
Camptothein-Based Anti-Cancer Therapies and Strategies to Improve Their Therapeutic Index


Simple Summary
Abstract
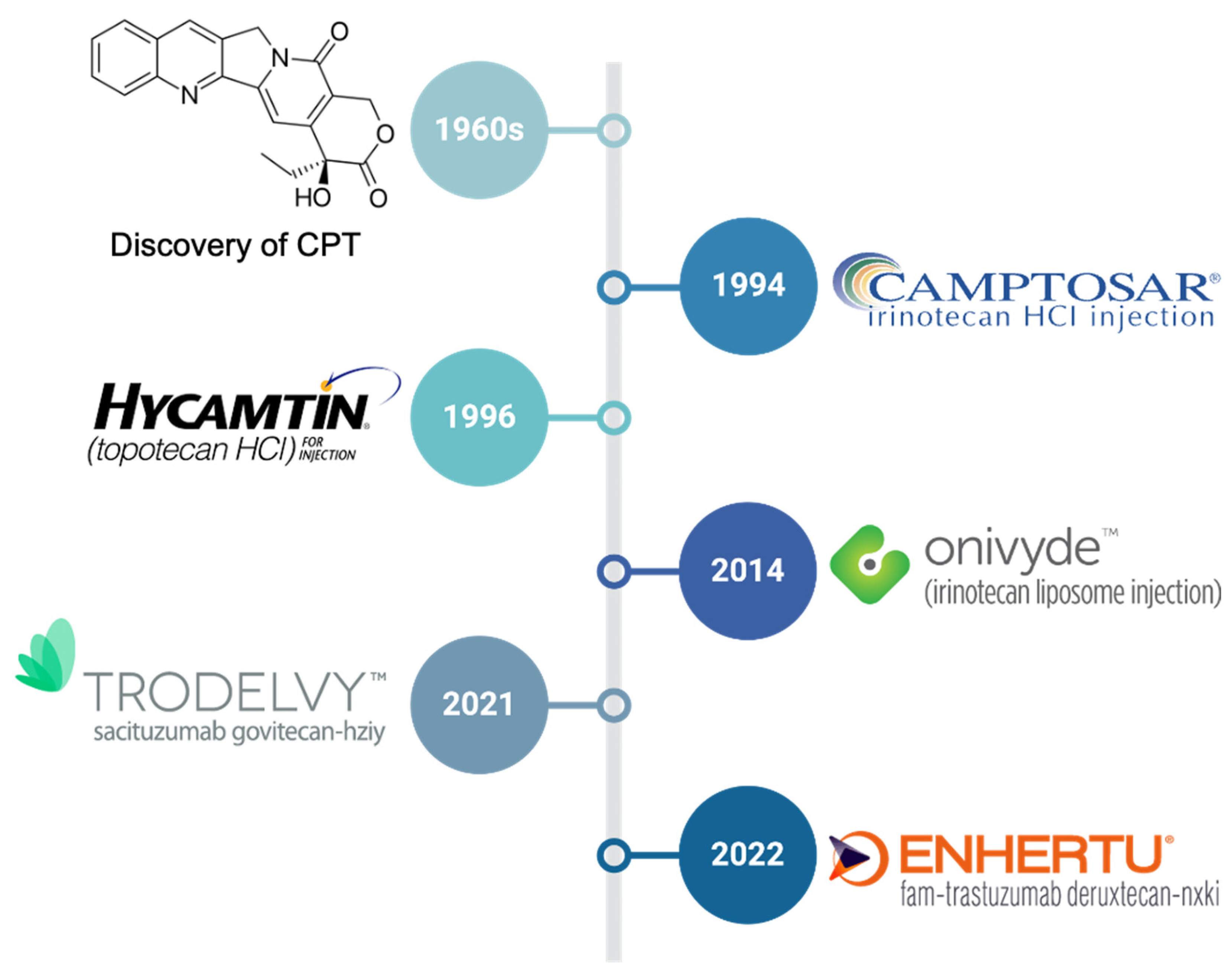
1. Introduction
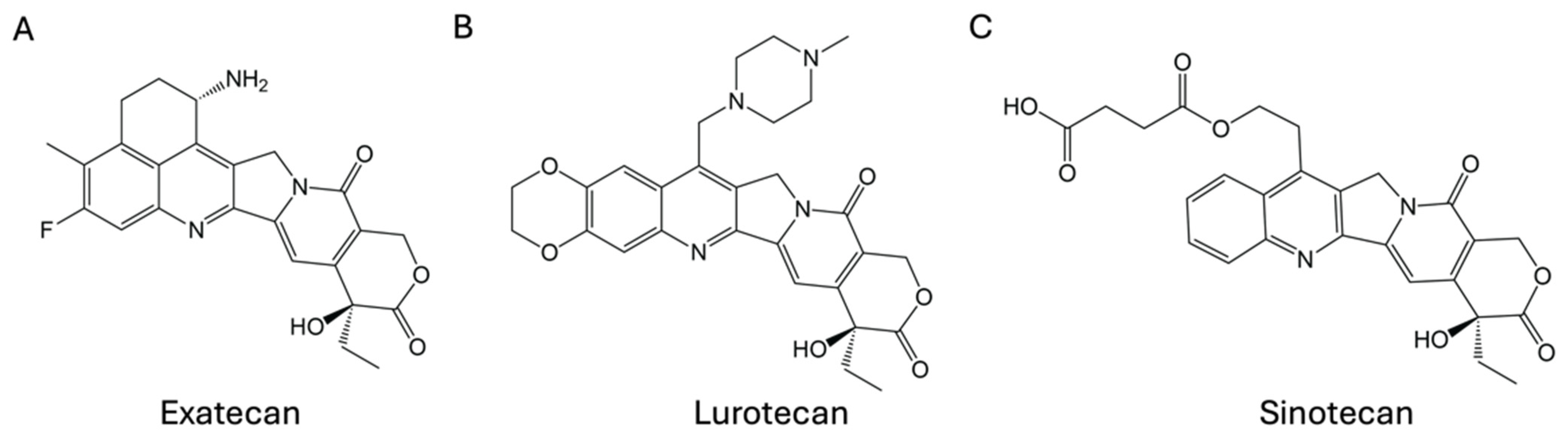
2. Small-Molecule CPT Derivatives
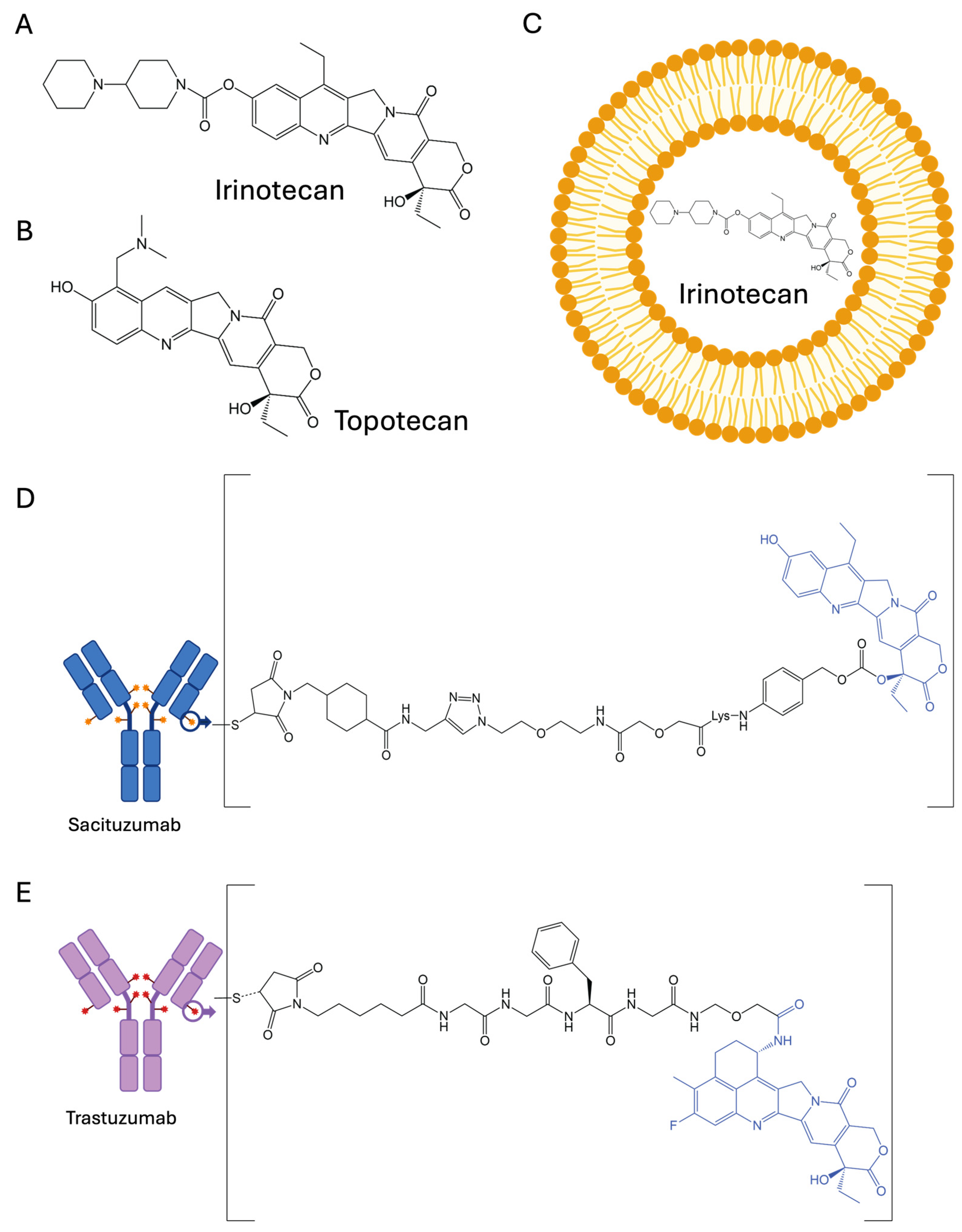
2.1. Irinotecan (CPT-11)
2.2. Topotecan
3. Application of Delivery Systems to Enhance Safety and Efficacy
3.1. Liposomes and Polymeric Nanoparticles
3.2. Antibody–Drug Conjugates
4. Optimization Strategies Under Development
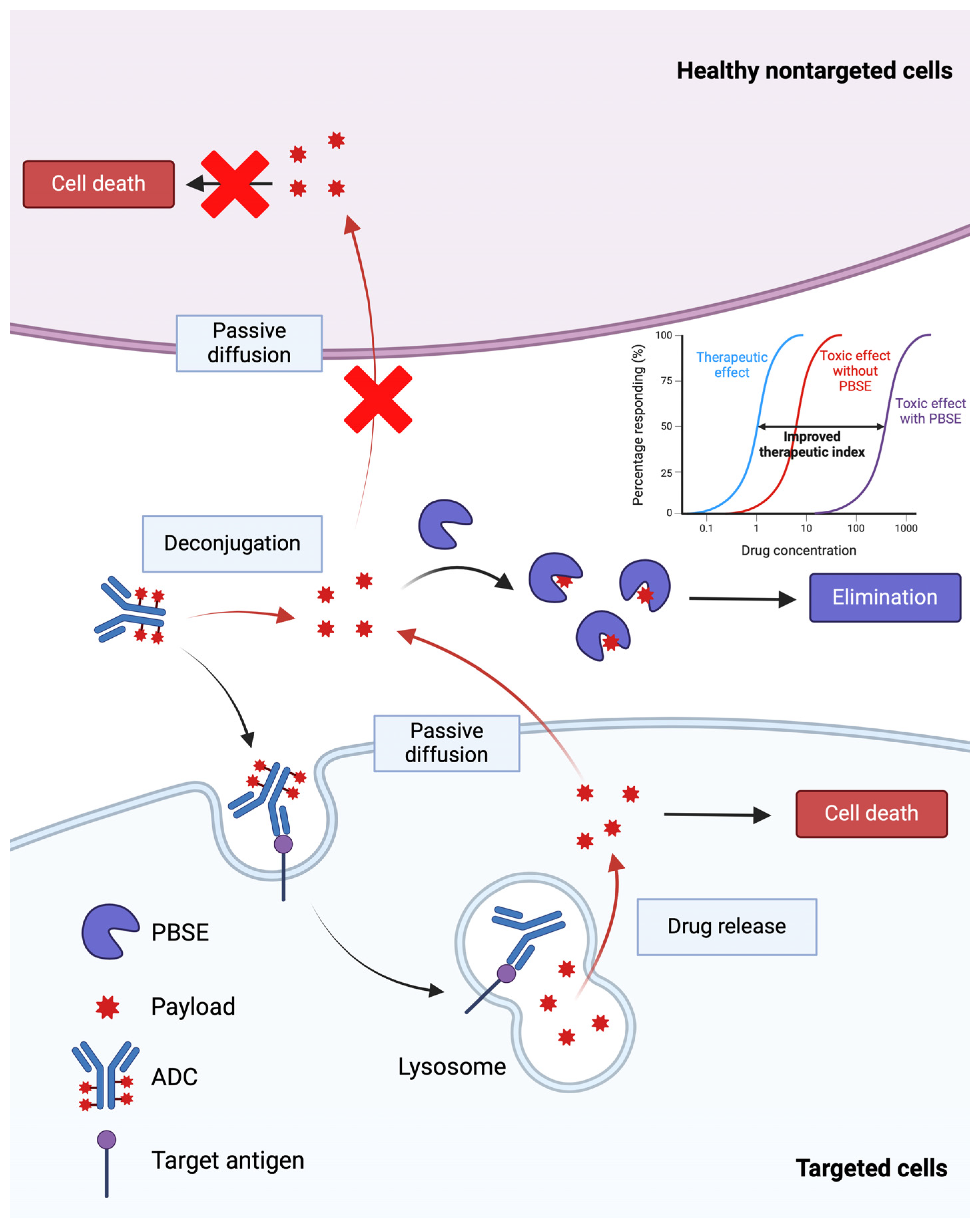
4.1. Inverse Targeting
4.1.1. Small-Molecule CPT Derivatives
4.1.2. Inverse Targeting to Enhance the Selectivity of ADC Therapy
4.2. Application of Adjuvants to Increase Tumor Distribution of ADCs
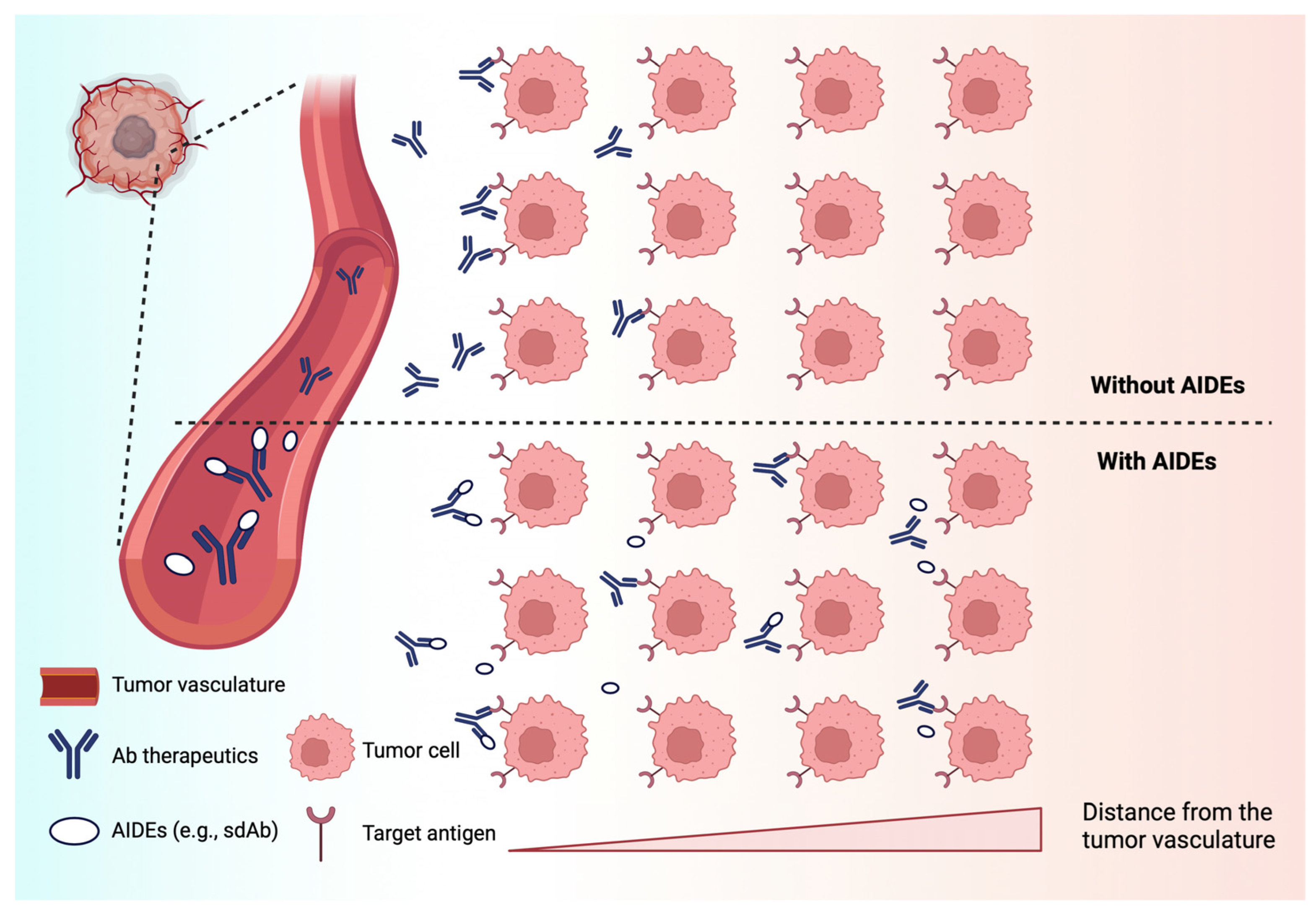
4.2.1. Anti-Idiotypic Distribution Enhancers
4.2.2. Co-Administration of “Naked” Antibody with ADCs
4.2.3. Engineering Fragment–Drug Conjugates (FDCs)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wall, M.E.; Wani, M.C.; Cook, C.E.; Palmer, K.H.; McPhail, A.T.; Sim, G.A. Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloidal Leukemia and Tumor Inhibitor from Camptotheca acuminata1,2. J. Am. Chem. Soc. 1966, 88, 3888–3890. [Google Scholar] [CrossRef]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Basili, S.; Moro, S. Novel camptothecin derivatives as topoisomerase I inhibitors. Expert Opin. Ther. Pat. 2009, 19, 555–574. [Google Scholar] [CrossRef]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985, 260, 14873–14878. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, Y.H.; Liu, L.F. Identification of mammalian DNA topoisomerase I as an intracellular target of the anticancer drug camptothecin. Cancer Res. 1988, 48, 1722–1726. [Google Scholar]
- Moertel, C.G.; Schutt, A.J.; Reitemeier, R.J.; Hahn, R.G. Phase II study of camptothecin (NSC-100880) in the treatment of advanced gastrointestinal cancer. Cancer Chemother. Rep. 1972, 56, 95–101. [Google Scholar]
- Li, F.; Jiang, T.; Li, Q.; Ling, X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: Did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am. J. Cancer Res. 2017, 7, 2350–2394. [Google Scholar]
- Chen, Z.; Liu, M.; Wang, N.; Xiao, W.; Shi, J. Unleashing the Potential of Camptothecin: Exploring Innovative Strategies for Structural Modification and Therapeutic Advancements. J. Med. Chem. 2024, 67, 3244–3273. [Google Scholar] [CrossRef]
- Fan, X.; Lin, X.; Ruan, Q.; Wang, J.; Yang, Y.; Sheng, M.; Zhou, W.; Kai, G.; Hao, X. Research progress on the biosynthesis and metabolic engineering of the anti-cancer drug camptothecin in Camptotheca acuminate. Ind. Crops Prod. 2022, 186, 115270. [Google Scholar] [CrossRef]
- ten Bokkel Huinink, W.; Gore, M.; Carmichael, J.; Gordon, A.; Malfetano, J.; Hudson, I.; Broom, C.; Scarabelli, C.; Davidson, N.; Spanczynski, M.; et al. Topotecan versus paclitaxel for the treatment of recurrent epithelial ovarian cancer. J. Clin. Oncol. 1997, 15, 2183–2193. [Google Scholar] [CrossRef]
- Cunningham, D.; Pyrhönen, S.; James, R.D.; Punt, C.J.; Hickish, T.F.; Heikkila, R.; Johannesen, T.B.; Starkhammar, H.; Topham, C.A.; Awad, L.; et al. Randomised trial of irinotecan plus supportive care versus supportive care alone after fluorouracil failure for patients with metastatic colorectal cancer. Lancet 1998, 352, 1413–1418. [Google Scholar] [CrossRef]
- Rougier, P.; Van Cutsem, E.; Bajetta, E.; Niederle, N.; Possinger, K.; Labianca, R.; Navarro, M.; Morant, R.; Bleiberg, H.; Wils, J.; et al. Randomised trial of irinotecan versus fluorouracil by continuous infusion after fluorouracil failure in patients with metastatic colorectal cancer. Lancet 1998, 352, 1407–1412. [Google Scholar] [CrossRef]
- Braybrooke, J.P.; Ranson, M.; Manegold, C.; Mattson, K.; Thatcher, N.; Cheverton, P.; Sekiguchi, M.; Suzuki, M.; Oyama, R.; Talbot, D.C. Phase II study of exatecan mesylate (DX-8951f) as first line therapy for advanced non-small cell lung cancer. Lung Cancer 2003, 41, 215–219. [Google Scholar] [CrossRef]
