
The Role of the Sirtuin Family Histone Deacetylases in Acute Myeloid Leukemia—A Promising Road Ahead

 , , , ,
, , , ,
Simple Summary
Abstract
1. Introduction
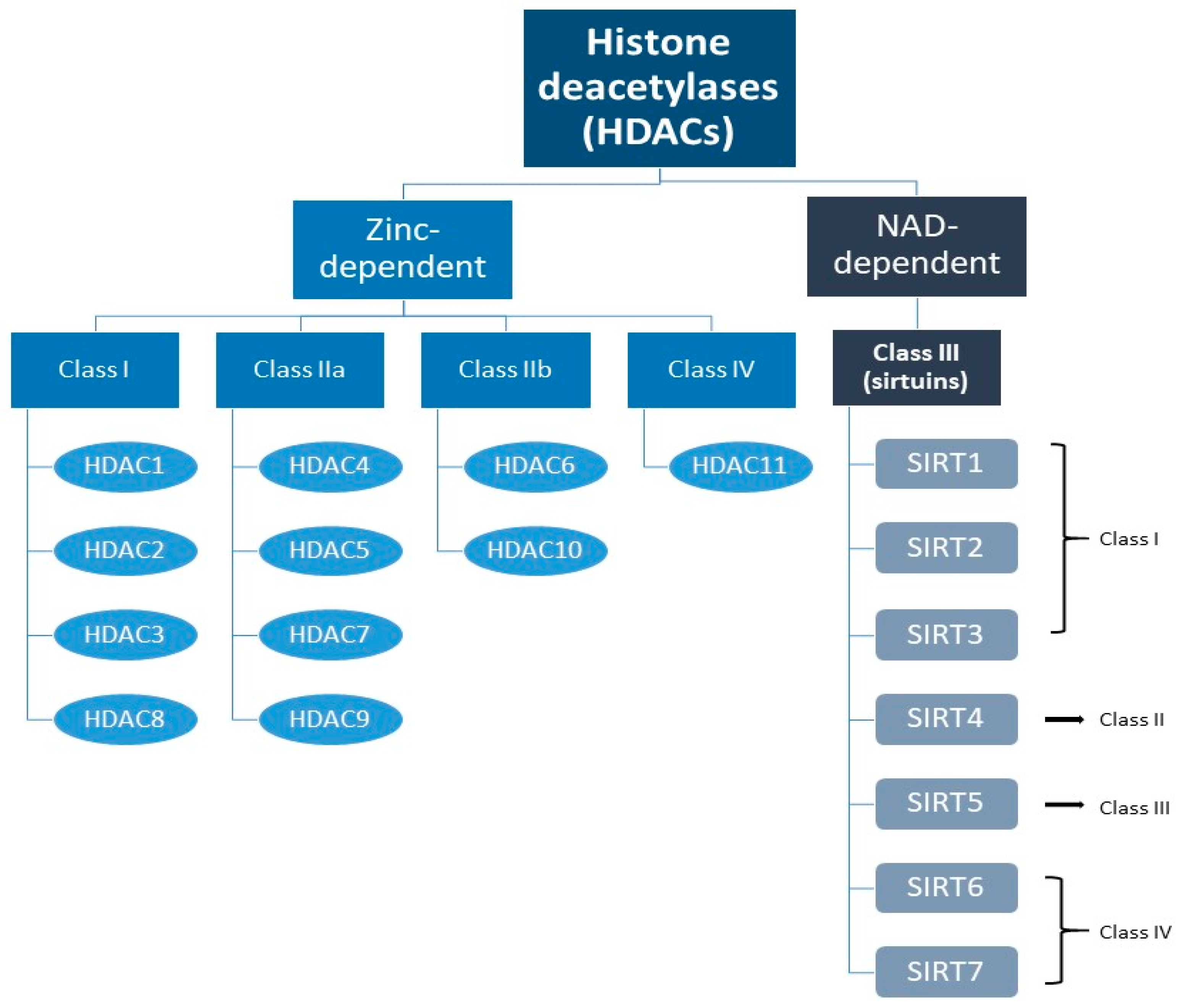
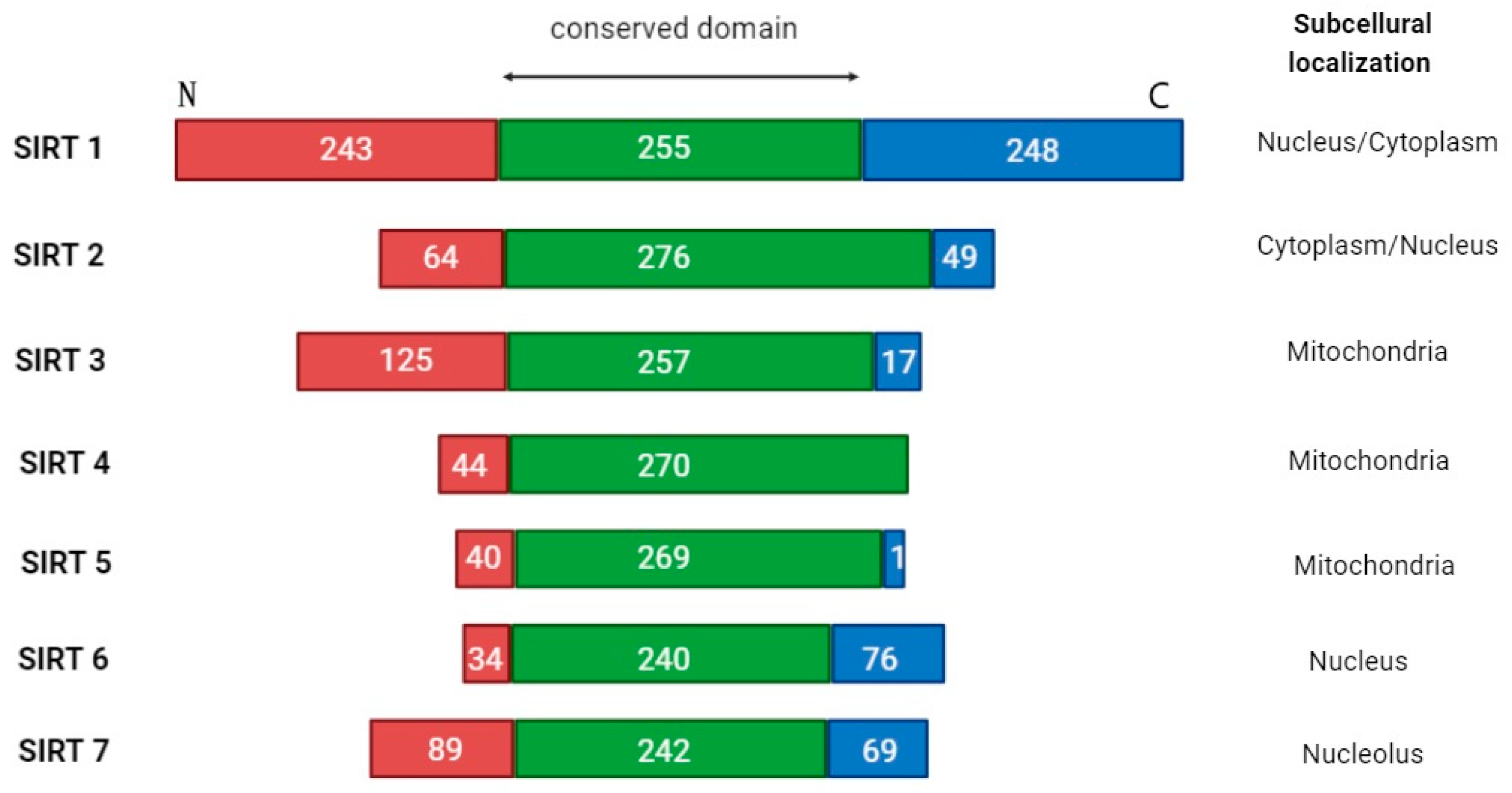
2. The Sirtuin Family
3. Classification of Sirtuins
4. Overview of the Role of the Sirtuin Family in AML
4.1. Sirtuin 1
4.2. Sirtuin 2
4.3. Sirtuin 3
4.4. Sirtuin 4
4.5. Sirtuin 5
4.6. Sirtuin 6
4.7. Sirtuin 7
5. Therapeutic Applications/Implications
6. Limitations, Hopes, and Future Challenges
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhou, Y.; Huang, G.; Cai, X.; Liu, Y.; Qian, B.; Li, D. Global, regional, and national burden of acute myeloid leukemia, 1990–2021: A systematic analysis for the global burden of disease study 2021. Biomark. Res. 2024, 12, 101. [Google Scholar] [CrossRef] [PubMed]
- Surveillance, Epidemiology, and End Results (SEER) Program (www.seer.cancer.gov) SEER*Stat Database: Incidence—SEER Research Data, 8 Registries, Nov 2023 Sub (1975–2021)—Linked To County Attributes—Time Dependent (1990–2022) Income/Rurality, 1969–2022 Counties, National Cancer Institute, DCCPS, Surveillance Research Program, Released April 2024, Based on the November 2023 Submission. Acute Myeloid Leukemia—Cancer Stat Facts (2024). Available online: https://seer.cancer.gov/statfacts/html/amyl.html (accessed on 14 February 2025).
