
Gene and Cell Therapy for Sarcomas: A Review

Simple Summary
Abstract
1. Introduction
Search Strategy
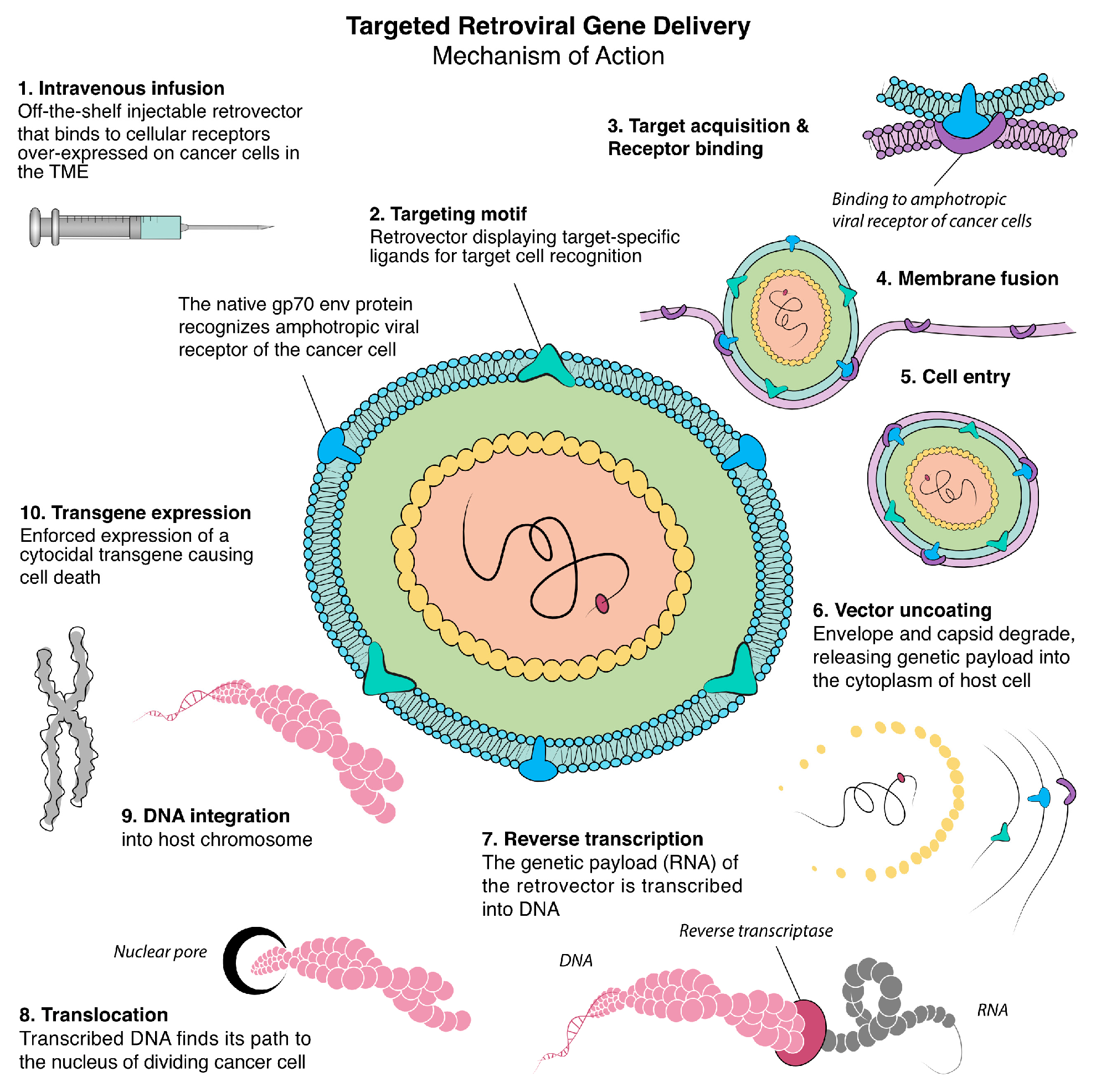
2. Retrovirus-Based Gene Therapy
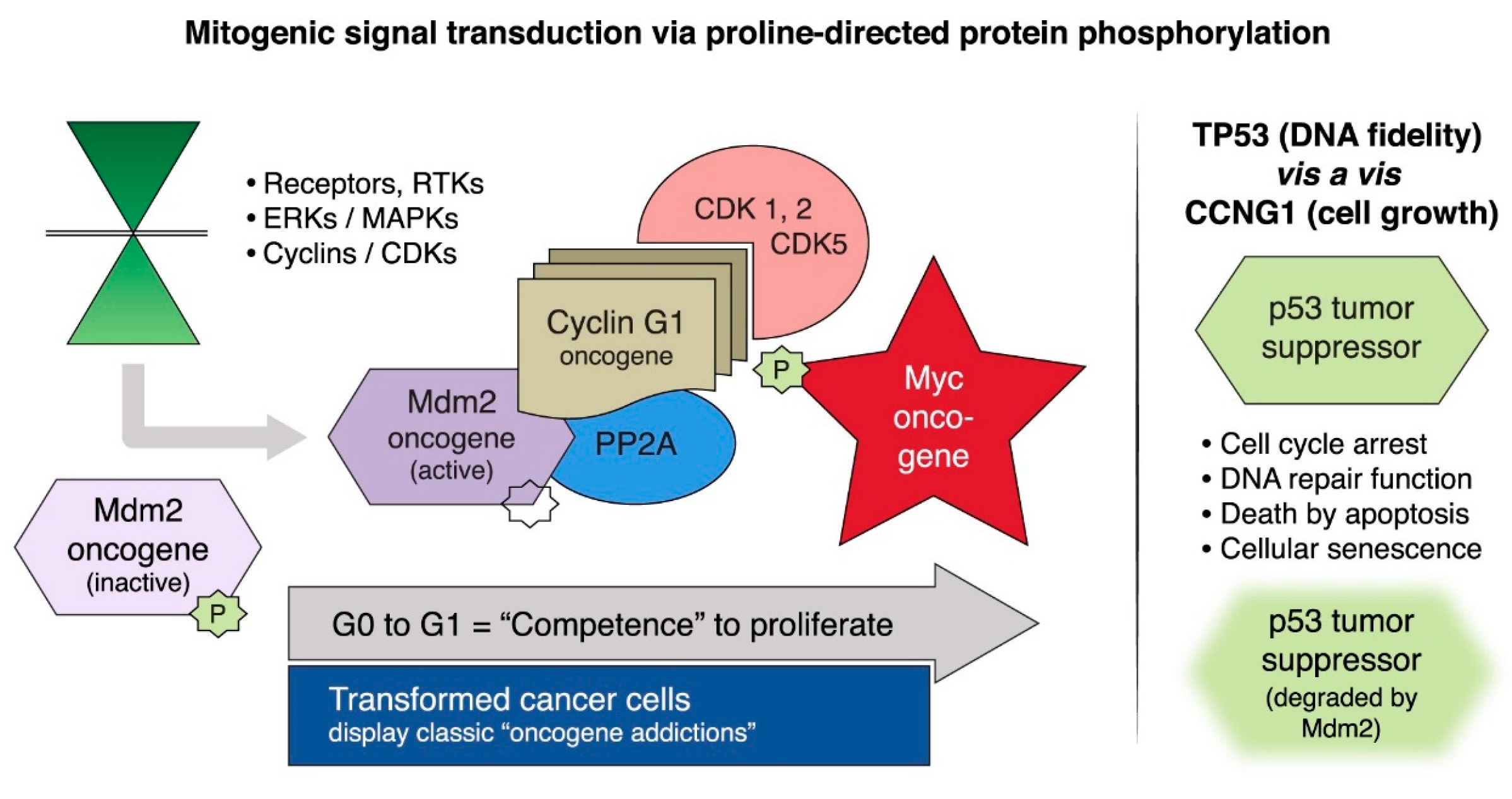
2.1. Targeting Executive Oncogenic Drivers
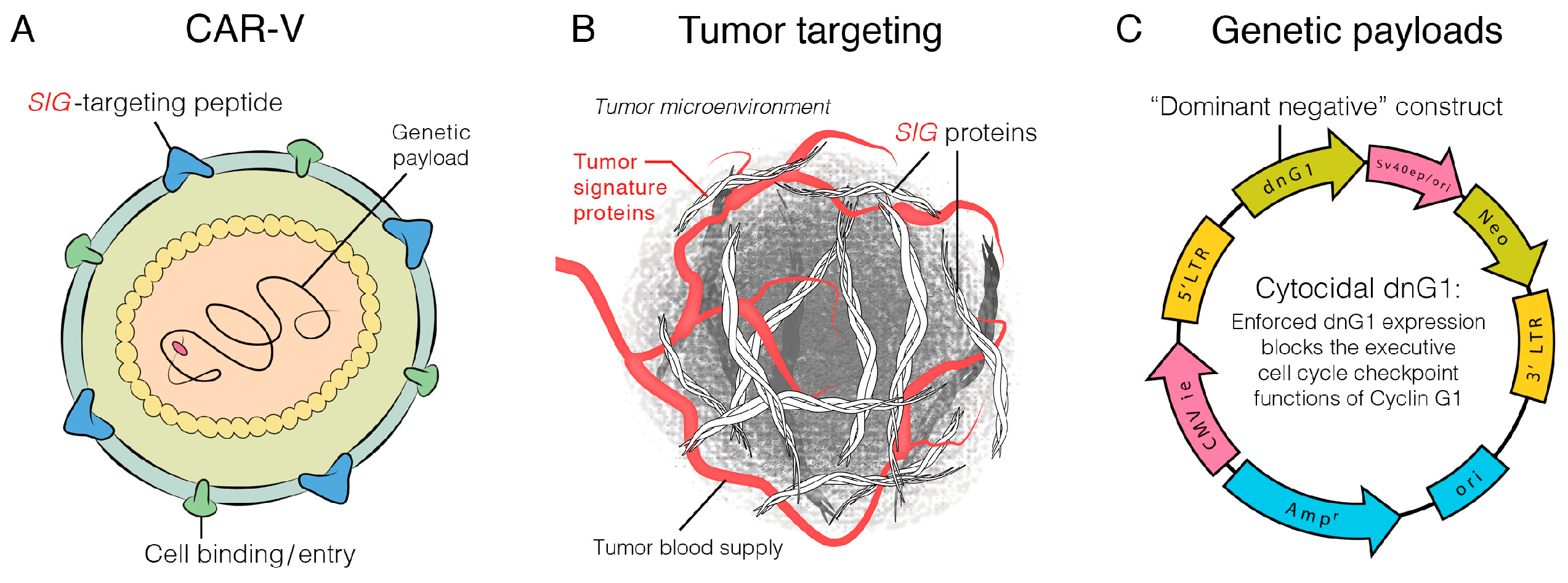
2.2. Rexin-G, the First Clinical Tumor-Targeted Gene Therapy Vector for Sarcoma
2.3. DNG64, a Paradigm Shift in Targeted Gene Delivery
2.4. The Genevieve Protocol: Rexin-G and Regulatable Reximmune-C
2.5. GEN2
2.6. Points to Consider with Retroviral-Based Gene Therapy
3. Oncolytic Virotherapy/Gene Therapy
3.1. Herpes Simplex Virus Type 1 Oncolytic Virus
3.2. Adenoviral-Based Oncolytic Virus
3.2.1. Gendicine
3.2.2. AdAPT-001
3.3. Points to Consider with Oncolytic Viruses
4. Cell Therapy for Sarcomas
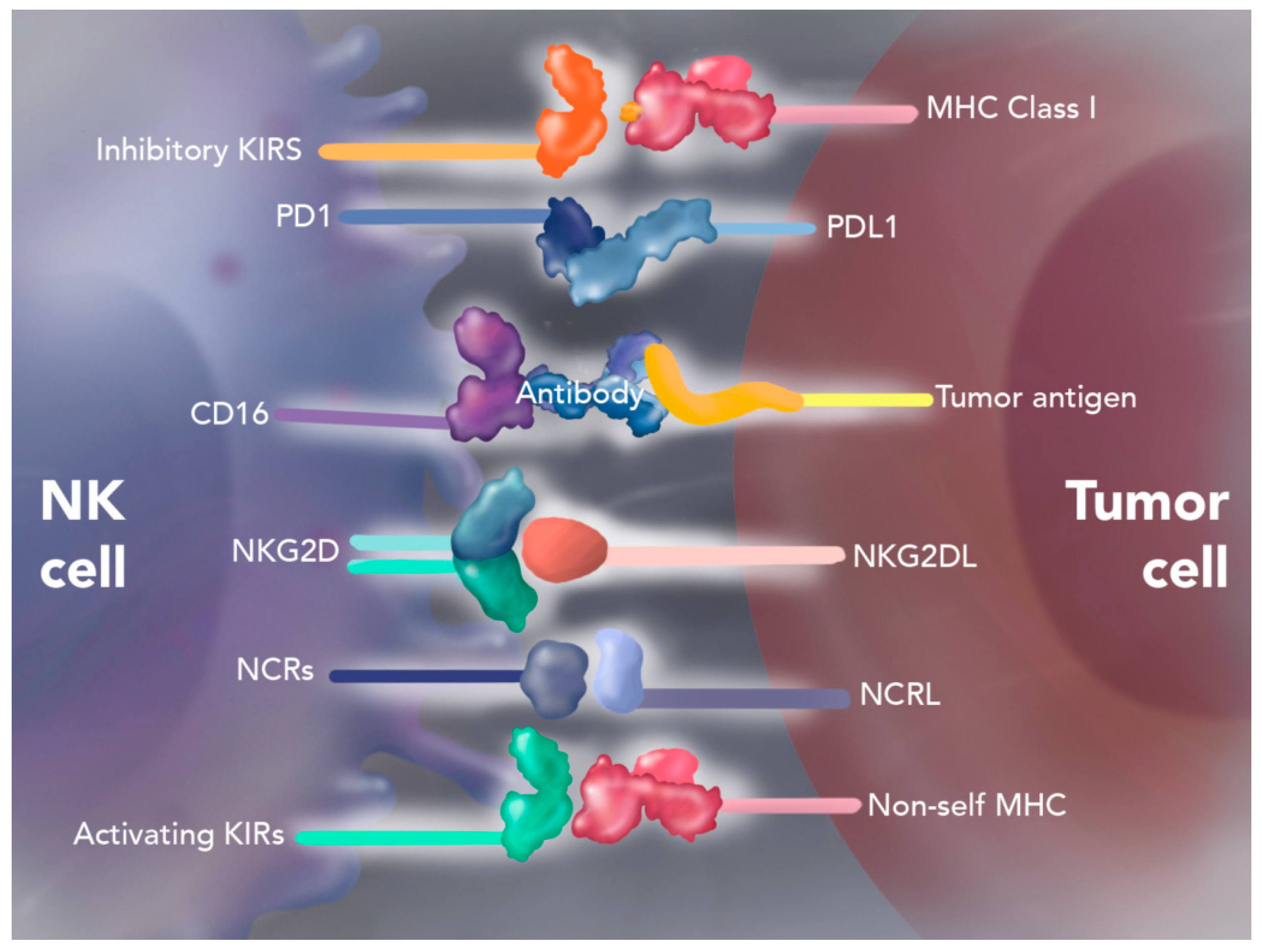
4.1. Natural Killer Cell Therapy
Autologous Enhanced Natural Killer Cell Therapy (SNK01)
4.2. Mesenchymal Stromal Cell Therapy
4.3. Chimeric Antigen Receptor (CAR)-T Cell Therapy
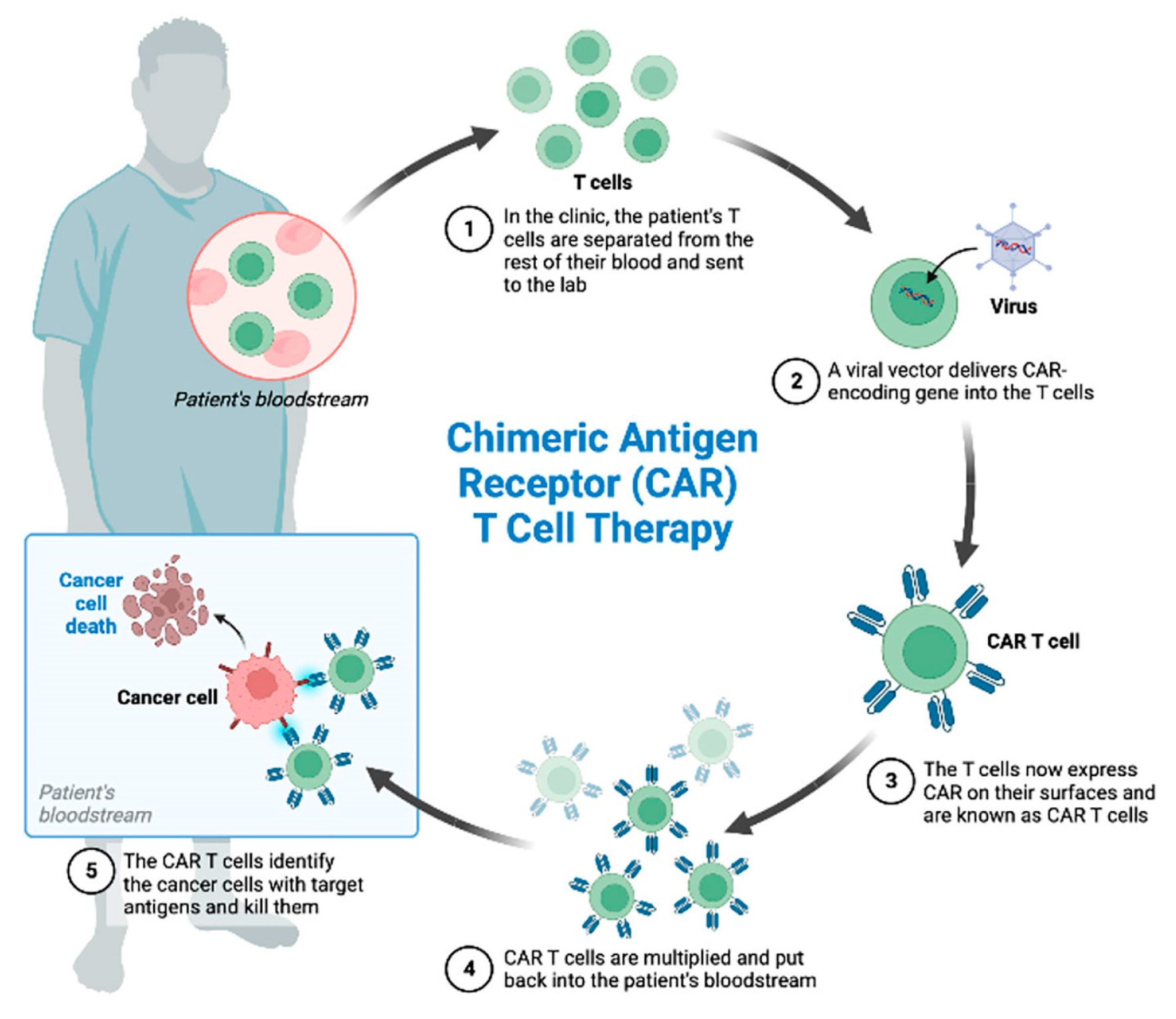
4.3.1. Afamitresgene Autoleucelis (Afami-Cel; TECELRA)
4.3.2. HER2-Specific CAR-T Cell Therapy
4.3.3. EGFR-Specific CAR-T Cell Therapy
4.4. T Cell Receptor (TCR) Therapy
Letetresgene Autoleucel (Lete-Cel, GSK3377794) Therapy
4.5. Dendritic Cell Therapy
4.6. Points to Consider with Gene Modified Cell Therapy
5. Other Gene and Cell Therapy Trials
6. Comparisons Between Gene and Cell Therapies and Current Therapies for Cancer
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADA | Adenosine deaminase |
| Ad | Adenovirus |
| Ad26 | Recombinant Adenovirus Serotype 26 |
| Ad 35 | Recombinant Adenovirus Serotype 35 |
| Ad5p53 | Adenovirus-mediated p53 |
| ASGCT | American Society of Gene and Cell Therapy |
| CAR | Chimeric antigen receptor |
| CAR-T | Chimeric antigen receptor-T |
| CBER | Center of Biologics Evaluation and Research |
| CBR | Clinical benefit rate |
| CCNG1 | Human cyclin G1 gene |
| CD | Clusters of differentiation |
| CDK | Cyclin-dependent protein kinase |
| CD-5FC | Cytosine deaminase-5-fluorouracil |
| Cfu | Colony-forming units |
| CI | Checkpoint inhibitor |
| CIV | Continuous intravenous infusion |
| CR | Complete remission |
| CRS | Cytokine release syndrome |
| ctDNA | Circulating tumor DNA |
| DCR | Disease control rate |
| DNA | Deoxyribonucleic acid |
| dnG1 | Dominant negative cyclin G1 |
| E1A | Early region 1A |
| EGFR | Epidermal growth factor receptor |
| ERK | Extracellular signal-regulated kinase |
| FDA | Food and Drug Administration |
| Gp70 | Glycoprotein 70 |
| GM-CSF | Granulocyte macrophage colony stimulating factor |
| HER2 | Human epidermal growth factor receptor 2 |
| HLA | Human Leukocyte Antigen |