- Duffaud, F.; Borner, M.; Chollet, P.; Vermorken, J.B.; Bloch, J.; Degardin, M.; Rolland, F.; Dittrich, C.; Baron, B.; Lacombe, D.; et al. Phase II study of OSI-211 (liposomal lurtotecan) in patients with metastatic or loco-regional recurrent squamous cell carcinoma of the head and neck: An EORTC New Drug Development Group Study. Eur. J. Cancer 2004, 40, 2748–2752. [Google Scholar] [CrossRef]
- Seiden, M.V.; Muggia, F.; Astrow, A.; Matulonis, U.; Campos, S.; Roche, M.; Sivret, J.; Rusk, J.; Barrett, E. A phase II study of liposomal lurtotecan (OSI-211) in patients with topotecan resistant ovarian cancer. Gynecol. Oncol. 2004, 93, 229–232. [Google Scholar] [CrossRef]
- Schöffski, P.; Wang, C.-C.; Schöffski, M.P.; Wozniak, A. Current role of topoisomerase I inhibitors for the treatment of mesenchymal malignancies and their potential future use as payload of sarcoma-specific antibody-drug conjugates. Oncol. Res. Treat. 2024, 47, 18–41. [Google Scholar] [CrossRef]
- Alshammari, M.K.; Alshehri, M.M.; Alshehri, A.M.; Alshlali, O.M.; Mahzari, A.M.; Almalki, H.H.; Kulaybi, O.Y.; Alghazwni, M.K.; Kamal, M.; Imran, M. Camptothecin loaded nano-delivery systems in the cancer therapeutic domains: A critical examination of the literature. J. Drug Deliv. Sci. Technol. 2023, 79, 104034. [Google Scholar] [CrossRef]
- Keam, S.J. Trastuzumab Deruxtecan: First Approval. Drugs 2020, 80, 501–508. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sacituzumab Govitecan: First Approval. Drugs 2020, 80, 1019–1025. [Google Scholar] [CrossRef]
- Ohe, Y.; Sasaki, Y.; Shinkai, T.; Eguchi, K.; Tamura, T.; Kojima, A.; Kunikane, H.; Okamoto, H.; Karato, A.; Ohmatsu, H.; et al. Phase I study and pharmacokinetics of CPT-11 with 5-day continuous infusion. J. Natl. Cancer Inst. 1992, 84, 972–974. [Google Scholar] [CrossRef]
- Negoro, S.; Fukuoka, M.; Masuda, N.; Takada, M.; Kusunoki, Y.; Matsui, K.; Takifuji, N.; Kudoh, S.; Niitani, H.; Taguchi, T. Phase I study of weekly intravenous infusions of CPT-11, a new derivative of camptothecin, in the treatment of advanced non-small-cell lung cancer. J. Natl. Cancer Inst. 1991, 83, 1164–1168. [Google Scholar] [CrossRef]
- Kawato, Y.; Aonuma, M.; Hirota, Y.; Kuga, H.; Sato, K. Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res. 1991, 51, 4187–4191. [Google Scholar]
- Gupta, E.; Lestingi, T.M.; Mick, R.; Ramirez, J.; Vokes, E.E.; Ratain, M.J. Metabolic fate of irinotecan in humans: Correlation of glucuronidation with diarrhea. Cancer Res. 1994, 54, 3723–3725. [Google Scholar]
- Iyer, L.; Das, S.; Janisch, L.; Wen, M.; Ramírez, J.; Karrison, T.; Fleming, G.F.; Vokes, E.E.; Schilsky, R.L.; Ratain, M.J. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002, 2, 43–47. [Google Scholar] [CrossRef]
- Conti, J.A.; Kemeny, N.E.; Saltz, L.B.; Huang, Y.; Tong, W.P.; Chou, T.-C.; Sun, M.; Pulliam, S.; Gonzalez, C. Irinotecan is an active agent in untreated patients with metastatic colorectal cancer. J. Clin. Oncol. 1996, 14, 709–715. [Google Scholar] [CrossRef]
- Akiyama, Y.; Fujita, K.; Nagashima, F.; Yamamoto, W.; Endo, H.; Sunakawa, Y.; Yamashita, K.; Ishida, H.; Mizuno, K.; Araki, K.; et al. Genetic testing for UGT1A1*28 and *6 in Japanese patients who receive irinotecan chemotherapy. Ann. Oncol. 2008, 19, 2089–2090. [Google Scholar] [CrossRef]
- Ando, Y.; Saka, H.; Ando, M.; Sawa, T.; Muro, K.; Ueoka, H.; Yokoyama, A.; Saitoh, S.; Shimokata, K.; Hasegawa, Y. Polymorphisms of UDP-Glucuronosyltransferase Gene and Irinotecan Toxicity: A Pharmacogenetic Analysis1. Cancer Res. 2000, 60, 6921–6926. [Google Scholar]
- Ychou, M.; Raoul, J.; Desseigne, F.; Borel, C.; Caroli-Bosc, F.; Jacob, J.; Seitz, J.; Kramar, A.; Hua, A.; Lefebvre, P. High-dose, single-agent irinotecan as first-line therapy in the treatment of metastatic colorectal cancer. Cancer Chemother. Pharmacol. 2002, 50, 383–391. [Google Scholar]