- Velten, L.; Story, B.A.; Hernández-Malmierca, P.; Raffel, S.; Leonce, D.R.; Milbank, J.; Paulsen, M.; Demir, A.; Szu-Tu, C.; Frömel, R.; et al. Identification of leukemic and pre-leukemic stem cells by clonal tracking from single-cell transcriptomics. Nat. Commun. 2021, 12, 1366. [Google Scholar] [CrossRef] [PubMed]
- Stelmach, P.; Trumpp, A. Leukemic stem cells and therapy resistance in acute myeloid leukemia. Haematologica 2023, 108, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.T.; Sun, J.; Zhang, L.; He, X.; Zhu, Y.H.; Dong, H.J.; Wang, H.Y.; Zhu, L.; Zou, J.Y.; Huang, J.W.; et al. Role of SIRT1 in hematologic malignancies. J. Zhejiang Univ. Sci. B 2019, 20, 391–398. [Google Scholar] [CrossRef]
- Batsivari, A.; Grey, W.; Bonnet, D. Understanding of the crosstalk between normal residual hematopoietic stem cells and the leukemic niche in acute myeloid leukemia. Exp. Hematol. 2021, 95, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, J.; Xu, L.; Găman, M.A.; Zou, Z. The genesis and evolution of acute myeloid leukemia stem cells in the microenvironment: From biology to therapeutic targeting. Cell Death Discov. 2022, 8, 397. [Google Scholar] [CrossRef]
- Shlush, L.I.; Mitchell, A.; Heisler, L.; Abelson, S.; Ng, S.W.K.; Trotman-Grant, A.; Medeiros, J.J.F.; Rao-Bhatia, A.; Jaciw-Zurakowsky, I.; Marke, R.; et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature 2017, 547, 104–108. [Google Scholar] [CrossRef]
- Khaldoyanidi, S.K.; Hindoyan, A.; Stein, A.; Subklewe, M. Leukemic stem cells as a target for eliminating acute myeloid leukemia: Gaps in translational research. Crit. Rev. Oncol. Hematol. 2022, 175, 103710. [Google Scholar] [CrossRef]
- Kumar, B.; Garcia, M.; Weng, L.; Jung, X.; Murakami, J.L.; Hu, X.; McDonald, T.; Lin, A.; Kumar, A.R.; DiGiusto, D.L.; et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia-permissive microenvironment through exosome secretion. Leukemia 2018, 32, 575–587. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Carafa, V.; Altucci, L.; Nebbioso, A. Dual Tumor Suppressor and Tumor Promoter Action of Sirtuins in Determining Malignant Phenotype. Front. Pharmacol. 2019, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Fang, D. The Roles of SIRT1 in Cancer. Genes. Cancer 2013, 4, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Chalkiadaki, A.; Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015, 15, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Zhang, X.; Yi, J.; Huang, J.; He, J.; Tao, Y. Sirtuins in metabolism; DNA repair and cancer. J. Exp. Clin. Cancer Res. 2016, 35, 182. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Cui, H. Function of Sirtuins in Cancer Stem Cells. Int. J. Stem Cell Res. Ther. 2016, 3, 024. [Google Scholar] [CrossRef]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef]
- Haq, M.F.U.; Hussain, M.Z.; Mahjabeen, I.; Akram, Z.; Saeed, N.; Shafique, R.; Abbasi, S.F.; Kayani, M.A. Oncometabolic role of mitochondrial sirtuins in glioma patients. PLoS ONE 2023, 18, e0281840. [Google Scholar] [CrossRef]
- Li, J.; Zhan, H.; Ren, Y.; Feng, M.; Wang, Q.; Jiao, Q.; Wang, Y.; Liu, X.; Zhang, S.; Du, L.; et al. Sirtuin 4 activates autophagy and inhibits tumorigenesis by upregulating the p53 signaling pathway. Cell Death Differ. 2023, 30, 313–326. [Google Scholar] [CrossRef]
- Shi, Q.; Liu, T.; Zhang, X.; Geng, J.; He, X.; Nu, M.; Pang, D. Decreased sirtuin 4 expression is associated with poor prognosis in patients with invasive breast cancer. Oncol. Lett. 2016, 12, 2606–2612. [Google Scholar] [CrossRef]
- Tang, X.; Li, Y.; Liu, L.; Guo, R.; Zhang, P.; Zhang, Y.; Zhang, Y.; Zhao, J.; Su, J.; Sun, L.; et al. Sirtuin 3 induces apoptosis and necroptosis by regulating mutant p53 expression in small-cell lung cancer. Oncol. Rep. 2020, 43, 591–600. [Google Scholar] [CrossRef]
- Wu, X.; Wang, S.; Zhao, X.; Lai, S.; Yuan, Z.; Zhan, Y.; Ni, K.; Liu, Z.; Liu, L.; Xin, R.; et al. Clinicopathological and prognostic value of SIRT6 in patients with solid tumors: A meta-analysis and TCGA data review. Cancer Cell Int. 2022, 22, 84. [Google Scholar] [CrossRef] [PubMed]
- Goes, J.V.C.; Carvalho, L.G.; de Oliveira, R.T.G.; Melo, M.M.L.; Novaes, L.A.C.; Moreno, D.A.; Gonçalves, P.G.; Montefusco-Pereira, C.V.; Pinheiro, R.F.; Ribeiro Junior, H.L. Role of Sirtuins in the Pathobiology of Onco-Hematological Diseases: A PROSPERO-Registered Study and In Silico Analysis. Cancers 2022, 14, 4611. [Google Scholar] [CrossRef]
- Costa-Machado, L.F.; Fernandez-Marcos, P.J. The sirtuin family in cancer. Cell Cycle 2019, 18, 2164–2196. [Google Scholar] [CrossRef] [PubMed]
- Sauve, A.A. Sirtuin chemical mechanisms. Biochim. Biophys. Acta 2010, 1804, 1591–1603. [Google Scholar] [CrossRef]
- Amaral, P.; Carbonell-Sala, S.; De La Vega, F.M.; Faial, T.; Frankish, A.; Gingeras, T.; Guigo, R.; Harrow, J.L.; Hatzigeorgiou, A.G.; Johnson, R.; et al. The status of the human gene catalogue. Nature 2023, 622, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A.V.; Mikheenko, A.; Vollger, M.R.; Altemose, N.; Uralsky, L.; Gershman, A.; et al. The complete sequence of a human genome. Science 2022, 376, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Agar, J.N.; Amster, I.J.; Baker, M.S.; Bertozzi, C.R.; Boja, E.S.; Costello, C.E.; Cravatt, B.F.; Fenselau, C.; Garcia, B.A.; et al. How many human proteoforms are there? Nat. Chem. Biol. 2018, 14, 206–214. [Google Scholar] [CrossRef]
- Ponomarenko, E.A.; Poverennaya, E.V.; Ilgisonis, E.V.; Pyatnitskiy, M.A.; Kopylov, A.T.; Zgoda, V.G.; Lisitsa, A.V.; Archakov, A.I. The Size of the Human Proteome: The Width and Depth. Int. J. Anal. Chem. 2016, 2016, 7436849. [Google Scholar] [CrossRef]
- Teixeira, C.S.S.; Cerqueira, N.M.F.S.A.; Gomes, P.; Sousa, S.F. A Molecular Perspective on Sirtuin Activity. Int. J. Mol. Sci. 2020, 21, 8609. [Google Scholar] [CrossRef]
- Towler, D.A.; Gordon, J.I.; Adams, S.P.; Glaser, L. The biology and enzymology of eukaryotic protein acylation. Annu. Rev. Biochem. 1988, 57, 69–99. [Google Scholar] [CrossRef]
- Mai, A.; Massa, S.; Rotili, D.; Cerbara, I.; Valente, S.; Pezzi, R.; Simeoni, S.; Ragno, R. Histone deacetylation in epigenetics: An attractive target for anticancer therapy. Med. Res. Rev. 2005, 25, 261–309. [Google Scholar] [CrossRef] [PubMed]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef]
- Brachmann, C.B.; Sherman, J.M.; Devine, S.E.; Cameron, E.E.; Pillus, L.; Boeke, J.D. The SIR2 gene family; conserved from bacteria to humans; functions in silencing; cell cycle progression; and chromosome stability. Genes. Dev. 1995, 9, 2888–2902. [Google Scholar] [CrossRef] [PubMed]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Rosato, R.R.; Grant, S. Histone deacetylase inhibitors: Insights into mechanisms of lethality. Expert. Opin. Ther. Targets 2005, 9, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Sawas, A.; Radeski, D.; O’Connor, O.A. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: A perspective review. Ther. Adv. Hematol. 2015, 6, 202–208. [Google Scholar] [CrossRef]