| hGM-CSF | Human granulocyte-macrophage colony-stimulating factor |
| HSV-ETK | Enhanced herpes simplex virus Type 1 thymidine kinase |
| HSV-tk | Herpes simplex virus Type 1 thymidine kinase |
| HSV-1 | Herpes simplex virus type-1 |
| ICI | Immune checkpoint inhibitor |
| IL | Interleukin |
| IC | Intracellular |
| Kb | Kilobase |
| MAGE-A4 | Human melanoma-associated antigen 4 |
| MAPK | Mitogen-activated protein kinase |
| MDM2 | Murine double minute 2 |
| MHC | Major Histocompatibility Complex |
| MSC | Mesenchymal stromal cell |
| NK | Natural killer |
| NY-ESO-1 | New York esophageal squamous cell carcinoma-1 |
| ORR | Objective response rate |
| OS | Overall survival |
| OVT | Oncolytic virotherapy |
| PET | Positron emission topography |
| PD | Progressive disease |
| PD-1 | Programmed death 1 programmed death ligand 1 inhibitors |
| PD-L1 | Programmed death ligand 1 inhibitors |
| PFS | Progression free survival |
| PP2A | Protein phosphatase 2A |
| PR | Partial response |
| rAd-p53 | Recombinant adenovirus-p53 |
| RANKL | Receptor activator of nuclear factor kappa ligand |
| RECIST | Response Evaluation Criteria in Solid Tumors |
| RNA | Ribonucleic acid |
| SCID | Severe combined immunodeficiency |
| SD | Stabilized disease |
| scFv | Single-chain variable fragment |
| SIG | Signature |
| TCR | T cell receptor |
| TGF | Transforming growth factor |
| TIL | Tumor infiltrating lymphocytes |
| TME | Tumor microenvironment |
| TNT | Talimogene laherparepvec, nivolumab, and trabectedin |
| TRAIL | Tumor necrosis factor-related apoptosis-inducing ligand |
| TVEC | Talimogene laherparepvec |
| USFDA | United States Food and Drug Administration |
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Mocellin, S.; Rossi, C.R.; Brandes, A.; Nitti, D. Adult soft tissue sarcomas: Conventional therapies and molecularly targeted approaches. Cancer Treat. Rev. 2006, 32, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Gervais, M.K.; Basile, G.; Dulude, J.P.; Mottard, S.; Gronchi, A. Histology-Tailored Approach to Soft Tissue Sarcoma. Ann. Surg. Oncol. 2024, 31, 7915–7929. [Google Scholar] [CrossRef] [PubMed]
- Judson, I.; Verweij, J.; Gelderblom, H.; Hartmann, J.T.; Schöffski, P.; Blay, J.Y.; Kerst, J.M.; Sufliarsky, J.; Whelan, J.; Hohenberger, P.; et al. Doxorubicin alone versus intensified doxorubicin plus ifosfamide for first-line treatment of advanced or metastatic soft-tissue sarcoma: A randomised controlled phase 3 trial. Lancet Oncol. 2014, 15, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, T.A. brief history of gene therapy. Nat. Genet. 1992, 2, 93–98. [Google Scholar] [CrossRef]