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E.; Marcello, J.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Sampson, J. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J. Clin. Oncol. 2007, 25, 4722–4729. [Google Scholar] [CrossRef]
- Kumanishi, R.; Mitani, S.; Kadowaki, S.; Matsushima, T.; Takahashi, N.; Ogata, T.; Yasui, H.; Ogata, M.; Satake, H.; Narita, Y. Efficacy and Safety of Nivolumab and Irinotecan as Third-Line Chemotherapy for Advanced Gastric Cancer: A Multi-Institutional Retrospective Study; American Society of Clinical Oncology: Alexandria, VA, USA, 2020. [Google Scholar]
- Kim, R.; Chaves, J.; Kavan, P.; Fakih, M.; Kortmansky, J.; Spencer, K.; Wong, L.; Tehfe, M.; Li, J.J.; Lee, M.; et al. 608P—Pembrolizumab (pembro) plus mFOLFOX or FOLFIRI in patients with metastatic colorectal cancer (mCRC): KEYNOTE-651 cohorts B and D. Ann. Oncol. 2019, 30, v229–v230. [Google Scholar] [CrossRef]
- Kümler, I.; Eefsen, R.L.; Sørensen, P.G.; Theile, S.; Fullerton, A.; Nielsen, P.G.; Jensen, B.V.; Nielsen, D.L. An open label phase 1 study evaluation safety, tolerability, and maximum tolerated dose of oral administration of irinotecan in combination with capecitabine. Cancer Chemother. Pharmacol. 2019, 84, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.H.; Kim, K.; Hutson, P.; DeVore, R.; Glick, J.; Stewart, J.; Johnson, D. Phase II study of topotecan in patients with extensive-stage small-cell carcinoma of the lung: An Eastern Cooperative Oncology Group Trial. J. Clin. Oncol. 1996, 14, 2345–2352. [Google Scholar] [CrossRef]
- Clarke-Pearson, D.L.; Van Le, L.; Iveson, T.; Whitney, C.W.; Hanjani, P.; Kristensen, G.; Malfetano, J.H.; Beckman, R.A.; Ross, G.A.; Lane, S.R.; et al. Oral topotecan as single-agent second-line chemotherapy in patients with advanced ovarian cancer. J. Clin. Oncol. 2001, 19, 3967–3975. [Google Scholar] [CrossRef]
- Vennepureddy, A.; Atallah, J.-P.; Terjanian, T. Role of topotecan in non-small cell lung cancer: A review of literature. World J. Oncol. 2015, 6, 429. [Google Scholar] [CrossRef]
- Masuda, N.; Matsui, K.; Negoro, S.; Takeda, K.; Kudoh, S.; Nakagawa, K.; Mukaiyama, A.; Arase, H.; Yoshida, P.; Ijima, T. Phase I and Pharmacologic Study of Weekly Bolus Topotecan for Advanced Non–Small-Cell Lung Cancer. Clin. Lung Cancer 2010, 11, 271–279. [Google Scholar] [CrossRef]
- Raymond, E.; Burris, H.; Rowinsky, E.; Eckardt, J.; Rodriguez, G.; Smith, L.; Weiss, G.; Von Hoff, D. Phase I study of daily times five topotecan and single injection of cisplatin in patients with previously untreated non-small-cell lung carcinoma. Ann. Oncol. 1997, 8, 1003–1008. [Google Scholar] [CrossRef]
- Armstrong, D.K. Topotecan Dosing Guidelines in Ovarian Cancer: Reduction and Management of Hematologic Toxicity. Oncologist 2004, 9, 33–42. [Google Scholar] [CrossRef]
- Hoskins, P.; Eisenhauer, E.; Beare, S.; Roy, M.; Drouin, P.; Stuart, G.; Bryson, P.; Grimshaw, R.; Capstick, V.; Zee, B. Randomized phase II study of two schedules of topotecan in previously treated patients with ovarian cancer: A National Cancer Institute of Canada Clinical Trials Group study. J. Clin. Oncol. 1998, 16, 2233–2237. [Google Scholar] [CrossRef]
- Callegaro-Filho, D.; Kavanagh, J.J.; Nick, A.M.; Ramirez, P.T.; Schmeler, K.M. Sustained complete response after maintenance therapy with topotecan and erlotinib for recurrent cervical cancer with distant metastases. Case Rep. Oncol. 2014, 7, 97–101. [Google Scholar] [CrossRef]
- Desai, P.A.; Takahashi, N.; Lissa, D.; Nichols, S.; Sciuto, L.; Abel, M.L.; Schroeder, B.; Schultz, C.; Steinberg, S.M.; Pinkiert, D. Tazemetostat in Combination with Topotecan and Pembrolizumab in Patients with Recurrent Small Cell Lung Cancer; American Society of Clinical Oncology: Alexandria, VA, USA, 2024. [Google Scholar]
- Hatefi, A.; Amsden, B. Camptothecin delivery methods. Pharm. Res. 2002, 19, 1389–1399. [Google Scholar] [CrossRef]