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol. Ther. 2014, 143, 323–336. [Google Scholar] [CrossRef]
- Zhao, E.; Hou, J.; Ke, X.; Abbas, M.N.; Kausar, S.; Zhang, L.; Cui, H. The Roles of Sirtuin Family Proteins in Cancer Progression. Cancers 2019, 11, 1949. [Google Scholar] [CrossRef]
- Giblin, W.; Lombard, D.B. Sirtuins, Healthspan, and Longevity in Mammals. In Handbook of the Biology of Aging, 8th ed.; Kaeberlein, M.R., Martin, M.G., Eds.; Academic Press: Cambridge, MA, USA, 2016; Chapter 3; pp. 83–132. [Google Scholar] [CrossRef]
- Kratz, E.M.; Sołkiewicz, K.; Kubis-Kubiak, A.; Piwowar, A. Sirtuins as Important Factors in Pathological States and the Role of Their Molecular Activity Modulators. Int. J. Mol. Sci. 2021, 22, 630. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Ng, E.E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.X. SIRT1; is it a tumor promoter or tumor suppressor? Int. J. Biol. Sci. 2009, 5, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef]
- Li, L.; Osdal, T.; Ho, Y.; Chun, S.; McDonald, T.; Agarwal, P.; Lin, A.; Chu, S.; Qi, J.; Li, L.; et al. SIRT1 activation by a c-MYC oncogenic network promotes the maintenance and drug resistance of human FLT3-ITD acute myeloid leukemia stem cells. Cell Stem Cell 2014, 15, 431–446. [Google Scholar] [CrossRef]
- O’Donnell, M.R.; Abboud, C.N.; Altman, J.; Appelbaum, F.R.; Arber, D.A.; Attar, E.; Borate, U.; Coutre, S.E.; Damon, L.E.; Goorha, S.; et al. NCCN Clinical Practice Guidelines Acute myeloid leukemia. J. Natl. Compr. Canc Netw. 2012, 10, 984–1021. [Google Scholar] [CrossRef] [PubMed]
- Sasca, D.; Hähnel, P.S.; Szybinski, J.; Khawaja, K.; Kriege, O.; Pante, S.V.; Bullinger, L.; Strand, S.; Strand, D.; Theobald, M.; et al. SIRT1 prevents genotoxic stress-induced p53 activation in acute myeloid leukemia. Blood 2014, 124, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef]
- Patel, J.P.; Gönen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef]
- Bradbury, C.A.; Khanim, F.L.; Hayden, R.; Bunce, C.M.; White, D.A.; Drayson, M.T.; Craddock, C.; Turner, B.M. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 2005, 19, 1751–1759. [Google Scholar] [CrossRef]
- Li, L.C.; Wang, J.D.; Yang, S.S.; Zhou, Z.; Zeng, Q.F.; Zheng, F. Establishment of Drug-Resistant Cell Lines of Acute Myeloid Leukemia and Correlation of Sirt1 and PGC-1α Expression Levels with Drug Resistance. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2022, 30, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.L.; Guo, R.; Wang, F.; Jiang, Z.X.; Tang, P.; Huang, Y.M.; Sun, L. The IRF9-SIRT1-P53 axis is involved in the growth of human acute myeloid leukemia. Exp. Cell Res. 2018, 365, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.W.; Koche, R.P.; Sinha, A.U.; Deshpande, A.J.; Zhu, N.; Eng, R.; Doench, J.G.; Xu, H.; Chu, S.H.; Qi, J.; et al. DOT1L inhibits SIRT1-mediated epigenetic silencing to maintain leukemic gene expression in MLL-rearranged leukemia. Nat. Med. 2015, 21, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Levavasseur, F.; Oussous, S.; Zubaidan, T.; Kosmider, O.; Pendino, F.; Rombaut, D.; Bouscary, D.; Fontenay, M.; Lauret, E.; Dusanter-Fourt, I. FOXP1 regulates oxidative stress; SIRT1 expression; and resistance to chemotherapies in acute myeloid leukemia cells. Blood Adv. 2023, 7, 3265–3275. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Wang, Z.; Li, L.; Zhang, H.; Modi, H.; Horne, D.; Bhatia, R.; Chen, W. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood 2012, 119, 1904–1914. [Google Scholar] [CrossRef]
- Van Etten, R.A. Aberrant cytokine signaling in leukemia. Oncogene 2007, 26, 6738–6749. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P.; Chen, C.Z. Micromanagers of gene expression: The potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet. 2004, 5, 396–400. [Google Scholar] [CrossRef]
- Zhou, L.; Fu, L.; Lv, N.; Chen, X.S.; Liu, J.; Li, Y.; Xu, Q.Y.; Huang, S.; Zhang, X.D.; Dou, L.P.; et al. A minicircuitry comprised of microRNA-9 and SIRT1 contributes to leukemogenesis in t(8;21) acute myeloid leukemia. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 786–794. [Google Scholar]
- Vaquero, A.; Scher, M.B.; Lee, D.H.; Sutton, A.; Cheng, H.L.; Alt, F.W.; Serrano, L.; Sternglanz, R.; Reinberg, D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes. Dev. 2006, 20, 1256–1261. [Google Scholar] [CrossRef]
- Harting, K.; Knöll, B. SIRT2-mediated protein deacetylation: An emerging key regulator in brain physiology and pathology. Eur. J. Cell Biol. 2010, 89, 262–269. [Google Scholar] [CrossRef]
- Ahmed, M.A.; O’Callaghan, C.; Chang, E.D.; Jiang, H.; Vassilopoulos, A. Context-Dependent Roles for SIRT2 and SIRT3 in Tumor Development Upon Calorie Restriction or High Fat Diet. Front. Oncol. 2020, 9, 1462. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, P.; Hu, C. The role of SIRT2 in cancer: A novel therapeutic target. Int. J. Cancer 2020, 147, 3297–3304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Sowers, M.L.; Cherryhomes, E.I.; Singh, V.K.; Mishra, A.; Restrepo, B.I.; Khan, A.; Jagannath, C. Sirtuin-dependent metabolic and epigenetic regulation of macrophages during tuberculosis. Front. Immunol. 2023, 14, 1121495. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Zou, S.W.; Liu, Y.; Zhou, X.; Mo, Y.; Wang, P.; Xu, Y.H.; Dong, B.; Xiong, Y.; Lei, Q.Y.; et al. Lysine-5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer Cell 2013, 23, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Kozako, T.; Mellini, P.; Ohsugi, T.; Aikawa, A.; Uchida, Y.I.; Honda, S.I.; Suzuki, T. Novel small molecule SIRT2 inhibitors induce cell death in leukemic cell lines. BMC Cancer 2018, 18, 791. [Google Scholar] [CrossRef]
- Dan, L.; Klimenkova, O.; Klimiankou, M.; Klusman, J.H.; van den Heuvel-Eibrink, M.M.; Reinhardt, D.; Welte, K.; Skokowa, J. The role of sirtuin 2 activation by nicotinamide phosphoribosyltransferase in the aberrant proliferation and survival of myeloid leukemia cells. Haematologica 2012, 97, 551–559. [Google Scholar] [CrossRef]
- Deng, A.; Ning, Q.; Zhou, L.; Liang, Y. SIRT2 is an unfavorable prognostic biomarker in patients with acute myeloid leukemia. Sci. Rep. 2016, 6, 27694. [Google Scholar] [CrossRef] [PubMed]
- McMahon, K.A.; Hiew, S.Y.; Hadjur, S.; Veiga-Fernandes, H.; Menzel, U.; Price, A.J.; Kioussis, D.; Williams, O.; Brady, H.J. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell 2007, 1, 338–345. [Google Scholar] [CrossRef]