- Sambrook, J.; Westphal, H.; Srinivasan, P.R.; Dulbecco, R. The integrated state of viral DNA in SV40-transformed cells. Proc. Natl. Acad. Sci. USA 1968, 60, 1288–1295. [Google Scholar] [CrossRef]
- Hill, M.; Hillova, J. Virus recovery in chicken cells tested with Rous sarcoma cell DNA. Nat. New Biol. 1972, 237, 35–39. [Google Scholar] [CrossRef]
- Friedmann, T.; Roblin, R. Gene therapy for human genetic disease? Science 1972, 175, 949–955. [Google Scholar] [CrossRef]
- Anderson, W.F. Prospects for human gene therapy. Science 1984, 226, 401–409. [Google Scholar] [CrossRef]
- Anson, D.S. The use of retroviral vectors for gene therapy-what are the risks? A review of retroviral pathogenesis and its relevance to retroviral vector-mediated gene delivery. Genet. Vaccines Ther. 2004, 2, 9. [Google Scholar] [CrossRef]
- Blaese, R.M.; Culver, K.W.; Miller, A.D.; Carter, C.S.; Fleisher, T.; Clerici, M.; Shearer, G.; Chang, L.; Chiang, Y.; Tolstoshev, P.; et al. T lymphocyte-directed gene therapy for ADA-SCID: Initial trial results after 4 years. Science 1995, 270, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Kurian, K.M.; Watson, C.J.; Wyllie, A.H. Retroviral vectors. Mol. Pathol. 2000, 53, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Waehler, R.; Russell, S.J.; Curiel, D.T. Engineering targeted viral vectors for gene therapy. Nat. Rev. Genet. 2007, 8, 573–587. [Google Scholar] [CrossRef]
- Russell, S.J.; Hawkins, R.E.; Winter, G. Retroviral vectors displaying functional antibody fragments. Nucleic Acids Res. 1993, 21, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Morse, M.A.; Chawla, S.P.; Wong, T.; Bruckner, H.W.; Hall, F.L.; Gordon, E.M. Tumor protein p53 mutation in archived tumor samples from a 12-year survivor of stage 4 pancreatic ductal adenocarcinoma may predict long-term survival with DeltaRex-G: A case report and review of the literature. Mol. Clin. Oncol. 2021, 15, 186. [Google Scholar] [CrossRef]
- Wu, L.; Liu, L.; Yee, A.; Carbonarohall, D.; Tolo, V.; Hall, F. Molecular-cloning of the human cycg1 gene encoding a g-type cyclin—Overexpression in human osteosarcoma cells. Oncol. Rep. 1994, 1, 705–711. [Google Scholar] [CrossRef]
- Gordon, E.M.; Liu, P.X.; Chen, Z.H.; Liu, L.; Whitley, M.D.; Gee, C.; Groshen, S.; Hinton, D.R.; Beart, R.W.; Hall, F.L. Inhibition of metastatic tumor growth in nude mice by portal vein infusions of matrix-targeted retroviral vectors bearing a cytocidal cyclin G1 construct. Cancer Res. 2000, 60, 3343–3347. [Google Scholar] [PubMed]
- Gordon, E.M.; Chen, Z.H.; Liu, L.; Whitley, M.; Liu, L.; Wei, D.; Groshen, S.; Hinton, D.R.; Anderson, W.F.; Beart, R.W.; et al. Systemic administration of a matrix-targeted retroviral vector is efficacious for cancer gene therapy in mice. Hum. Gene Ther. 2001, 12, 193–204. [Google Scholar] [CrossRef]
