). Previous studies demonstrated that the C-terminal domain of AIF is necessary and sufficient to induce chromatin condensation and large-scale DNA fragmentation leading to caspase-independent apoptosis [
34]. Therefore, we constructed additional GnRH-AIF chimeric proteins variants. The GnRH-AIFct chimeric protein, including the only AIF C-terminal domain, was indeed able to cause cell death, however, less effectively than the original GnRH-AIF, full-length, chimeric protein. In contrast, the GnRH-AIFnt chimeric protein, containing the first 352 amino acids of the N-terminal domain, did not kill the targeted cells (
Figure 5E,F). These results confirmed that the C-terminal domain is the apoptotic domain of the AIF protein and confirmed that the AIF component of the chimeric proteins is the killing moiety responsible for the killing activity of the chimeric proteins. Further co-immunoprecipitation experiments should be performed to determine the exact region in the C-terminal domain by which AIF binds to the DNA.
Most importantly, the activity of GnRH-AIF was remarkable on primary organoid cultures from human colorectal cancer, and the specificity was proven using organoids originating from normal colon culture. In recent years, tumor organoid models have been widely used for cancer drug testing [
43]. Produced by using stem cell-derived tissues and cultivated under specific conditions, organoids mimic the heterogeneity and the complexity of the original tumor, therefore representing a promising tool for drug testing for human cancer therapies [
44]. This result further demonstrates the potential of the GnRH-AIF chimeric protein as a new highly effective modality for the treatment of solid tumors (
Figure 9).
In this work, we showed further proof of the efficacity of the GnRH peptide as a targeting moiety for GnRH-R overexpressing cells. We used the GnRH/GnRH-receptor system to direct the AIF into cancer cells expressing the receptor [
19,
20]. Previously, using the bacterial toxin
Pseudomonas exotoxin A (PE) as well as human apoptotic proteins, such as Bax, Bak, Bik, and DFF40 [
16,
19,
20], we constructed a number chimeric proteins based on GnRH as the “targeting” component. Testing these GnRH-based chimeric proteins on different cancer cell lines, primary cultures, as well as in colon cancer models in mice [
13,
45], we were the first to report that a wide variety of solid tumors that are not part of the reproductive system, such as colon cancer, kidney cancer, and many other solid tumors, overexpress the GnRH-R/binding sites [
16,
28,
33].
AIF is involved in other cellular contexts such as parthanatos [
46], a form of programmed cell death linked to neurodegenerative diseases such as Alzheimer’s and Parkinson’s. Parthanatos involves a cascade of signaling steps that cause cell death, induced by PARP-1 overactivation, and involves AIF and a macrophage migration inhibitory factor (MIF) [
47]. Therefore, manipulating AIF activity opens up new opportunities for developing therapeutic strategies for treating various diseases such as Alzheimer’s and Parkinson’s [
48].