- Jude, C.D.; Climer, L.; Xu, D.; Artinger, E.; Fisher, J.K.; Ernst, P. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell 2007, 1, 324–337. [Google Scholar] [CrossRef]
- Hao, C.; Shao, X.; Song, J.; Peng, M.; Lao, Y.; Mack, R.; Zhang, L.; Wei, W.; Liu, N.; Wang, T.; et al. SIRT2 regulates proliferation and chemotherapy response of MLL-ENL-driven acute myeloid leukemia. Biochem. Biophys. Res. Commun. 2022, 596, 36–42. [Google Scholar] [CrossRef]
- Xu, H.; Li, Y.; Chen, L.; Wang, C.; Wang, Q.; Zhang, H.; Lin, Y.; Li, Q.; Pang, T. SIRT2 mediates multidrug resistance in acute myelogenous leukemia cells via ERK1/2 signaling pathway. Int. J. Oncol. 2016, 48, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Yip, P.Y. Phosphatidylinositol 3-kinase-AKT-mammalian target of rapamycin (PI3K-Akt-mTOR) signaling pathway in non-small cell lung cancer. Transl. Lung Cancer Res. 2015, 4, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhao, H.; Zhu, J.; Zhang, L.; Zha, J.; Zhang, L.; Ding, Y.; Jian, X.; Xia, J.; Xu, B.; et al. SIRT2 inhibitor SirReal2 enhances anti-tumor effects of PI3K/mTOR inhibitor VS-5584 on acute myeloid leukemia cells. Cancer Med. 2023, 12, 18901–18917. [Google Scholar] [CrossRef] [PubMed]
- Dan, L.; Gigina, A.; Welte, K.; Skokowa, J. Nampt/SIRT2 Hyperactivation Lead to Activation of β-Catenin: A Possible Mechanism of Leukomogenic Transformation inCN Patients. Blood 2010, 116, 1486. [Google Scholar] [CrossRef]
- Xu, S.N.; Wang, T.S.; Li, X.; Wang, Y.P. SIRT2 activates G6PD to enhance NADPH production and promote leukaemia cell proliferation. Sci. Rep. 2016, 6, 32734. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.; Maugeri, A.; De Luca, L.; Gitto, R.; Lombardo, G.E.; Musumeci, L.; De Sarro, G.; Cirmi, S.; Navarra, M. The SIRT2 Pathway Is Involved in the Antiproliferative Effect of Flavanones in Human Leukemia Monocytic THP-1 Cells. Biomedicines 2022, 10, 2383. [Google Scholar] [CrossRef]
- Zhao, L.; Qi, Y.; Xu, L.; Tao, X.; Han, X.; Yin, L.; Peng, J. MicroRNA-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biol. 2018, 15, 284–296. [Google Scholar] [CrossRef]
- Havelange, V.; Stauffer, N.; Heaphy, C.C.; Volinia, S.; Andreeff, M.; Marcucci, G.; Croce, C.M.; Garzon, R. Functional implications of microRNAs in acute myeloid leukemia by integrating microRNA and messenger RNA expression profiling. Cancer 2011, 117, 4696–4706. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Chen, Y.; Fu, L.L.; Wen, X.; Wang, X.Y.; Liu, J.; Cheng, Y.; Huang, J. Sirtuin-3 (SIRT3); a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014, 5, e1047. [Google Scholar] [CrossRef]
- Zhao, K.; Zhou, Y.; Qiao, C.; Ni, T.; Li, Z.; Wang, X.; Guo, Q.; Lu, N.; Wei, L. Oroxylin A promotes PTEN-mediated negative regulation of MDM2 transcription via SIRT3-mediated deacetylation to stabilize p53 and inhibit glycolysis in wt-p53 cancer cells. J. Hematol. Oncol. 2015, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Zhou, Y.; Dai, Q.; Qiao, C.; Zhao, L.; Hui, H.; Lu, N.; Guo, Q.L. Oroxylin A induces dissociation of hexokinase II from the mitochondria and inhibits glycolysis by SIRT3-mediated deacetylation of cyclophilin D in breast carcinoma. Cell Death Dis. 2013, 4, e601. [Google Scholar] [CrossRef] [PubMed]
- Bergaggio, E.; Riganti, C.; Garaffo, G.; Vitale, N.; Mereu, E.; Bandini, C.; Pellegrino, E.; Pullano, V.; Omedè, P.; Todoerti, K.; et al. IDH2 inhibition enhances proteasome inhibitor responsiveness in hematological malignancies. Blood 2019, 133, 156–167. [Google Scholar] [CrossRef]
- O’Brien, C.; Berman, J.; Culp-Hill, R.; Reisz, J.; Ling, T.; Rondeau, V.; Li, M.; Yang, M.; Hong, J.Y.; Lin, H.; et al. Sirtuin 3 Inhibition Targets AML Stem Cells through Perturbation of Fatty Acid Oxidation. Blood 2021, 138 (Suppl. S1), 2240. [Google Scholar] [CrossRef]
- O’Brien, C.; Ling, T.; Berman, J.M.; Culp-Hill, R.; Reisz, J.A.; Rondeau, V.; Jahangiri, S.; St-Germain, J.; Macwan, V.; Astori, A.; et al. Simultaneous inhibition of Sirtuin 3 and cholesterol homeostasis targets acute myeloid leukemia stem cells by perturbing fatty acid β-oxidation and inducing lipotoxicity. Haematologica 2023, 108, 2343–2357. [Google Scholar] [CrossRef]
- Ma, J.; Liu, B.; Yu, D.; Zuo, Y.; Cai, R.; Yang, J.; Cheng, J. SIRT3 deacetylase activity confers chemoresistance in AML via regulation of mitochondrial oxidative phosphorylation. Br. J. Haematol. 2019, 187, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jiang, Z.; Yang, Y.; Zhang, C.; Liu, H.; Wan, J. The functions and mechanisms of post-translational modification in protein regulators of RNA methylation: Current status and future perspectives. Int. J. Biol. Macromol. 2023, 253, 126773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shen, Y.; Wei, W.; Wang, W.; Jiang, D.; Ren, Y.; Peng, Z.; Fan, Q.; Cheng, J.; Ma, J. Dysregulation of SIRT3 SUMOylation Confers AML Chemoresistance via Controlling HES1-Dependent Fatty Acid Oxidation. Int. J. Mol. Sci. 2022, 23, 8282. [Google Scholar] [CrossRef]
- Ma, J.; Liu, B.; Yu, D.; Tam, W.; Yang, J.; Cheng, J. SIRT3 Sumoylation Contributes to Chemoresistance in AML. Blood 2018, 132 (Suppl. S1), 3929. [Google Scholar] [CrossRef]
- Strzałka, P.; Krawiec, K.; Jarych, D.; Wiśnik, A.; Soin, M.; Góralska, M.; Mikulski, D.; Czemerska, M.; Zawlik, I.; Pluta, A.; et al. Assessment of SIRT1-SIRT7 and TP53 Genes Expression in Patients with Acute Myeloid Leukemia. Blood 2023, 142 (Suppl. S1), 6048. [Google Scholar] [CrossRef]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 2006, 126, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Zhu, G. Sirtuin-4 (SIRT4), a therapeutic target with oncogenic and tumor-suppressive activity in cancer. Onco Targets Ther. 2018, 11, 3395–3400. [Google Scholar] [CrossRef]
- Frye, R.A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 1999, 260, 273–279. [Google Scholar] [CrossRef]
- Jeong, S.M.; Xiao, C.; Finley, L.W.; Lahusen, T.; Souza, A.L.; Pierce, K.; Li, Y.H.; Wang, X.; Laurent, G.; German, N.J.; et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell 2013, 23, 450–463. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.M.; Lee, A.; Lee, J.; Haigis, M.C. SIRT4 protein suppresses tumor formation in genetic models of Myc-induced B cell lymphoma. J. Biol. Chem. 2014, 289, 4135–4144. [Google Scholar] [CrossRef] [PubMed]
- Csibi, A.; Fendt, S.M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Yu, Z.; Chen, X.; Huang, G. SIRT4 is upregulated in Chinese patients with esophageal cancer. Int. J. Clin. Exp. Pathol. 2016, 9, 10543–10549. [Google Scholar]
- Strzałka, P.; Krawiec, K.; Jarych, D.; Wiśnik, A.; Góralska, M.; Soin, M.; Mikulski, D.; Czemerska, M.; Zawlik, I.; Pluta, A.; et al. Evaluation of Sirtuins and TP53 Gene Expression and Their Impact on Prognosis in Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2023, 23 (Suppl. S1), S314–S315. [Google Scholar] [CrossRef]