- Gordon, E.M.; Ravicz, J.R.; Liu, S.; Chawla, S.P.; Hall, F.L. Cell cycle checkpoint control: The cyclin G1/Mdm2/p53 axis emerges as a strategic target for broad-spectrum cancer gene therapy—A review of molecular mechanisms for oncologists. Mol. Clin. Oncol. 2018, 9, 115–134. [Google Scholar] [CrossRef]
- Chawla, S.P.; Chawla, N.S.; Quon, D.; Chua-Alcala, V.; Blackwelder, W.C.; Hall, F.L.; Gordon, E.M. An advanced phase 1/2 study using an XC-targeted gene therapy vector for chemotherapy resistant sarcoma. Sarcoma Res. Int. 2016, 3, 1024. [Google Scholar]
- Chawla, S.P.; Bruckner, H.; Morse, M.A.; Assudani, N.; Hall, F.L.; Gordon, E.M. A Phase I-II Study Using Rexin-G Tumor-Targeted Retrovector Encoding a Dominant-Negative Cyclin G1 Inhibitor for Advanced Pancreatic Cancer. Mol. Ther. Oncolytics 2018, 12, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Al-Shihabi, A.; Chawla, S.P.; Hall, F.L.; Gordon, E.M. Exploiting Oncogenic Drivers along the CCNG1 Pathway for Cancer Therapy and Gene Therapy. Mol. Ther. Oncolytics 2018, 11, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Chawla, S.P.; van Tine, B.; Federman, N.; Schwab, J.; Jones, R.; Subbiah, V.; Chawla, N.S.; Afshar, N.; Hoos, W.; Feldman, N.; et al. Proceedings of the Think Tank for Osteosarcoma Medical Advisory Board. Anticancer Res. 2024, 44, 2765–2768. [Google Scholar] [CrossRef]
- Chawla, S.P.; Wong, S.; Quon, D.; Moradkhani, A.; Chua, V.S.; Brigham, D.A.; Reed, R.A.; Swaney, W.; Hall, F.L.; Gordon, E.M. Three year results of Blessed: Expanded access for DNG64 for an intermediate size population with advanced pancreatic cancer and sarcoma (NCT04091295) and individual patient use of DNG64 for solid malignancies (IND# 19130). Front. Mol. Med. 2022, 2, 1092286. [Google Scholar] [CrossRef]
- Jeffrey, S.L.; Agarwal, D.A.; Reed, R.; Haroun, G.; Guryeva, V.; Afuta, L.; Kusakawa, H.; Carter, R.; Swaney, W.; Hall, F.L.; et al. DNG64, a tumor targeted CCNG1 inhibitor, demonstrates clinical benefit in advanced chemoresistant sarcoma following FDA CBER authorization for use as platform therapy with FDA approved drugs. In Proceedings of the Connective Tissue Oncology Society Annual Meetings, San Diego, CA, USA, 13–16 November 2024. [Google Scholar]
- Ignacio, J.G.; San Juan, F.; Manalo, R.; Nategh, E.S.; Tamhane, J.; Kantamnei, L.; Chawla, S.P.; Hall, F.L.; Gordon, E.M. The Genevieve Protocol: Phase I/II Evaluation of a Dual Targeted Approach to Cancer Gene Therapy/Immunotherapy. Clin. Oncol. 2018, 3, 1537. [Google Scholar]
- ClinicalTrials.gov. Phase 1 Study of GEN2 in Patients with Advanced Solid Tumors. 2024. Available online: https://clinicaltrials.gov/study/NCT06391918 (accessed on 3 January 2025).