- Liu, P.; Chen, G.; Zhang, J. A Review of Liposomes as a Drug Delivery System: Current Status of Approved Products, Regulatory Environments, and Future Perspectives. Molecules 2022, 27, 1372. [Google Scholar] [CrossRef] [PubMed]
- Cortesi, R.; Esposito, E.; Maietti, A.; Menegatti, E.; Nastruzzi, C. Formulation study for the antitumor drug camptothecin: Liposomes, micellar solutions and a microemulsion. Int. J. Pharm. 1997, 159, 95–103. [Google Scholar] [CrossRef]
- Maruyama, K. Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv. Drug Deliv. Rev. 2011, 63, 161–169. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; Li, C.-P.; Bodoky, G.; Dean, A.; Shan, Y.-S.; Jameson, G.; Macarulla, T.; Lee, K.-H.; Cunningham, D.; Blanc, J.F. Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): A global, randomised, open-label, phase 3 trial. Lancet 2016, 387, 545–557. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Melisi, D.; Macarulla, T.; Cid, R.P.; Chandana, S.R.; De La Fouchardière, C.; Dean, A.; Kiss, I.; Lee, W.J.; Goetze, T.O. NALIRIFOX versus nab-paclitaxel and gemcitabine in treatment-naive patients with metastatic pancreatic ductal adenocarcinoma (NAPOLI 3): A randomised, open-label, phase 3 trial. Lancet 2023, 402, 1272–1281. [Google Scholar] [CrossRef]
- Kalra, A.V.; Kim, J.; Klinz, S.G.; Paz, N.; Cain, J.; Drummond, D.C.; Nielsen, U.B.; Fitzgerald, J.B. Preclinical Activity of Nanoliposomal Irinotecan Is Governed by Tumor Deposition and Intratumor Prodrug Conversion. Cancer Res. 2014, 74, 7003–7013. [Google Scholar] [CrossRef]
- Roy, A.C.; Park, S.R.; Cunningham, D.; Kang, Y.K.; Chao, Y.; Chen, L.T.; Rees, C.; Lim, H.Y.; Tabernero, J.; Ramos, F.J.; et al. A randomized phase II study of PEP02 (MM-398), irinotecan or docetaxel as a second-line therapy in patients with locally advanced or metastatic gastric or gastro-oesophageal junction adenocarcinoma. Ann. Oncol. 2013, 24, 1567–1573. [Google Scholar] [CrossRef]
- Passero, F.C., Jr.; Grapsa, D.; Syrigos, K.N.; Saif, M.W. The safety and efficacy of Onivyde (irinotecan liposome injection) for the treatment of metastatic pancreatic cancer following gemcitabine-based therapy. Expert Rev. Anticancer Ther. 2016, 16, 697–703. [Google Scholar] [CrossRef]
- Adiwijaya, B.; Kim, J.; Lang, I.; Csõszi, T.; Cubillo, A.; Chen, J.S.; Wong, M.; Park, J.O.; Kim, J.S.; Rau, K.M. Population pharmacokinetics of liposomal irinotecan in patients with cancer. Clin. Pharmacol. Ther. 2017, 102, 997–1005. [Google Scholar] [CrossRef]
- Kraut, E.H.; Fishman, M.N.; Lorusso, P.M.; Gordon, M.S.; Rubin, E.H.; Haas, A.; Fetterly, G.J.; Cullinan, P.; Dul, J.L.; Steinberg, J. Final results of a phase I study of liposome encapsulated SN-38 (LE-SN38): Safety, pharmacogenomics, pharmacokinetics, and tumor response. J. Clin. Oncol. 2005, 23, 2017. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Janku, F.; Moser, J.C.; Fu, S.; Wages, D.S.; Wheeler, C.A.; Mori, M.; Shimoyama, S.; Yamada, N.; Subach, R.A. A First-in-Human Phase 1 Dose Escalation Study of FF-10850 (Liposomal Topotecan) in Patients with Advanced Solid Tumors; American Society of Clinical Oncology: Alexandria, VA, USA, 2022. [Google Scholar]
- Kang, J.; Kumar, V.; Yang, D.; Chowdhury, P.R.; Hohl, R.J. Cyclodextrin complexation: Influence on the solubility, stability, and cytotoxicity of camptothecin, an antineoplastic agent. Eur. J. Pharm. Sci. 2002, 15, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical Applications of Cyclodextrins. 1. Drug Solubilization and Stabilization. J. Pharm. Sci. 1996, 85, 1017–1025. [Google Scholar] [CrossRef]
- González-Ruiz, V.; Cores, Á.; Martín-Cámara, O.; Orellana, K.; Cervera-Carrascón, V.; Michalska, P.; Olives, A.I.; León, R.; Martín, M.A.; Menéndez, J.C. Enhanced Stability and Bioactivity of Natural Anticancer Topoisomerase I Inhibitors through Cyclodextrin Complexation. Pharmaceutics 2021, 13, 1609. [Google Scholar] [CrossRef]