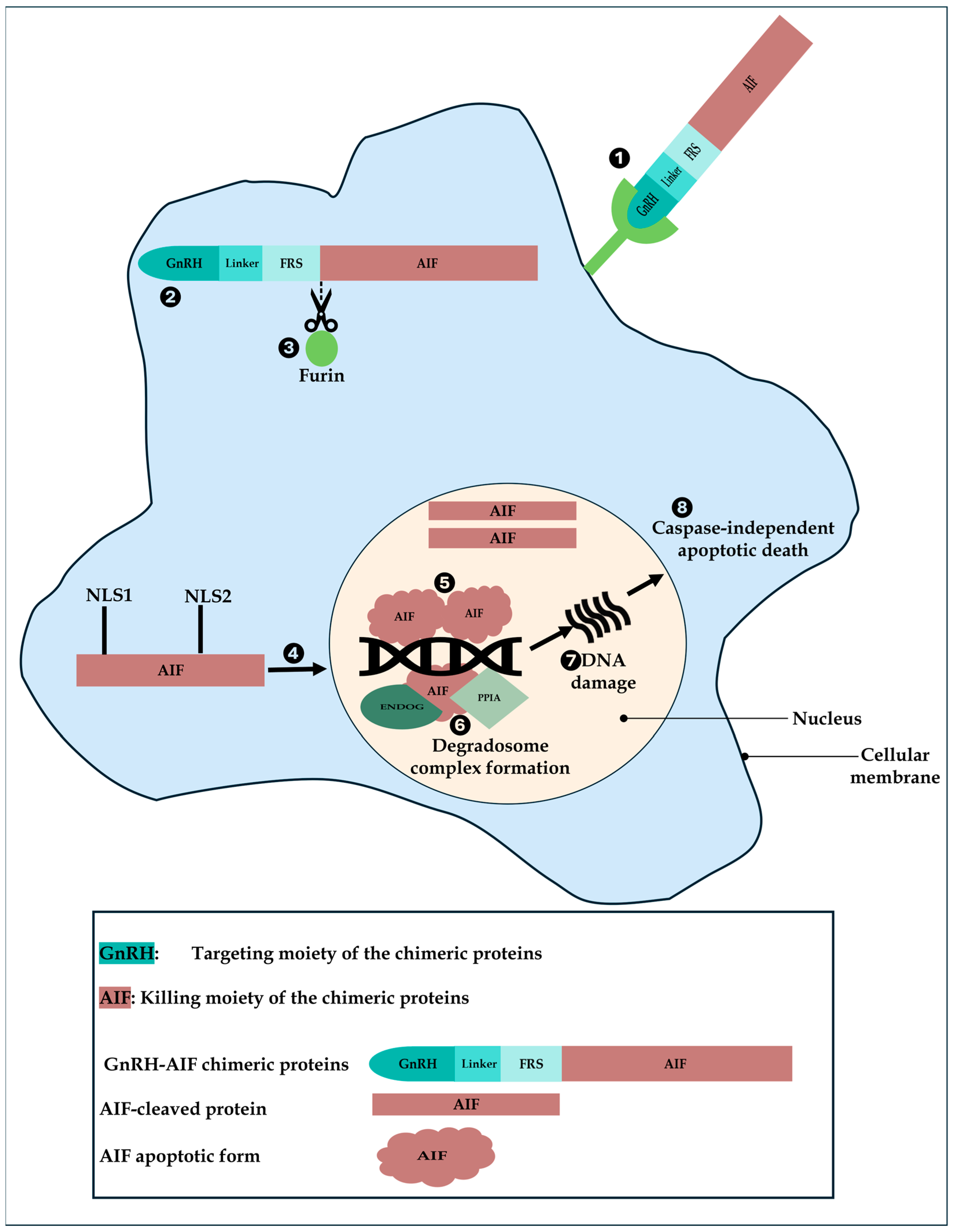
Based on our results, we propose a model of action of GnRH-AIF in GnRH-R overexpressing cancer cells (
Figure 10). First, GnRH-AIF enters the cells and localizes primarily in the cytosol where it is cleaved at the Furin site. The AIF-cleaved apoptotic protein translocates into the nucleus, binds to DNA, recruits ENDOG and PPIA among other unknown proteins, causing DNA fragmentation, and finally leads to caspase-independent apoptotic death.
GnRH-AIF is a new chimeric protein designed to bypass the classical caspase-dependent apoptotic pathways. The highly encouraging results described here regarding GnRH-AIF chimeric protein’s killing specificity and efficacy in vitro against human target cancer cells lines and ex vitro in human colon cancer organoid models suggest it as a novel treatment for solid cancers overexpressing the GnRH-R. Solid tumors account for a large and diverse group of cancers that can form metastases, are hard to treat, and have a high rate of mortality [
49]. For example, colorectal cancer was the fourth-leading cause of cancer death in both men and women younger than 50 years in the late 1990s but is now first in men and second in women [
50]. In addition, we suggest that AIF in the form of chimeric proteins can be applied to other types of tumors, using the appropriate targeting moiety so expanding therapy based on AIF proteins to a variety of other tumors.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}