- Muravyeva, A.; Smirnikhina, S. Strategies for Modifying Adenoviral Vectors for Gene Therapy. Int. J. Mol. Sci. 2024, 25, 12461. [Google Scholar] [CrossRef]
- Chawla, S.P.; Tellez, W.A.; Chomoyan, H.; Valencia, C.; Ahari, A.; Omelchenko, N.; Makrievski, S.; Brigham, D.A.; Chua-Alcala, V.; Quon, D.; et al. Activity of TNT: A phase 2 study using talimogene laherparepvec, nivolumab and trabectedin for previously treated patients with advanced sarcomas (NCT# 03886311). Front. Oncol. 2023, 13, 1116937. [Google Scholar] [CrossRef]
- Milas, M.; Yu, D.; Lang, A.; Ge, T.; Feig, B.; El-Naggar, A.K.; Pollock, R.E. Adenovirus-mediated p53 gene therapy inhibits human sarcoma tumorigenicity. Cancer Gene Ther. 2000, 7, 422–429. [Google Scholar] [CrossRef]
- Xia, Y.; Du, Z.; Wang, X.; Li, X. Treatment of Uterine Sarcoma with rAd-p53 (Gendicine) Followed by Chemotherapy: Clinical Study of TP53 Gene Therapy. Hum. Gene Ther. 2018, 29, 242–250. [Google Scholar] [CrossRef]
- Zhang, W.W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, J.M.; Chandler, L.A.; Lin, H.; et al. The First Approved Gene Therapy Product for Cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef]
- Xiao, S.W.; Xu, Y.Z.; Xiao, B.F.; Jiang, J.; Liu, C.Q.; Fang, Z.W.; Li, D.M.; Li, X.F.; Cai, Y.; Li, Y.H.; et al. Recombinant Adenovirus-p53 Gene Therapy for Advanced Unresectable Soft-Tissue Sarcomas. Hum. Gene Ther. 2018, 29, 699–707. [Google Scholar] [CrossRef] [PubMed]
- Kesari, S.; Bessudo, A.; Gastman, B.R.; Conley, A.P.; Villaflor, V.M.; Nabell, L.M.; Madere, D.; Chacon, E.; Spencer, C.; Li, L.; et al. BETA PRIME: Phase I study of AdAPT-001 as monotherapy and combined with a checkpoint inhibitor in superficially accessible, treatment-refractory solid tumors. Future Oncol. 2022, 18, 3245–3254. [Google Scholar] [CrossRef] [PubMed]
- Conley, A.P.; Ravi, V.; Roland, C.L.; Reid, T.R.; Larson, C.; Caroen, S.; Williams, J.A.; Oronsky, B.; Stirn, M.; Abrouk, N.; et al. Phase 1/2 study of the TGF-β-trap-enhanced oncolytic adenovirus, AdAPT-001, plus an immune checkpoint inhibitor for patients with immune refractory cancers. J. Clin. Oncol. 2024, 42, 2506. [Google Scholar] [CrossRef]
- Reid, T.R.; Oronsky, B.; Williams, J.; Caroen, S.; Conley, A. TGF-β trap of AdAPT-001 turns up the heat on tumors and turns down checkpoint blocker resistance. J. Immunother. Cancer 2024, 12, e009613. [Google Scholar] [CrossRef]
- Gene, Cell, + RNA Therapy Landscape Report, American Society of Gene & Cell Therapy and Citeline. 2024. Available online: https://www.asgct.org/global/documents/asgct-citeline-q2-2024-report.aspx (accessed on 14 December 2024).
- Shin, M.H.; Kim, J.; Lim, S.A.; Kim, J.; Kim, S.J.; Lee, K.M. NK Cell-Based Immunotherapies in Cancer. Immune Netw. 2020, 20, e14. [Google Scholar] [CrossRef]
- Lachota, M.; Vincenti, M.; Winiarska, M.; Boye, K.; Zagożdżon, R.; Malmberg, K.J. Prospects for NK Cell Therapy of Sarcoma. Cancers 2020, 12, 3719. [Google Scholar] [CrossRef]
- NK Gen Biotech, Inc. SNK Technology. 2022. Available online: https://nkgenbiotech.com/our-technology/#snk (accessed on 6 March 2025).
- Wood, G.E.; Meyer, C.; Petitprez, F.; D’Angelo, S.P. Immunotherapy in Sarcoma: Current Data and Promising Strategies. American Society of Clinical Oncology educational book. Am. Soc. Clin. Oncol. Annu. Meet. 2024, 44, e432234. [Google Scholar] [CrossRef]
- Poznanski, S.M.; Ritchie, T.M.; Fan, I.Y.; El-Sayes, A.; Portillo, A.L.; Ben-Avi, R.; Rojas, E.A.; Chew, M.V.; Shargall, Y.; Ashkar, A.A. Expanded human NK cells from lung cancer patients sensitize patients’ PDL1-negative tumors to PD1-blockade therapy. J. Immunother. Cancer 2021, 9, e001933. [Google Scholar] [CrossRef]
- Gordon, E.M.; Chawla, S.P.; Chua-Alcala, V.S.; Brigham, D.A.; Ahari, A.; Younesi, E.; Valencia, C.; Chang, P.Y.; Song, P.Y. Case Reports, Literature Review and Future Perspectives. J. Cancer Res. Cell Ther. 2022, 6, 126. [Google Scholar] [CrossRef]
- Chawla, S.P.; Chua, V.S.; Gordon, E.M.; Kim, T.T.; Feske, W.; Gibson, B.L.; Chang, P.Y.; Robinson, D.; Song, P.Y. Interim analysis of a phase I study of SNK01 (Autologous Nongenetically Modified Natural Killer Cells with Enhanced Cytotoxicity) and avelumab in advanced refractory sarcoma. J. Clin. Oncol. 2022, 40, 11517. [Google Scholar] [CrossRef]
- Stamatopoulos, A.; Stamatopoulos, T.; Gamie, Z.; Kenanidis, E.; Ribeiro, R.D.C.; Rankin, K.S.; Gerrand, C.; Dalgarno, K.; Tsiridis, E. Mesenchymal stromal cells for bone sarcoma treatment: Roadmap to clinical practice. J. Bone Oncol. 2019, 16, 100231. [Google Scholar] [CrossRef] [PubMed]
- Spaeth, E.; Klopp, A.; Dembinski, J.; Andreeff, M.; Marini, F. Inflammation and tumor microenvironments: Defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008, 15, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Loebinger, M.R.; Eddaoudi, A.; Davies, D.; Janes, S.M. Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer. Cancer Res. 2009, 69, 4134–4142. [Google Scholar] [CrossRef] [PubMed]
- Theoleyre, S.; Wittrant, Y.; Tat, S.K.; Fortun, Y.; Redini, F.; Heymann, D. The molecular triad OPG/RANK/RANKL: Involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004, 15, 457–475. [Google Scholar] [CrossRef]
- Kucerova, L.; Altanerova, V.; Matuskova, M.; Tyciakova, S.; Altaner, C. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res. 2007, 67, 6304–6313. [Google Scholar] [CrossRef]
- Zhou, Z.; Lafleur, E.A.; Koshkina, N.V.; Worth, L.L.; Lester, M.S.; Kleinerman, E.S. Interleukin-12 up-regulates Fas expression in human osteosarcoma and Ewing’s sarcoma cells by enhancing its promoter activity. Mol. Cancer Res. 2005, 3, 685–691. [Google Scholar] [CrossRef]