- Vaquero, A.; Reinberg, D. Sirtuins in Biology and Disease. In Epigenetics in Biology and Medicine, 1st ed.; Esteller, M., Ed.; CRC Press: Boca Raton, FL, USA, 2009; Chapter 6; pp. 73–104. [Google Scholar] [CrossRef]
- Yue, X.; Shi, Y.; Luo, Q. Advances of SIRT4 in cancer metabolism and therapy. Pediatr. Discov. 2023, 1, e17. [Google Scholar] [CrossRef]
- Jeong, S.M.; Hwang, S.; Seong, R.H. SIRT4 regulates cancer cell survival and growth after stress. Biochem. Biophys. Res. Commun. 2016, 470, 251–256. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Rardin, M.J.; Carrico, C.; He, W.; Sahu, A.K.; Gut, P.; Najjar, R.; Fitch, M.; Hellerstein, M.; Gibson, B.W.; et al. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Mol. Cell 2015, 59, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef]
- Park, J.; Chen, Y.; Tishkoff, D.X.; Peng, C.; Tan, M.; Dai, L.; Xie, Z.; Zhang, Y.; Zwaans, B.M.; Skinner, M.E.; et al. SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol. Cell 2013, 50, 919–930. [Google Scholar] [CrossRef]
- Wątroba, M.; Szukiewicz, D. Sirtuins in the biology of aging. In Sirtuin Biology in Medicine; Maiese, K., Ed.; Academic Press: Cambridge, MA, USA, 2021; Chapter 5; pp. 79–90. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, J.; Hu, K.; He, X.; Yun, D.; Tong, T.; Han, L. Sirtuins and their Biological Relevance in Aging and Age-Related Diseases. Aging Dis. 2020, 11, 927–945. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Che, W.; Zheng, C.; Liu, W.; Wen, J.; Fu, H.; Tang, K.; Zhang, J.; Xu, Y. SIRT5: A safeguard against oxidative stress-induced apoptosis in cardiomyocytes. Cell Physiol. Biochem. 2013, 32, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Wang, X.; Ow, S.H.; Chen, W.; Ong, W.C. Sirtuin 5 is Anti-apoptotic and Anti-oxidative in Cultured SH-EP Neuroblastoma Cells. Neurotox. Res. 2017, 31, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, Y.; Li, Y.; Zhao, Y.; Jiang, H. Sirt5 Attenuates Cisplatin-Induced Acute Kidney Injury through Regulation of Nrf2/HO-1 and Bcl-2. Biomed. Res. Int. 2019, 2019, 4745132. [Google Scholar] [CrossRef]
- Krawiec, K.; Strzałka, P.; Czemerska, M.; Wiśnik, A.; Zawlik, I.; Wierzbowska, A.; Pluta, A. Targeting Apoptosis in AML: Where Do We Stand? Cancers 2022, 14, 4995. [Google Scholar] [CrossRef]
- Li, M.; Melnick, A.M. Non-oncogene Addiction to SIRT5 in Acute Myeloid Leukemia. Blood Cancer Discov. 2021, 2, 198–200. [Google Scholar] [CrossRef]
- Yan, D.; Franzini, A.; Pomicter, A.D.; Halverson, B.J.; Antelope, O.; Mason, C.C.; Ahmann, J.M.; Senina, A.V.; Vellore, N.A.; Jones, C.L.; et al. SIRT5 is a druggable metabolic vulnerability in acute myeloid leukemia. Blood Cancer Discov. 2021, 2, 266–287. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Pomicter, A.D.; Heaton, W.L.; Mason, C.C.; Ahmann, J.; Senina, A.; Franzini, A.; Halverson, B.; Than, H.; Clair, P.M.; et al. SIRT5 As a Therapeutic Target in Acute Myeloid Leukemia. Blood 2018, 132 (Suppl. S1), 907. [Google Scholar] [CrossRef]
- Rajabi, N.; Hansen, T.N.; Nielsen, A.L.; Nguyen, H.T.; Bæk, M.; Bolding, J.E.; Bahlke, O.Ø.; Petersen, S.E.G.; Bartling, C.R.O.; Strømgaard, K.; et al. Investigation of carboxylic acid isosteres and prodrugs for inhibition of the human SIRT5 lysine deacylase enzyme. Angew. Chem. Int. Ed. 2022, 61, e202115805. [Google Scholar] [CrossRef]
- Arévalo, C.M.; Cruz-Rodriguez, N.; Quijano, S.; Fiorentino, S. Plant-derived extracts and metabolic modulation in leukemia: A promising approach to overcome treatment resistance. Front. Mol. Biosci. 2023, 10, 1229760. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Shi, W.; Zhu, R.; Small, D.; Li, L.; Ma, S.; Hu, Y. Targeting SIRT5 By Succinylating Hadha Synergizes with Venetoclax in Acute Myeloid Leukemia. Blood 2023, 142 (Suppl. S1), 4164. [Google Scholar] [CrossRef]
- Zhang, J.; Luo, C.; Long, H. Sirtuin 5 regulates acute myeloid leukemia cell viability and apoptosis by succinylation modification of glycine decarboxylase. Open Life Sci. 2024, 19, 20220832. [Google Scholar] [CrossRef] [PubMed]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef]
- Ozkan, T.; Koc, A.; Karabay, A.Z.; Hekmatshoar, Y.; Sunguroglu, A. An investigation on the effects of SIRT5 modulators on SIRT5 and Cytochrome C protein expressions in K562 chronic myeloid leukemia cell line. J. Fac. Pharm. 2022, 46, 805–816. [Google Scholar] [CrossRef]
- Bateman, B.; Bates, B.; Deininger, M.; Zhao, H.G.G. Chronic Mitochondrial Dysfunction Renders Acute Myeloid Leukemia Resistant to Multipronged Inhibition of Mitochondrial Metabolism. Blood 2023, 142 (Suppl. S1), 2739. [Google Scholar] [CrossRef]
- Li, F.; He, X.; Ye, D.; Lin, Y.; Yu, H.; Yao, C.; Huang, L.; Zhang, J.; Wang, F.; Xu, S.; et al. NADP(+)-IDH Mutations Promote Hypersuccinylation that Impairs Mitochondria Respiration and Induces Apoptosis Resistance. Mol. Cell 2015, 60, 661–675. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Fabbrizi, E.; Fiorentino, F.; Carafa, V.; Altucci, L.; Mai, A.; Rotili, D. Emerging Roles of SIRT5 in Metabolism, Cancer, and SARS-CoV-2 Infection. Cells 2023, 12, 852. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.A. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Beauharnois, J.M.; Bolívar, B.E.; Welch, J.T. Sirtuin 6: A review of biological effects and potential therapeutic properties. Mol. Biosyst. 2013, 9, 1789–1806. [Google Scholar] [CrossRef]
- Klein, M.A.; Denu, J.M. Biological and catalytic functions of sirtuin 6 as targets for small-molecule modulators. J. Biol. Chem. 2020, 295, 11021–11041. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Pavane, M.S.; Banu, L.H.; Gopikar, A.S.R.; Elizabeth, K.R.; Pathak, S. Traditional medicine for aging-related disorders: Implications for drug discovery. In Stem Cells and Aging; Pathak, S., Banerjee, A., Eds.; Academic Press: Cambridge, MA, USA, 2021; Chapter 20; pp. 281–297. [Google Scholar] [CrossRef]
- Zhang, J.; Yin, X.J.; Xu, C.J.; Ning, Y.X.; Chen, M.; Zhang, H.; Chen, S.F.; Yao, L.Q. The histone deacetylase SIRT6 inhibits ovarian cancer cell proliferation via down-regulation of Notch 3 expression. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 818–824. [Google Scholar] [PubMed]
- Choe, M.; Brusgard, J.L.; Chumsri, S.; Bhandary, L.; Zhao, X.F.; Lu, S.; Goloubeva, O.G.; Polster, B.M.; Fiskum, G.M.; Girnun, G.D.; et al. The RUNX2 Transcription Factor Negatively Regulates SIRT6 Expression to Alter Glucose Metabolism in Breast Cancer Cells. J. Cell Biochem. 2015, 116, 2210–2226. [Google Scholar] [CrossRef]
- Qi, J.; Cui, C.; Deng, Q.; Wang, L.; Chen, R.; Zhai, D.; Xie, L.; Yu, J. Downregulated SIRT6 and upregulated NMNAT2 are associated with the presence; depth and stage of colorectal cancer. Oncol. Lett. 2018, 16, 5829–5837. [Google Scholar] [CrossRef]