- Ünal, S.; Aktaş, Y.; Benito, J.M.; Bilensoy, E. Cyclodextrin nanoparticle bound oral camptothecin for colorectal cancer: Formulation development and optimization. Int. J. Pharm. 2020, 584, 119468. [Google Scholar] [CrossRef]
- Voss, M.H.; Hussain, A.; Vogelzang, N.; Lee, J.L.; Keam, B.; Rha, S.Y.; Vaishampayan, U.; Harris, W.B.; Richey, S.; Randall, J.M.; et al. A randomized phase II trial of CRLX101 in combination with bevacizumab versus standard of care in patients with advanced renal cell carcinoma. Ann. Oncol. 2017, 28, 2754–2760. [Google Scholar] [CrossRef]
- Weiss, G.J.; Chao, J.; Neidhart, J.D.; Ramanathan, R.K.; Bassett, D.; Neidhart, J.A.; Choi, C.H.J.; Chow, W.; Chung, V.; Forman, S.J.; et al. First-in-human phase 1/2a trial of CRLX101, a cyclodextrin-containing polymer-camptothecin nanopharmaceutical in patients with advanced solid tumor malignancies. Investig. New Drugs 2013, 31, 986–1000. [Google Scholar] [CrossRef]
- Shenderova, A.; Burke, T.G.; Schwendeman, S.P. The Acidic Microclimate in Poly(lactide-co-glycolide) Microspheres Stabilizes Camptothecins. Pharm. Res. 1999, 16, 241–248. [Google Scholar] [CrossRef]
- Dadashzadeh, S.; Derakhshandeh, K.; Shirazi, F.H. 9-Nitrocamptothecin polymeric nanoparticles: Cytotoxicity and pharmacokinetic studies of lactone and total forms of drug in rats. Anti-Cancer Drugs 2008, 19, 805–811. [Google Scholar] [CrossRef]
- Xiao, B.; Si, X.; Han, M.K.; Viennois, E.; Zhang, M.; Merlin, D. Co-delivery of camptothecin and curcumin by cationic polymeric nanoparticles for synergistic colon cancer combination chemotherapy. J. Mater. Chem. B 2015, 3, 7724–7733. [Google Scholar] [CrossRef]
- Zolot, R.S.; Basu, S.; Million, R.P. Antibody–drug conjugates. Nat. Rev. Drug Discov. 2013, 12, 259–260. [Google Scholar] [CrossRef]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody-drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Mecklenburg, L. A Brief Introduction to Antibody–Drug Conjugates for Toxicologic Pathologists. Toxicol. Pathol. 2018, 46, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, E.; Balthasar, J. Monoclonal Antibody Pharmacokinetics and Pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Rugo, H.S.; Bardia, A.; Tolaney, S.M.; Arteaga, C.; Cortes, J.; Sohn, J.; Marmé, F.; Hong, Q.; Delaney, R.J.; Hafeez, A. TROPiCS-02: A Phase III study investigating sacituzumab govitecan in the treatment of HR+/HER2-metastatic breast cancer. Future Oncol. 2020, 16, 705–715. [Google Scholar] [CrossRef]
- Dum, D.; Taherpour, N.; Menz, A.; Höflmayer, D.; Völkel, C.; Hinsch, A.; Gorbokon, N.; Lennartz, M.; Hube-Magg, C.; Fraune, C.; et al. Trophoblast Cell Surface Antigen 2 Expression in Human Tumors: A Tissue Microarray Study on 18,563 Tumors. Pathobiology 2022, 89, 245–258. [Google Scholar] [CrossRef]
- Inamura, K.; Yokouchi, Y.; Kobayashi, M.; Ninomiya, H.; Sakakibara, R.; Subat, S.; Nagano, H.; Nomura, K.; Okumura, S.; Shibutani, T.; et al. Association of tumor TROP2 expression with prognosis varies among lung cancer subtypes. Oncotarget 2017, 8, 28725–28735. [Google Scholar] [CrossRef]
- Trerotola, M.; Cantanelli, P.; Guerra, E.; Tripaldi, R.; Aloisi, A.L.; Bonasera, V.; Lattanzio, R.; Lange, R.d.; Weidle, U.H.; Piantelli, M.; et al. Upregulation of Trop-2 quantitatively stimulates human cancer growth. Oncogene 2013, 32, 222–233. [Google Scholar] [CrossRef]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016, 8, 659–671. [Google Scholar] [CrossRef]
- Masubuchi, N.; May, R.D.; Atsumi, R. A predictive model of human myelotoxicity using five camptothecin derivatives and the in vitro colony-forming unit granulocyte/macrophage assay. Clin. Cancer Res. 2004, 10, 6722–6731. [Google Scholar] [CrossRef]
- Kumazawa, E.; Jimbo, T.; Ochi, Y.; Tohgo, A. Potent and broad antitumor effects of DX-8951f, a water-soluble camptothecin derivative, against various human tumors xenografted in nude mice. Cancer Chemother. Pharmacol. 1998, 42, 210–220. [Google Scholar] [CrossRef]