- Uscanga-Palomeque, A.C.; Chávez-Escamilla, A.K.; Alvizo-Báez, C.A.; Saavedra-Alonso, S.; Terrazas-Armendáriz, L.D.; Tamez-Guerra, R.S.; Rodríguez-Padilla, C.; Alcocer-González, J.M. CAR-T Cell Therapy: From the Shop to Cancer Therapy. Int. J. Mol. Sci. 2023, 24, 15688. [Google Scholar] [CrossRef]
- Prommersberger, S.; Hudecek, M.; Nerreter, T. Antibody-Based CAR T Cells Produced by Lentiviral Transduction. Curr. Protoc. Immunol. 2020, 128, e93. [Google Scholar] [CrossRef]
- Tomasik, J.; Jasiński, M.; Basak, G.W. Next generations of CAR-T cells—New therapeutic opportunities in hematology? Front. Immunol. 2022, 13, 1034707. [Google Scholar] [CrossRef]
- Gatto, L.; Ricciotti, I.; Tosoni, A.; Di Nunno, V.; Bartolini, S.; Lucia Ranieri, L.; Franceschi, E. CAR-T cells neurotoxicity from consolidated practice in hematological malignancies to fledgling experience in CNS tumors: Fill the gap. Front. Oncol. 2023, 13, 1206983. [Google Scholar] [CrossRef]
- Wang, J.Y.; Wang, L. CAR-T cell therapy: Where are we now, and where are we heading? Blood Sci. 2023, 5, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Dagar, G.; Gupta, A.; Masoodi, T.; Nisar, S.; Merhi, M.; Hashem, S.; Chauhan, R.; Dagar, M.; Mirza, S.; Bagga, P.; et al. Correction: Harnessing the potential of CAR-T cell therapy: Progress, challenges, and future directions in hematological and solid tumor treatments. J. Transl. Med. 2023, 21, 571. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, J.; Che, S.; Zhao, H. CAR-T cell therapy for hematological malignancies: History, status and promise. Heliyon 2023, 9, e21776. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Afamitresgene Autoleucel: First Approval. Mol. Diagn. Ther. 2024, 28, 861–866. [Google Scholar] [CrossRef]
- Hong, D.S.; Van Tine, B.A.; Biswas, S.; McAlpine, C.; Johnson, M.L.; Olszanski, A.J.; Clarke, J.M.; Araujo, D.; Blumenschein, G.R.; Kebriaei, P.; et al. Autologous T cell therapy for MAGE-A4+ solid cancers in HLA-A*02+ patients: A phase 1 trial. Nat. Med. 2023, 29, 104–114. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Araujo, D.M.; Abdul Razak, A.R.; Agulnik, M.; Attia, S.; Blay, J.Y.; Carrasco Garcia, I.; Charlson, J.A.; Choy, E.; Demetri, G.D.; et al. Afamitresgene autoleucel for advanced synovial sarcoma and myxoid round cell liposarcoma (SPEARHEAD-1): An international, open-label, phase 2 trial. Lancet 2024, 403, 1460–1471. [Google Scholar] [CrossRef]
- Hegde, M.; Navai, S.; DeRenzo, C.; Joseph, S.K.; Sanber, K.; Wu, M.; Gad, A.Z.; Janeway, K.A.; Campbell, M.; Mullikin, D.; et al. Autologous HER2-specific CAR T cells after lymphodepletion for advanced sarcoma: A phase 1 trial. Nat. Cancer 2024, 5, 880–894. [Google Scholar] [CrossRef]
- Ebb, D.; Meyers, P.; Grier, H.; Bernstein, M.; Gorlick, R.; Lipshultz, S.E.; Krailo, M.; Devidas, M.; Barkauskas, D.A.; Siegal, G.P.; et al. Phase II trial of trastuzumab in combination with cytotoxic chemotherapy for treatment of metastatic osteosarcoma with human epidermal growth factor receptor 2 overexpression: A report from the children’s oncology group. J. Clin. Oncol. 2012, 30, 2545–2551. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. HER2 Chimeric Antigen Receptor (CAR) T Cells in Combination with Checkpoint Blockade in Patients with Advanced Sarcoma. 2024. Available online: https://clinicaltrials.gov/study/NCT04995003 (accessed on 14 December 2024).
- Hattinger, C.M.; Patrizio, M.P.; Magagnoli, F.; Luppi, S.; Serra, M. An update on emerging drugs in osteosarcoma: Towards tailored therapies? Expert Opin. Emerg. Drugs 2019, 24, 153–171. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. C7R-GD2.CART Cells for Patients with Relapsed or Refractory Neuroblastoma and Other GD2 Positive Cancers (GAIL-N). 2024. Available online: https://clinicaltrials.gov/study/NCT03635632 (accessed on 14 December 2024).
- Morandi, F.; Sabatini, F.; Podestà, M.; Airoldi, I. Immunotherapeutic Strategies for Neuroblastoma: Present, Past and Future. Vaccines 2021, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Tumino, N.; Weber, G.; Besi, F.; Del Bufalo, F.; Bertaina, V.; Paci, P.; Quatrini, L.; Antonucci, L.; Sinibaldi, M.; Quintarelli, C.; et al. Polymorphonuclear myeloid-derived suppressor cells impair the anti-tumor efficacy of GD2.CAR T-cells in patients with neuroblastoma. J. Hematol. Oncol. 2021, 14, 191. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Study of CAR T-Cells Targeting the GD2 with IL-15+iCaspase9 for Relapsed/Refractory Neuroblastoma or Relapsed/Refractory Osteosarcoma. 2024. Available online: https://clinicaltrials.gov/study/NCT03721068 (accessed on 14 December 2024).
- Flugel, C.L.; Majzner, R.G.; Krenciute, G.; Dotti, G.; Riddell, S.R.; Wagner, D.L.; Abou-El-Enein, M. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nature reviews. Clin. Oncol. 2023, 20, 49–62. [Google Scholar] [CrossRef]
- Haroun, G.; Gordon, E.M. DNG64, tumor targeted retrovector encoding a CCNG1 inhibitor, for CAR-T cell therapy induced cytokine release syndrome. Front. Mol. Med. 2024, 4, 1461151. [Google Scholar] [CrossRef]
- Roddie, C.; O’Reilly, M.; Dias Alves Pinto, J.; Vispute, K.; Lowdell, M. Manufacturing chimeric antigen receptor T cells: Issues and challenges. Cytotherapy 2019, 21, 327–340. [Google Scholar] [CrossRef]
- Gyurdieva, A.; Zajic, S.; Chang, Y.F.; Houseman, E.A.; Zhong, S.; Kim, J.; Nathenson, M.; Faitg, T.; Woessner, M.; Turner, D.C.; et al. Biomarker correlates with response to NY-ESO-1 TCR T cells in patients with synovial sarcoma. Nat. Commun. 2022, 13, 5296. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Master Protocol to Assess the Safety and Antitumor Activity of Genetically Engineered T Cells in NY-ESO-1 and/or LAGE-1a Positive Solid Tumors (IGNYTE-ESO). 2024. Available online: https://clinicaltrials.gov/study/NCT03967223 (accessed on 3 January 2025).