- Min, L.; Ji, Y.; Bakiri, L.; Qiu, Z.; Cen, J.; Chen, X.; Chen, L.; Scheuch, H.; Zheng, H.; Qin, L.; et al. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat. Cell Biol. 2012, 14, 1203–1211. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Lai, C.C.; Lin, P.M.; Lin, S.F.; Hsu, C.H.; Lin, H.C.; Hu, M.L.; Hsu, C.M.; Yang, M.Y. Altered expression of SIRT gene family in head and neck squamous cell carcinoma. Tumour Biol. 2013, 34, 1847–1854. [Google Scholar] [CrossRef]
- Lefort, K.; Brooks, Y.; Ostano, P.; Cario-André, M.; Calpini, V.; Guinea-Viniegra, J.; Albinger-Hegyi, A.; Hoetzenecker, W.; Kolfschoten, I.; Wagner, E.F.; et al. A miR-34a-SIRT6 axis in the squamous cell differentiation network. EMBO J. 2013, 32, 2248–2263. [Google Scholar] [CrossRef] [PubMed]
- Cea, M.; Cagnetta, A.; Adamia, S.; Acharya, C.; Tai, Y.T.; Fulciniti, M.; Ohguchi, H.; Munshi, A.; Acharya, P.; Bhasin, M.K.; et al. Evidence for a role of the histone deacetylase SIRT6 in DNA damage response of multiple myeloma cells. Blood 2016, 127, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C.; Kafeel, M.I.; Avezbakiyev, B.; Chen, C.; Sun, Y.; Rathnasabapathy, C.; Kalavar, M.; He, Z.; Burton, J.; Lichter, S. Histone deacetylase in chronic lymphocytic leukemia. Oncology 2011, 81, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Cagnetta, A.; Soncini, D.; Orecchioni, S.; Talarico, G.; Minetto, P.; Guolo, F.; Retali, V.; Colombo, N.; Carminati, E.; Clavio, M.; et al. Depletion of SIRT6 enzymatic activity increases acute myeloid leukemia cells’ vulnerability to DNA-damaging agents. Haematologica 2018, 103, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Cea, M.; Cagnetta, A.; Soncini, D.; Minetto, P.; Lovera, D.; Guolo, F.; Colombo, N.; Ballerini, F.; Clavio, M.; Miglino, M.; et al. SIRT6 Inhibition As a Novel Approach for Treating Acute Myeloid Leukemia. Blood 2016, 128, 5222. [Google Scholar] [CrossRef]
- Cea, M.; Cagnetta, A.; Lovera, D.; Grasso, R.; Colombo, N.; Bergamaschi, M.; Aquino, S.; Guolo, F.; Minetto, P.; Ballerini, F.; et al. A Novel Synthetic Lethal Approach Targeting SIRT6 in Acute Myeloid Leukemia. Blood 2015, 126, 1375. [Google Scholar] [CrossRef]
- Hubner, S.E.; de Camargo Magalhães, E.S.; Hoff, F.W.; Brown, B.D.; Qiu, Y.; Horton, T.M.; Kornblau, S.M. DNA Damage Response-Related Proteins Are Prognostic for Outcome in Both Adult and Pediatric Acute Myelogenous Leukemia Patients: Samples from Adults and from Children Enrolled in a Children’s Oncology Group Study. Int. J. Mol. Sci. 2023, 24, 5898. [Google Scholar] [CrossRef]
- Carraway, H.E.; Malkaram, S.A.; Cen, Y.; Shatnawi, A.; Fan, J.; Ali, H.E.A.; Abd Elmageed, Z.Y.; Buttolph, T.; Denvir, J.; Primerano, D.A.; et al. Activation of SIRT6 by DNA hypomethylating agents and clinical consequences on combination therapy in leukemia. Sci. Rep. 2020, 10, 10325. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, Z.; Sheng, F.; Yin, Z. MYC upregulated LINC00319 promotes human acute myeloid leukemia (AML) cells growth through stabilizing SIRT6. Biochem. Biophys. Res. Commun. 2019, 509, 314–321. [Google Scholar] [CrossRef]
- Zhang, P.; Brinton, L.T.; Williams, K.; Sher, S.; Orwick, S.; Tzung-Huei, L.; Mims, A.S.; Coss, C.C.; Kulp, S.K.; Youssef, Y.; et al. Targeting DNA Damage Repair Functions of Two Histone Deacetylases; HDAC8 and SIRT6; Sensitizes Acute Myeloid Leukemia to NAMPT Inhibition. Clin. Cancer Res. 2021, 27, 2352–2366. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Jiang, Y.; Kitano, A.; Hu, T.; Murdaugh, R.L.; Li, Y.; Hoegenauer, K.A.; Chen, R.; Takahashi, K.; Nakada, D. Nuclear NAD+ homeostasis governed by NMNAT1 prevents apoptosis of acute myeloid leukemia stem cells. Sci. Adv. 2021, 7, eabf3895. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, X.; Yang, Y.; Li, Z.; Chen, Y.; Shang, S.; Wang, Y. DNMT3A mutation promotes leukemia development through NAM-NAD metabolic reprogramming. J. Transl. Med. 2023, 21, 481. [Google Scholar] [CrossRef] [PubMed]
- Blank, M.F.; Grummt, I. The seven faces of SIRT7. Transcription 2017, 8, 67–74. [Google Scholar] [CrossRef]
- Vazquez, B.N.; Thackray, J.K.; Simonet, N.G.; Kane-Goldsmith, N.; Martinez-Redondo, P.; Nguyen, T.; Bunting, S.; Vaquero, A.; Tischfield, J.A.; Serrano, L. SIRT7 promotes genome integrity and modulates non-homologous end joining DNA repair. EMBO J. 2016, 35, 1488–1503. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shi, L.; Yang, S.; Yan, R.; Zhang, D.; Yang, J.; He, L.; Li, W.; Yi, X.; Sun, L.; et al. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat. Commun. 2016, 7, 12235. [Google Scholar] [CrossRef]
- Ianni, A.; Hoelper, S.; Krueger, M.; Braun, T.; Bober, E. Sirt7 stabilizes rDNA heterochromatin through recruitment of DNMT1 and Sirt1. Biochem. Biophys. Res. Commun. 2017, 492, 434–440. [Google Scholar] [CrossRef]
- Ianni, A.; Kumari, P.; Tarighi, S.; Braun, T.; Vaquero, A. SIRT7: A novel molecular target for personalized cancer treatment? Oncogene 2024, 43, 993–1006. [Google Scholar] [CrossRef]
- Barber, M.F.; Michishita-Kioi, E.; Xi, Y.; Tasselli, L.; Kioi, M.; Moqtaderi, Z.; Tennen, R.I.; Paredes, S.; Young, N.L.; Chen, K.; et al. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature 2012, 487, 114–118. [Google Scholar] [CrossRef]
- Li, L.; Bhatia, R. The controversial role of Sirtuins in tumorigenesis—SIRT7 joins the debate. Cell Res. 2013, 23, 10–12. [Google Scholar] [CrossRef]
- Ford, E.; Voit, R.; Liszt, G.; Magin, C.; Grummt, I.; Guarente, L. Mammalian Sir2 homolog SIRT7 is an activator of RNA polymerase I transcription. Genes Dev. 2006, 20, 1075–1080. [Google Scholar] [CrossRef]
- Malik, S.; Villanova, L.; Tanaka, S.; Aonuma, M.; Roy, N.; Berber, E.; Pollack, J.R.; Michishita-Kioi, E.; Chua, K.F. SIRT7 inactivation reverses metastatic phenotypes in epithelial and mesenchymal tumors. Sci. Rep. 2015, 5, 9841. [Google Scholar] [CrossRef] [PubMed]
- Poniewierska-Baran, A.; Warias, P.; Zgutka, K. Sirtuins (SIRTs) As a Novel Target in Gastric Cancer. Int. J. Mol. Sci. 2022, 23, 15119. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Noh, J.H.; Jung, K.H.; Eun, J.W.; Bae, H.J.; Kim, M.G.; Chang, Y.G.; Shen, Q.; Park, W.S.; Lee, J.Y.; et al. Sirtuin7 oncogenic potential in human hepatocellular carcinoma and its regulation by the tumor suppressors MiR-125a-5p and MiR-125b. Hepatology 2013, 57, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Lu, R.Q.; Xie, S.H.; Zheng, H.; Wen, X.M.; Gao, X.; Guo, L. SIRT7 Exhibits Oncogenic Potential in Human Ovarian Cancer Cells. Asian Pac. J. Cancer Prev. 2015, 16, 3573–3577. [Google Scholar] [CrossRef] [PubMed]