- Rowinsky, E.K.; Johnson, T.R.; Geyer, C.E., Jr.; Hammond, L.A.; Eckhardt, S.G.; Drengler, R.; Smetzer, L.; Coyle, J.; Rizzo, J.; Schwartz, G.; et al. DX-8951f, a hexacyclic camptothecin analog, on a daily-times-five schedule: A phase I and pharmacokinetic study in patients with advanced solid malignancies. J. Clin. Oncol. 2000, 18, 3151–3163. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2019, 382, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Jänne, P.A.; Baik, C.; Su, W.-C.; Johnson, M.L.; Hayashi, H.; Nishio, M.; Kim, D.-W.; Koczywas, M.; Gold, K.A.; Steuer, C.E.; et al. Efficacy and Safety of Patritumab Deruxtecan (HER3-DXd) in EGFR Inhibitor–Resistant, EGFR-Mutated Non–Small Cell Lung Cancer. Cancer Discov. 2022, 12, 74–89. [Google Scholar] [CrossRef]
- Yu, H.A.; Goto, Y.; Hayashi, H.; Felip, E.; Yang, J.C.-H.; Reck, M.; Yoh, K.; Lee, S.-H.; Paz-Ares, L.; Besse, B.; et al. HERTHENA-Lung01, a Phase II Trial of Patritumab Deruxtecan (HER3-DXd) in Epidermal Growth Factor Receptor–Mutated Non–Small-Cell Lung Cancer After Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Therapy and Platinum-Based Chemotherapy. J. Clin. Oncol. 2023, 41, 5363–5375. [Google Scholar] [CrossRef]
- Bardia, A.; Krop, I.E.; Kogawa, T.; Juric, D.; Tolcher, A.W.; Hamilton, E.P.; Mukohara, T.; Lisberg, A.; Shimizu, T.; Spira, A.I.; et al. Datopotamab Deruxtecan in Advanced or Metastatic HR+/HER2– and Triple-Negative Breast Cancer: Results From the Phase I TROPION-PanTumor01 Study. J. Clin. Oncol. 2024, 42, 2281–2294. [Google Scholar] [CrossRef]
- Okajima, D.; Yasuda, S.; Maejima, T.; Karibe, T.; Sakurai, K.; Aida, T.; Toki, T.; Yamaguchi, J.; Kitamura, M.; Kamei, R.; et al. Datopotamab Deruxtecan, a Novel TROP2-directed Antibody–drug Conjugate, Demonstrates Potent Antitumor Activity by Efficient Drug Delivery to Tumor Cells. Mol. Cancer Ther. 2021, 20, 2329–2340. [Google Scholar] [CrossRef]
- Ahn, M.J.; Lisberg, A.; Paz-Ares, L.; Cornelissen, R.; Girard, N.; Pons-Tostivint, E.; Vicente Baz, D.; Sugawara, S.; Cobo Dols, M.; Pérol, M.; et al. LBA12 Datopotamab deruxtecan (Dato-DXd) vs docetaxel in previously treated advanced/metastatic (adv/met) non-small cell lung cancer (NSCLC): Results of the randomized phase III study TROPION-Lung01. Ann. Oncol. 2023, 34, S1305–S1306. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.-T.; et al. Molecular Basis of Valine-Citrulline-PABC Linker Instability in Site-Specific ADCs and Its Mitigation by Linker Design. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers 2023, 15, 713. [Google Scholar] [CrossRef]
- Hickey, A.R.; Wenger, T.L.; Carpenter, V.P.; Tilson, H.H.; Hlatky, M.A.; Furberg, C.D.; Kirkpatrick, C.H.; Strauss, H.C.; Smith, T.W. Digoxin Immune Fab therapy in the management of digitalis intoxication: Safety and efficacy results of an observational surveillance study. J. Am. Coll. Cardiol. 1991, 17, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Pollack, C.V.; Reilly, P.A.; Eikelboom, J.; Glund, S.; Verhamme, P.; Bernstein, R.A.; Dubiel, R.; Huisman, M.V.; Hylek, E.M.; Kamphuisen, P.W.; et al. Idarucizumab for Dabigatran Reversal. N. Engl. J. Med. 2015, 373, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Lobo, E.D.; Balthasar, J.P. Application of anti-methotrexate Fab fragments for the optimization of intraperitoneal methotrexate therapy in a murine model of peritoneal cancer. J. Pharm. Sci. 2005, 94, 1957–1964. [Google Scholar] [CrossRef] [PubMed]
- Balsari, A.; Menard, S.; Colnaghi, M.; Ghione, M. Anti-drug monoclonal antibodies antagonize toxic effect more than anti-tumor activity of doxorubicin. Int. J. Cancer 1991, 47, 889–892. [Google Scholar] [CrossRef]
- Gutowski, M.C.; Fix, D.V.; Corvalan, J.R.; Johnson, D.A. Reduction of toxicity of a vinca alkaloid by an anti-vinca alkaloid antibody. Cancer Investig. 1995, 13, 370–374. [Google Scholar] [CrossRef]
- Balthasar, J.; Fung, H.L. Utilization of antidrug antibody fragments for the optimization of intraperitoneal drug therapy: Studies using digoxin as a model drug. J. Pharmacol. Exp. Ther. 1994, 268, 734–739. [Google Scholar] [CrossRef]