- D’Angelo, S.P.; Furness, A.J.S.; Thistlethwaite, F.; Burgess, M.A.; Riedel, R.F.; Haanen, J.; Noujaim, J.; Chalmers, A.W.; Pousa, A.L.; Chugh, R.; et al. Lete-cel in patients with synovial sarcoma or myxoid/round cell liposarcoma: Planned interim analysis of the pivotal IGNYTE-ESO trial. J. Clin. Oncol. 2024, 42, 2500. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Noujaim, J.C.; Thistlethwaite, F.; Razak, A.R.A.; Stacchiotti, S.; Chow, W.A.; Haanen, J.B.A.G.; Chalmers, A.W.; Robinson, S.I.; Van Tine, B.A.; et al. IGNYTE-ESO: A master protocol to assess safety and activity of letetresgene autoleucel (lete-cel; GSK3377794) in HLA-A*02+ patients with synovial sarcoma or myxoid/round cell liposarcoma (Substudies 1 and 2). J. Clin. Oncol. 2021, 39, TPS11582. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Pilot Study of Tumor Cell Vaccine for High-risk Solid Tumor Patients Following Stem Cell Transplantation. 2006. Available online: https://clinicaltrials.gov/study/NCT00405327 (accessed on 14 December 2024).
- ClinicalTrials.gov. Dendritic Cell-based Immunotherapy for Advanced Solid Tumours of Children and Young Adults (DEND/TIA). 2014. Available online: https://clinicaltrials.gov/study/NCT02496520 (accessed on 14 December 2024).
- Berneman, Z.N.; Germonpre, P.; Huizing, M.T.; Van de Valede, A.; Nijs, G.; Stein, B.; Van Tendeloo, V.F.; Lion, E.; Smits, E.L.; Anguille, S. Dendritic cell vaccination in malignant pleural mesothelioma: A phase I/II study. J. Clin. Oncol. 2014, 32, 7583. [Google Scholar] [CrossRef]
- Raj, S.; Bui, M.M.; Springett, G.; Conley, A.; Lavilla-Alonso, S.; Zhao, X.; Chen, D.; Haysek, R.; Gonzalez, R.; Letson, G.D.; et al. Long-Term Clinical Responses of Neoadjuvant Dendritic Cell Infusions and Radiation in Soft Tissue Sarcoma. Sarcoma 2015, 2015, 614736. [Google Scholar] [CrossRef]
- Mackall, C.L.; Rhee, E.H.; Read, E.J.; Khuu, H.M.; Leitman, S.F.; Bernstein, D.; Tesso, M.; Long, L.M.; Grindler, D.; Merino, M.; et al. A pilot study of consolidative immunotherapy in patients with high-risk pediatric sarcomas. Clin. Cancer Res. 2008, 14, 4850–4858. [Google Scholar] [CrossRef] [PubMed]
- Dhir, A.; Koru-Sengul, T.; Grosso, J.; D’Amato, G.Z.; Trucco, M.M.; Rosenberg, A.; Gilboa, E.; Goldberg, J.M.; Trent, J.C.; Wilky, B.A. Phase 1 trial of autologous dendritic cell vaccination with imiquimod immunomodulation in children and adults with refractory sarcoma. J. Clin. Oncol. 2021, 39, 11542. [Google Scholar] [CrossRef]
- Mackall, C.; Baird, K.; Bernstein, D.B.; Wood, B.J.; Steinberg, S.B.; Beq, S.; Croughs, T.; Sabatino, M.; Stroncek, D.; Merchant, M.S.; et al. Survival in metastatic Ewing sarcoma (EWS) and rhabdomyosarcoma (RMS) following consolidation immunotherapy with autologous lymphocyte infusion, dendritic cell vaccines ± CYT107 (rhIL-7). J. Clin. Oncol. 2013, 31, 10013. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCT Number | Status/Phase | Sarcoma/Histologic Subtype | Therapy | Type of Therapy |
|---|---|---|---|---|
| NCT06126510 | Active, Recruiting Phase 2 | Soft Tissue Sarcoma | Recombinant Oncolytic Herpes Simplex Virus Type 1 (HSV-1) | Oncolytic Therapy |
| NCT06171282 | Active, Recruiting Phase 1 | Osteosarcoma, Sarcoma, Soft Tissue Sarcoma, Bone Tumor | Recombinant Herpes Simplex Virus I (Oncolytic Virus) | Oncolytic Therapy |
| NCT05851456 | Active, Recruiting Phase 1 | Osteosarcoma, Sarcoma, Soft Tissue Sarcoma, Bone Tumor | Recombinant Herpes Simplex Virus I (Oncolytic Virus) | Oncolytic Therapy |
| NCT05296564 | Active, Recruiting Phase ½ | Synovial Sarcoma, Soft Tissue Sarcoma, Melanoma Stage IV, Triple Negative Breast Cancer, Metastatic Cancer | Anti-NY-ESO-1 TCR-Gene Engineered Lymphocytes | Gene-Modified Cell Therapy |
| NCT05621668 | Active, Recruiting Phase 1 | Soft Tissue Sarcoma, Bone Sarcoma | T-Cell Membrane-Anchored Tumor Targeted IL-12 (Attil-12) | Gene-Modified Cell Therapy |
| NCT05549921 | Active, Recruiting Phase 2 | Soft Tissue Sarcoma | TAEST16001 cells (T cells targeting NY-ESO-1) | Gene-Modified Cell Therapy |
| NCT04897321 | Active, Recruiting Phase 1 | Ewing Sarcoma, Soft Tissue Sarcoma, Clear Cell Sarcoma, Pediatric Solid Tumor, Osteosarcoma | Autologous T cells genetically engineered to express B7-H3-CARs | Gene-Modified Cell Therapy |
| NCT04044768 | Active, Recruiting Phase 2 | Advanced Synovial Sarcoma, Myxoid/Round Cell Liposarcoma | ADP-A2M4 (Afamitresgene autoleucel SPEAR™ T cells) | Gene-Modified Cell Therapy |
| NCT05642455 | Active, Recruiting Phase ½ | Synovial Sarcoma, Malignant Peripheral Nerve Sheath Tumor (MPNST), Osteosarcoma | Afamitresgene autoleucel | Gene-Modified Cell Therapy |
| NCT04995003 | Active, Recruiting Phase 1 | Sarcoma with HER-2 Overexpression, including Osteosarcoma, Rhabdomyosarcoma, Ewing Sarcoma, Synovial Sarcoma, Soft Tissue Sarcoma, Undifferentiated Sarcoma | HER2-CAR T cells + Immune Checkpoint Inhibitor (Pembrolizumab/Nivolumab) | Gene-Modified Cell Therapy |
| NCT05607095 | Active, Recruiting Phase 1 | Undifferentiated Pleomorphic Sarcoma, Dedifferentiated Liposarcoma | Autologous Tumor Infiltrating Lymphocytes (LN-144 or LN-145) | Cellular Therapy |
| NCT05952310 | Active, Recruiting Phase 1/2 | Malignant Sarcoma | Allogeneic Haploidentical NK Cells + Chemotherapy/Radiotherapy | Cellular Therapy |
| NCT04074564 | Active, Recruiting Phase 1 | Soft Tissue Sarcoma, Osteosarcoma | Multi-antigen Autologous Immune Cell Injection | Cellular Therapy |
| NCT05634369 | Active, Recruiting Phase 1/2 | Refractory or Relapsed Pediatric Sarcoma | Infusion of TGFβ Imprinted NK cells | Cellular Therapy |
| NCT Number | Study Status | Clinical Indication | Therapy | Type of Therapy |
|---|---|---|---|---|
| NCT02736565 | COMPLETED | Metastatic Ewing’s tumor, Ewing Family of Tumors, Recurrent and Advanced Ewing’s Sarcoma, Sarcomas | pbi-shRNA‚ EWS/FLI1 Type 1 Lipoplex | Gene Therapy |