- Aljada, A.; Saleh, A.M.; Alkathiri, M.; Shamsa, H.B.; Al-Bawab, A.; Nasr, A. Altered Sirtuin 7 Expression is Associated with Early Stage Breast Cancer. Breast Cancer 2015, 9, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Ji, Y.; Zhang, D.; Liu, Y.; Fang, P. MicroRNA-3666-induced suppression of SIRT7 inhibits the growth of non-small cell lung cancer cells. Oncol. Rep. 2016, 36, 3051–3057. [Google Scholar] [CrossRef]
- Li, W.; Zhu, D.; Qin, S. SIRT7 suppresses the epithelial-to-mesenchymal transition in oral squamous cell carcinoma metastasis by promoting SMAD4 deacetylation. J. Exp. Clin. Cancer Res. 2018, 37, 148. [Google Scholar] [CrossRef] [PubMed]
- Raza, U.; Tang, X.; Liu, Z.; Liu, B. SIRT7: The seventh key to unlocking the mystery of aging. Physiol. Rev. 2024, 104, 253–280. [Google Scholar] [CrossRef]
- Wang, Y.; Maruichi, A.; Song, Z.; Greenberg, P.; Chen, D. SIRT7 improves hematopoiesis in myelodysplastic syndrome through regulating mitochondrial stress. Exp. Hematol. 2023, 124, S157. [Google Scholar]
- Nowicki, M.; Wierzbowska, A.; Stec-Martyna, E.; Kulczycka-Wojdala, D.; Nowicki, G.; Szmigielska-Kapłon, A. SIRT1-SIRT7 Expression in Patients with Lymphoproliferative Disorders Undergoing Hematopoietic Stem Cell Mobilization. Cancers 2022, 14, 1213. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.; Schmidt, M.; Huber, O.; Frietsch, J.J.; Scholl, S.; Heidel, F.H.; Hochhaus, A.; Müller, J.P.; Ernst, T. SIRT7: An influence factor in healthy aging and the development of age-dependent myeloid stem-cell disorders. Leukemia 2020, 34, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Metzeler, K.H.; Hummel, M.; Bloomfield, C.D.; Spiekermann, K.; Braess, J.; Sauerland, M.-C.; Heinecke, A.; Radmacher, M.; Marcucci, G.; Whitman, S.P.; et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood 2008, 112, 4193–4201. [Google Scholar] [CrossRef]
- Vakhrusheva, O.; Smolka, C.; Gajawada, P.; Kostin, S.; Boettger, T.; Kubin, T.; Braun, T.; Bober, E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res. 2008, 102, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Kim, D.K.; Kim, E.S.; Park, S.J.; Kwon, J.H.; Shin, J.; Park, S.M.; Moon, Y.H.; Wang, H.J.; Gho, Y.S.; et al. Comparative interactomes of SIRT6 and SIRT7: Implication of functional links to aging. Proteomics 2014, 14, 1610–1622. [Google Scholar] [CrossRef] [PubMed]
- Ianni, A.; Kumari, P.; Tarighi, S.; Simonet, N.G.; Popescu, D.; Guenther, S.; Hölper, S.; Schmidt, A.; Smolka, C.; Yue, S.; et al. SIRT7-dependent deacetylation of NPM promotes p53 stabilization following UV-induced genotoxic stress. Proc. Natl. Acad. Sci. USA 2021, 118, e2015339118. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Tian, J.; Zheng, G.; Zhao, J. Sirtuin7 is involved in protecting neurons against oxygen-glucose deprivation and reoxygenation-induced injury through regulation of the p53 signaling pathway. J. Biochem. Mol. Toxicol. 2017, 31, e21955. [Google Scholar] [CrossRef]
- Sun, M.; Zhai, M.; Zhang, N.; Wang, R.; Liang, H.; Han, Q.; Jia, Y.; Jiao, L. MicroRNA-148b-3p is involved in regulating hypoxia/reoxygenation-induced injury of cardiomyocytes in vitro through modulating SIRT7/p53 signaling. Chem. Biol. Interact. 2018, 296, 211–219. [Google Scholar] [CrossRef]
- Yu, M.; Shi, X.; Ren, M.; Liu, L.; Qi, H.; Zhang, C.; Zou, J.; Qiu, X.; Zhu, W.G.; Zhang, Y.E.; et al. SIRT7 Deacetylates STRAP to Regulate p53 Activity and Stability. Int. J. Mol. Sci. 2020, 21, 4122. [Google Scholar] [CrossRef]
- Olsen, E.A.; Kim, Y.H.; Kuzel, T.M.; Pacheco, T.R.; Foss, F.M.; Parker, S.; Frankel, S.R.; Chen, C.; Ricker, J.L.; Arduino, J.M.; et al. Phase IIb multicenter trial of vorinostat in patients with persistent; progressive; or treatment refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2007, 25, 3109–3115. [Google Scholar] [CrossRef]
- O’Connor, O.A.; Horwitz, S.; Masszi, T.; Van Hoof, A.; Brown, P.; Doorduijn, J.; Hess, G.; Jurczak, W.; Knoblauch, P.; Chawla, S.; et al. Belinostat in Patients with Relapsed or Refractory Peripheral T-Cell Lymphoma: Results of the Pivotal Phase II BELIEF (CLN-19) Study. J. Clin. Oncol. 2015, 33, 2492–2499. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Yang, H.; Bueso-Ramos, C.; Ferrajoli, A.; Cortes, J.; Wierda, W.G.; Faderl, S.; Koller, C.; Morris, G.; Rosner, G.; et al. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood 2008, 111, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Hou, Y.; Dovat, K.; Dovat, S.; Song, C.; Ge, Z. Synergistic effect of HDAC inhibitor Chidamide with Cladribine on cell cycle arrest and apoptosis by targeting HDAC2/c-Myc/RCC1 axis in acute myeloid leukemia. Exp. Hematol. Oncol. 2023, 12, 23. [Google Scholar] [CrossRef] [PubMed]
- Maher, K.R.; Schafer, D.; Schaar, D.; Sabo, R.; Bandyopadhyay, D.; Grant, S. Pevonedistat plus belinostat in relapsed/refractory acute myeloid leukemia or myelodysplastic syndrome: A phase I multicenter study. JCO 2023, 41, e19015. [Google Scholar] [CrossRef]
- Maher, K.R.; Shafer, D.; Schaar, D.; Bandyopadhyay, D.; Deng, X.; Wright, J.; Piekarz, R.; Rudek, M.A.; Harvey, R.D.; Grant, S. A phase I study of MLN4924 and belinostat in relapsed/refractory acute myeloid leukemia or myelodysplastic syndrome. Cancer Chemother. Pharmacol. 2025, 95, 24. [Google Scholar] [CrossRef]
- Li, L.; Osdal, T.; Ho, Y.W.; McDonald, T.; Chun, S.; Chu, S.; Lin, A.; Dos Santos, C.; Popa, M.L.; Gjertsen, B.T.; et al. Pharmacological Inhibition of the SIRT1 Deacetylase with The Small Molecule Inhibitor Tenovin-6 Enhances Ablation of FLT3-ITD+ LSC in Combination with TKI Treatment. Blood 2013, 122, 2685. [Google Scholar] [CrossRef]
- Westerberg, G.; Chiesa, J.A.; Andersen, C.A.; Diamanti, D.; Magnoni, L.; Pollio, G.; Darpo, B.; Zhou, M. Safety, pharmacokinetics, pharmacogenomics and QT concentration-effect modelling of the SirT1 inhibitor selisistat in healthy volunteers. Br. J. Clin. Pharmacol. 2015, 79, 477–491. [Google Scholar] [CrossRef]
- Bitterman, K.J.; Anderson, R.M.; Cohen, H.Y.; Latorre-Esteves, M.; Sinclair, D.A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002, 277, 45099–45107. [Google Scholar] [CrossRef]
- Hwang, E.S.; Song, S.B. Nicotinamide is an inhibitor of SIRT1 in vitro, but can be a stimulator in cells. Cell. Mol. Life Sci. 2017, 74, 3347–3362. [Google Scholar] [CrossRef]
- Hwang, E.S.; Song, S.B. Possible Adverse Effects of High-Dose Nicotinamide: Mechanisms and Safety Assessment. Biomolecules 2020, 10, 687. [Google Scholar] [CrossRef]
- Gryniukova, A.; Kaiser, F.; Myziuk, I.; Alieksieieva, D.; Leberecht, C.; Heym, P.P.; Tarkhanova, O.O.; Moroz, Y.S.; Borysko, P.; Haupt, V.J. AI-Powered Virtual Screening of Large Compound Libraries Leads to the Discovery of Novel Inhibitors of Sirtuin-1. J. Med. Chem. 2023, 66, 10241–10251. [Google Scholar] [CrossRef] [PubMed]