- Balthasar, J.P.; Fung, H.L. Inverse targeting of peritoneal tumors: Selective alteration of the disposition of methotrexate through the use of anti-methotrexate antibodies and antibody fragments. J. Pharm. Sci. 1996, 85, 1035–1043. [Google Scholar] [CrossRef]
- Shah, D.K.; Balthasar, J.P. Predicting the effects of 8C2, a monoclonal anti-topotecan antibody, on plasma and tissue disposition of topotecan. J. Pharmacokinet. Pharmacodyn. 2014, 41, 55–69. [Google Scholar] [CrossRef]
- Shah, D.K.; Balthasar, J.P. PK/TD modeling for prediction of the effects of 8C2, an anti-topotecan mAb, on topotecan-induced toxicity in mice. Int. J. Pharm. 2014, 465, 228–238. [Google Scholar] [CrossRef]
- Ocean, A.J.; Starodub, A.N.; Bardia, A.; Vahdat, L.T.; Isakoff, S.J.; Guarino, M.; Messersmith, W.A.; Picozzi, V.J.; Mayer, I.A.; Wegener, W.A.; et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: Safety and pharmacokinetics. Cancer 2017, 123, 3843–3854. [Google Scholar] [CrossRef] [PubMed]
- Powell, C.A.; Modi, S.; Iwata, H.; Takahashi, S.; Smit, E.F.; Siena, S.; Chang, D.Y.; Macpherson, E.; Qin, A.; Singh, J.; et al. Pooled analysis of drug-related interstitial lung disease and/or pneumonitis in nine trastuzumab deruxtecan monotherapy studies. ESMO Open 2022, 7, 100554. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, K.; Wang, K.; Zhu, H. Treatment-related adverse events of antibody–drug conjugates in clinical trials: A systematic review and meta-analysis. Cancer 2023, 129, 283–295. [Google Scholar] [CrossRef]
- Bordeau, B.M.; Nguyen, T.D.; Polli, J.R.; Chen, P.; Balthasar, J.P. Payload-Binding Fab Fragments Increase the Therapeutic Index of MMAE Antibody–Drug Conjugates. Mol. Cancer Ther. 2023, 22, 459–470. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Use of Payload Binding Selectivity Enhancers to Improve Therapeutic Index of Maytansinoid–Antibody–Drug Conjugates. Mol. Cancer Ther. 2023, 22, 1332–1342. [Google Scholar] [CrossRef]
- Bordeau, B.M.; Abuqayyas, L.; Nguyen, T.D.; Chen, P.; Balthasar, J.P. Development and Evaluation of Competitive Inhibitors of Trastuzumab-HER2 Binding to Bypass the Binding-Site Barrier. Front. Pharmacol. 2022, 13, 837744. [Google Scholar] [CrossRef]
- Bordeau, B.M.; Balthasar, J.P. Strategies to enhance monoclonal antibody uptake and distribution in solid tumors. Cancer Biol. Med. 2021, 18, 649–664. [Google Scholar] [CrossRef]
- Chen, P.; Bordeau, B.M.; Zhang, Y.; Balthasar, J.P. Transient Inhibition of Trastuzumab–Tumor Binding to Overcome the “Binding-Site Barrier” and Improve the Efficacy of a Trastuzumab–Gelonin Immunotoxin. Mol. Cancer Ther. 2022, 21, 1573–1582. [Google Scholar] [CrossRef]
- Bordeau, B.M.; Yang, Y.; Balthasar, J.P. Transient Competitive Inhibition Bypasses the Binding Site Barrier to Improve Tumor Penetration of Trastuzumab and Enhance T-DM1 Efficacy. Cancer Res. 2021, 81, 4145–4154. [Google Scholar] [CrossRef]
- Ponte, J.F.; Lanieri, L.; Khera, E.; Laleau, R.; Ab, O.; Espelin, C.; Kohli, N.; Matin, B.; Setiady, Y.; Miller, M.L.; et al. Antibody Co-Administration Can Improve Systemic and Local Distribution of Antibody-Drug Conjugates to Increase In Vivo Efficacy. Mol. Cancer Ther. 2021, 20, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Guo, L.; Verma, A.; Wong, G.G.-L.; Thurber, G.M.; Shah, D.K. Antibody Coadministration as a Strategy to Overcome Binding-Site Barrier for ADCs: A Quantitative Investigation. AAPS J. 2020, 22, 28. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; Thurber, G.M. Design of high avidity and low affinity antibodies for in situ control of antibody drug conjugate targeting. Sci. Rep. 2022, 12, 7677. [Google Scholar] [CrossRef]
- Li, Z.; Krippendorff, B.-F.; Shah, D.K. Influence of molecular size on the clearance of antibody fragments. Pharm. Res. 2017, 34, 2131–2141. [Google Scholar] [CrossRef]