| NCT05578820 | COMPLETED | Sarcoma and other solid tumors | Stimotimagene copolymerplasmid | Gene Therapy |
| NCT00258687 | COMPLETED | Clear cell sarcoma, Alveolar Soft Part Sarcoma, and other solid tumors | GM-CSF gene-transduced tumor vaccine (GVAX) | Gene Therapy |
| NCT03725605 | COMPLETED | Soft Tissue Sarcoma | LTX-315 and Tumor-infiltrating lymphocytes | Oncolytic-peptide Therapy |
| NCT04318964 | ACTIVE_NOT_RECRUITING | Soft Tissue Sarcoma | TCR affinity-enhanced specific T cell therapy (TAEST16001 cells) | Gene-Modified Cell Therapy |
| NCT02319824 | COMPLETED | Sarcoma | Autologous NY-ESO-1-specific CD8-positive T Lymphocytes | Gene-Modified Cell Therapy |
| NCT03250325 | COMPLETED | Synovial Sarcoma | TBI-1301 gene-transduced T cell therapy | Gene-Modified Cell Therapy |
| NCT01477021 | COMPLETED | Liposarcoma, Synovial Sarcoma, Recurrent and Stage III-IV Adult Soft Tissue Sarcoma | NY-ESO-1-specific T cells | Gene-Modified Cell Therapy |
| NCT03132922 | ACTIVE_NOT_RECRUITING | Synovial Sarcoma and Myxoid Round Cell Liposarcoma | Autologous genetically modified MAGE-A4 c1032 T cells | Gene-Modified Cell Therapy |
| NCT02650986 | ACTIVE_NOT_RECRUITING | Advanced Synovial Sarcoma, Unresectable Synovial Sarcoma, and other solid tumors | Autologous NY-ESO-1 TCR/dnTGFbetaRII transgenic T cells | Gene-Modified Cell Therapy |
| NCT02992743 | COMPLETED | Neoplasms | Letetresgene autoleucel (GSK3377794) | Gene-Modified Cell Therapy |
| NCT05993299 | ACTIVE_NOT_RECRUITING | Neoplasms | Letetresgene autoleucel | Gene-Modified Cell Therapy |
| NCT03462316 | ACTIVE_NOT_RECRUITING | Bone Sarcoma, Soft Tissue Sarcoma | NY-ESO-1 TCR Affinity Enhancing Specific T cell Therapy | Gene-Modified Cell Therapy |
| NCT01241162 | COMPLETED | Neuroblastoma, Ewing’s Sarcoma, Osteogenic Sarcoma, Rhabdomyosarcoma, Synovial Sarcoma | Autologous dendritic cell vaccine with adjuvant | Cellular Therapy |
| NCT00003081 | COMPLETED | Sarcoma | Peripheral blood stem cell transplantation | Cellular Therapy |
| NCT04802070 | COMPLETED | Sarcoma | Autologous cytokine-induced killer cells | Cellular Therapy |
| NCT04052334 | COMPLETED | Sarcoma | Tumor-infiltrating lymphocytes | Cellular Therapy |
| NCT00357396 | COMPLETED | Sarcoma | T-Cell Depleted Allogeneic Hematopoietic Stem Cell Transplant | Cellular Therapy |
| NCT00043979 | COMPLETED | Sarcoma | Therapeutic allogeneic lymphocytes, peripheral blood stem cell transplantation | Cellular Therapy |
| NCT00245011 | COMPLETED | Sarcoma | Sm-EDTMP, peripheral blood stem cell transplantation | Cellular Therapy |
| NCT01347034 | COMPLETED | Soft Tissue Sarcoma | Autologous Dendritic Cells | Cellular Therapy |
| NCT02472392 | COMPLETED | Ewing Sarcoma | Allogeneic Stem Cell Transplantation | Cellular Therapy |
| NCT02100891 | COMPLETED | Ewing Sarcoma, Neuroblastoma, Rhabdomyosarcoma, Osteosarcoma, and CNS Tumors | HLA-haploidentical hematopoietic cell transplantation and Donor NK Cell Infusion | Cellular Therapy |
| NCT02890758 | COMPLETED | Soft Tissue Sarcoma, Ewing’s Sarcoma, Rhabdomyosarcoma, and other hematologic and solid tumors | Natural Killer (NK) Cells and ALT803 | Cellular Therapy |
| NCT02849366 | COMPLETED | Recurrent Adult Soft Tissue Sarcoma | Natural Killer Immunotherapy | Cellular Therapy |
| NCT01522820 | COMPLETED | Sarcoma and other tumors. | DEC-205/NY-ESO-1 Fusion Protein CDX-1401 | Cellular Therapy |
| NCT00948961 | COMPLETED | Advanced Malignancies | CDX 1401 dendritic cell | Cellular Therapy |
| Gene Therapy | Cell Therapy | Chemotherapy | Targeted Therapy | Immunotherapy | Radiation Therapy | |
|---|---|---|---|---|---|---|
| Mechanism of action | Transfers a cytocidal gene into cancer cells | Transplants healthy cells to enhance immunity, bone marrow recovery | Kills both healthy and normal cells | Targets specific molecules or receptors overexpressed in cancer cells | Stimulates immune system to attack cancer cells | Kills cancer cells using high-energy radiation |
| Specificity, Yes/No | Yes | Yes | No | Yes | Yes | No |
| Adverse Reactions/Short-term/Long Term | Short Term: None to minimal (Retroviral-based) to moderate to severe (CRS, neurotoxicity, cytopenias in CAR-T, TCR-T) Long Term: None to Unknown | Short Term: Minimal, infusional reactions to Severe: Cytopenias if conditioning regimens are used Long Term: Second malignancies | Short Term: Moderate to severe bone marrow suppression, organ dysfunction Long Term: Second malignancies, irreversible organ damage | Short Term: Usually mild to moderate, to severe (Interstitial lung disease) Long Term: Unknown | Short Term: Mild to severe, immune-related Long Term: Autoimmune disorders, Irreversible organ | Short Term: Mild to severe organ damage Long Term: Second malignancies Irreversible organ damage |
| Relative Cost ($) | $$$ to $$$$ | $$$ | $ | $$$ | $$$ | $$ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chawla, S.P.; Pang, S.S.; Jain, D.; Jeffrey, S.; Chawla, N.S.; Song, P.Y.; Hall, F.L.; Gordon, E.M. Gene and Cell Therapy for Sarcomas: A Review. Cancers 2025, 17, 1125. https://doi.org/10.3390/cancers17071125
Chawla SP, Pang SS, Jain D, Jeffrey S, Chawla NS, Song PY, Hall FL, Gordon EM. Gene and Cell Therapy for Sarcomas: A Review. Cancers. 2025; 17(7):1125. https://doi.org/10.3390/cancers17071125
Chicago/Turabian StyleChawla, Sant P., Skyler S. Pang, Darshit Jain, Samantha Jeffrey, Neal S. Chawla, Paul Y. Song, Frederick L. Hall, and Erlinda M. Gordon. 2025. "Gene and Cell Therapy for Sarcomas: A Review" Cancers 17, no. 7: 1125. https://doi.org/10.3390/cancers17071125
APA StyleChawla, S. P., Pang, S. S., Jain, D., Jeffrey, S., Chawla, N. S., Song, P. Y., Hall, F. L., & Gordon, E. M. (2025). Gene and Cell Therapy for Sarcomas: A Review. Cancers, 17(7), 1125. https://doi.org/10.3390/cancers17071125