- Schmidt, M.M.; Wittrup, K.D. A modeling analysis of the effects of molecular size and binding affinity on tumor targeting. Mol. Cancer Ther. 2009, 8, 2861–2871. [Google Scholar] [CrossRef]
- Sun, Q.; Ojha, T.; Kiessling, F.; Lammers, T.; Shi, Y. Enhancing Tumor Penetration of Nanomedicines. Biomacromolecules 2017, 18, 1449–1459. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Bordeau, B.M.; Zhang, Y.; Mattle, A.G.; Balthasar, J.P. Half-Life Extension and Biodistribution Modulation of Biotherapeutics via Red Blood Cell Hitch-Hiking with Novel Anti-Band 3 Single-Domain Antibodies. Int. J. Mol. Sci. 2023, 24, 475. [Google Scholar] [CrossRef]
- Schulte, S. Half-life extension through albumin fusion technologies. Thromb. Res. 2009, 124 (Suppl. S2), S6–S8. [Google Scholar] [CrossRef]
- Trüssel, S.; Dumelin, C.; Frey, K.; Villa, A.; Buller, F.; Neri, D. New Strategy for the Extension of the Serum Half-Life of Antibody Fragments. Bioconjugate Chem. 2009, 20, 2286–2292. [Google Scholar] [CrossRef]
- Dennis, M.S.; Zhang, M.; Meng, Y.G.; Kadkhodayan, M.; Kirchhofer, D.; Combs, D.; Damico, L.A. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J. Biol. Chem. 2002, 277, 35035–35043. [Google Scholar] [CrossRef]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- De Vos, J.; Devoogdt, N.; Lahoutte, T.; Muyldermans, S. Camelid single-domain antibody-fragment engineering for (pre)clinical in vivo molecular imaging applications: Adjusting the bullet to its target. Expert Opin. Biol. Ther. 2013, 13, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Iezzi, M.E.; Policastro, L.; Werbajh, S.; Podhajcer, O.; Canziani, G.A. Single-Domain Antibodies and the Promise of Modular Targeting in Cancer Imaging and Treatment. Front. Immunol. 2018, 9, 273. [Google Scholar] [CrossRef] [PubMed]
- Nessler, I.; Khera, E.; Vance, S.; Kopp, A.; Qiu, Q.; Keating, T.A.; Abu-Yousif, A.O.; Sandal, T.; Legg, J.; Thompson, L.; et al. Increased Tumor Penetration of Single-Domain Antibody-Drug Conjugates Improves In Vivo Efficacy in Prostate Cancer Models. Cancer Res. 2020, 80, 1268–1278. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Composition | Current Status | Indications |
|---|---|---|---|
| CRLX101 | cyclodextrin-based polymer linked to CPT | Phase II | solid tumor |
| LY01610 | liposomal irinotecan | Phase II | small-cell lung cancer |
| U3-1402 (patritumab deruxtecan) | anti-HER3-DXd ADC | Phase II | breast cancer, non-small-cell lung cancer |
| DS-1062 (datopotamab deruxtecan) | anti-Trop2-DXd ADC | Phase III | breast cancer, non-small-cell lung cancer |
| DS-6000 (raludotatug deruxtecan) | anti-CDH6-DXd ADC | Phase II/III | ovarian tumor, renal cell carcinoma |
| DS-7300 (ifinatamab deruxtecan) | anti-B7H3-DXd ADC | phase III | solid tumor |
| Abbv-400 | anti-Met-CPT derivative ADC | Phase II | solid tumor |
| HS-20093 | anti-B7H3-HS-9265 ADC | Phase II | solid tumor |
| SKB264 | anti-TROP2-KL610023 ADC | Phase II | breast cancer, non-small-cell lung cancer |
| IBI343 | anti-CLDN18.2-exatecan ADC | Phase II | gastric cancer |
| DB-1311 | anti-B7H3-P1021 ADC | Phase I/II | solid tumor |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gong, J.; Zhang, W.; Balthasar, J.P. Camptothein-Based Anti-Cancer Therapies and Strategies to Improve Their Therapeutic Index. Cancers 2025, 17, 1032. https://doi.org/10.3390/cancers17061032
Gong J, Zhang W, Balthasar JP. Camptothein-Based Anti-Cancer Therapies and Strategies to Improve Their Therapeutic Index. Cancers. 2025; 17(6):1032. https://doi.org/10.3390/cancers17061032
Chicago/Turabian StyleGong, Jue, Wenqiu Zhang, and Joseph P. Balthasar. 2025. "Camptothein-Based Anti-Cancer Therapies and Strategies to Improve Their Therapeutic Index" Cancers 17, no. 6: 1032. https://doi.org/10.3390/cancers17061032
APA StyleGong, J., Zhang, W., & Balthasar, J. P. (2025). Camptothein-Based Anti-Cancer Therapies and Strategies to Improve Their Therapeutic Index. Cancers, 17(6), 1032. https://doi.org/10.3390/cancers17061032