- Warda, E.T.; El-Ashmawy, M.B.; Habib, E.E.; Abdelbaky, M.S.M.; Garcia-Granda, S.; Thamotharan, S.; El-Emam, A.A. Synthesis and in vitro antibacterial, antifungal, anti-proliferative activities of novel adamantane-containing thiazole compounds. Sci. Rep. 2022, 12, 21058. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Wang, Y.; Zhang, J.; Wei, Z.; Yan, T.; Feng, C.; Xu, Z.; Zhou, A.; Wu, Y. Discovery of Novel SIRT1/2 Inhibitors with Effective Cytotoxicity against Human Leukemia Cells. J. Chem. Inf. Model. 2023, 63, 4780–4790. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Wu, W.; Zhang, J.; Li, L.; Wan, Y.; Tang, X.; Chen, X.; Liu, S.; Yao, X. Tenovin-1 inhibited dengue virus replication through SIRT2. Eur. J. Pharmacol. 2021, 907, 174264. [Google Scholar] [CrossRef] [PubMed]
- Lain, S.; Hollick, J.J.; Campbell, J.; Staples, O.D.; Higgins, M.; Aoubala, M.; McCarthy, A.; Appleyard, V.; Murray, K.E.; Baker, L.; et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008, 13, 454–463. [Google Scholar] [CrossRef]
- Ladds, M.J.G.W.; Popova, G.; Pastor-Fernández, A.; Kannan, S.; van Leeuwen, I.M.M.; Håkansson, M.; Walse, B.; Tholander, F.; Bhatia, R.; Verma, C.S.; et al. Exploitation of dihydroorotate dehydrogenase (DHODH) and p53 activation as therapeutic targets: A case study in polypharmacology. J. Biol. Chem. 2020, 295, 17935–17949. [Google Scholar] [CrossRef]
- George, J.; Nihal, M.; Singh, C.K.; Ahmad, N. 4′-Bromo-resveratrol, a dual Sirtuin-1 and Sirtuin-3 inhibitor, inhibits melanoma cell growth through mitochondrial metabolic reprogramming. Mol. Carcinog. 2019, 58, 1876–1885. [Google Scholar] [CrossRef]
- Chen, P.T.; Yeong, K.Y. New sirtuin modulators: Their uncovering, pharmacophore, and implications in drug discovery. Med. Chem. Res. 2024, 33, 1064–1078. [Google Scholar] [CrossRef]
- Li, R.; Yan, L.; Sun, X.; Zheng, W. A bicyclic pentapeptide-based highly potent and selective pan-SIRT1/2/3 inhibitor harboring Nε-thioacetyl-lysine. Bioorg. Med. Chem. 2020, 28, 115356. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.-L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Yang, J.; Li, J.; Wei, T.T.; Pang, J.Y.; Du, Y.H. Marine Compound Exerts Antiaging Effect in Human Endothelial Progenitor Cells via Increasing Sirtuin1 Expression. ACS Pharmacol. Transl. Sci. 2023, 6, 1673–1680. [Google Scholar] [CrossRef]
- Ciarlo, C.; Kaufman, C.K.; Kinikoglu, B.; Michael, J.; Yang, S.; D’Amato, C.; Blokzijl-Franke, S.; den Hertog, J.; Schlaeger, T.M.; Zhou, Y.; et al. A chemical screen in zebrafish embryonic cells establishes that Akt activation is required for neural crest development. elife 2017, 6, e29145. [Google Scholar] [CrossRef] [PubMed]
- Shankar, U.; Jain, N.; Majee, P.; Mishra, S.K.; Rathi, B.; Kumar, A. Potential Drugs Targeting Nsp16 Protein May Corroborates a Promising Approach to Combat SARSCoV-2 Virus. ChemRxiv 2020. [Google Scholar] [CrossRef]
- Schiedel, M.; Rumpf, T.; Karaman, B.; Lehotzky, A.; Gerhardt, S.; Ovádi, J.; Sippl, W.; Einsle, O.; Jung, M. Structure-Based Development of an Affinity Probe for Sirtuin 2. Angew. Chem. Int. Ed. Engl. 2016, 55, 2252–2256. [Google Scholar] [CrossRef] [PubMed]
- Kalbas, D.; Meleshin, M.; Liebscher, S.; Zessin, M.; Melesina, J.; Schiene-Fischer, C.; Bülbül, E.F.; Bordusa, F.; Sippl, W.; Schutkowski, M. Small Changes Make the Difference for SIRT2: Two Different Binding Modes for 3-Arylmercapto-Acylated Lysine Derivatives. Biochemistry 2022, 61, 1705–1722. [Google Scholar] [CrossRef]
- Mazur, G.; Pańczyk-Straszak, K.; Krysińska, K.; Niemiec, K.; Waszkielewicz, A. Novel xanthone derivatives as potent sirtuin 2 inhibitors. Bioorg Med. Chem. Lett. 2024, 100, 129620. [Google Scholar] [CrossRef] [PubMed]
- Sinatra, L.; Vogelmann, A.; Friedrich, F.; Tararina, M.A.; Neuwirt, E.; Colcerasa, A.; König, P.; Toy, L.; Yesiloglu, T.Z.; Hilscher, S.; et al. Development of First-in-Class Dual Sirt2/HDAC6 Inhibitors as Molecular Tools for Dual Inhibition of Tubulin Deacetylation. J. Med. Chem. 2023, 66, 14787–14814. [Google Scholar] [CrossRef] [PubMed]
- Abbotto, E.; Casini, B.; Piacente, F.; Scarano, N.; Cerri, E.; Tonelli, M.; Astigiano, C.; Millo, E.; Sturla, L.; Bruzzone, S.; et al. Novel Thiazole-Based SIRT2 Inhibitors Discovered via Molecular Modelling Studies and Enzymatic Assays. Pharmaceuticals 2023, 16, 1316. [Google Scholar] [CrossRef] [PubMed]
- Parenti, M.D.; Grozio, A.; Bauer, I.; Galeno, L.; Damonte, P.; Millo, E.; Sociali, G.; Franceschi, C.; Ballestrero, A.; Bruzzone, S.; et al. Discovery of novel and selective SIRT6 inhibitors. J. Med. Chem. 2014, 57, 4796–4804. [Google Scholar] [CrossRef]
- Pannek, M.; Alhalabi, Z.; Tomaselli, D.; Menna, M.; Fiorentino, F.; Robaa, D.; Weyand, M.; Puhlmann, M.; Tomassi, S.; Barreca, F.; et al. Specific Inhibitors of Mitochondrial Deacylase Sirtuin 4 Endowed with Cellular Activity. J. Med. Chem. 2024, 67, 1843–1860. [Google Scholar] [CrossRef]
- Polletta, L.; Vernucci, E.; Carnevale, I.; Arcangeli, T.; Rotili, D.; Palmerio, S.; Steegborn, C.; Nowak, T.; Schutkowski, M.; Pellegrini, L.; et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 2015, 11, 253–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.L.; Chen, Y.; Wang, Y.Q.; Tao, E.W.; Tan, J.; Liu, Q.Q.; Li, C.M.; Tong, X.M.; Gao, Q.Y.; Hong, J.; et al. Sirtuin5 protects colorectal cancer from DNA damage by keeping nucleotide availability. Nat. Commun. 2022, 13, 6121. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hu, L.; Deng, J.; Hou, S.; Mou, L.; Lei, P.; Chen, X.; Liu, J.; Jiang, Y.; Xiong, R.; et al. Design, synthesis and biological evaluation of 2,4,6- trisubstituted triazine derivatives as new nonpeptide small-molecule SIRT5 inhibitors. Bioorg Med. Chem. 2023, 93, 117455. [Google Scholar] [CrossRef] [PubMed]
- Mou, L.; Yang, L.; Hou, S.; Wang, B.; Wang, X.; Hu, L.; Deng, J.; Liu, J.; Chen, X.; Jiang, Y.; et al. Structure-Activity Relationship Studies of 2,4,5-Trisubstituted Pyrimidine Derivatives Leading to the Identification of a Novel and Potent Sirtuin 5 Inhibitor against Sepsis-Associated Acute Kidney Injury. J. Med. Chem. 2023, 66, 11517–11535. [Google Scholar] [CrossRef] [PubMed]
- Damonte, P.; Sociali, G.; Parenti, M.D.; Soncini, D.; Bauer, I.; Boero, S.; Grozio, A.; Holtey, M.V.; Piacente, F.; Becherini, P.; et al. SIRT6 inhibitors with salicylate-like structure show immunosuppressive and chemosensitizing effects. Bioorg Med. Chem. 2017, 25, 5849–5858. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Zhang, Z.; Hu, H.; Zhang, Y.; Xiong, Y. SIRT6 Activator UBCS039 Inhibits Thioacetamide-Induced Hepatic Injury In Vitro and In Vivo. Front. Pharmacol. 2022, 13, 837544. [Google Scholar] [CrossRef]
- Ding, Y.N.; Wang, H.Y.; Chen, X.F.; Tang, X.; Chen, H.Z. Roles of Sirtuins in Cardiovascular Diseases: Mechanisms and Therapeutics. Circ. Res. 2025, 136, 524–550. [Google Scholar] [CrossRef]
- Chung, S.; Yao, H.; Caito, S.; Hwang, J.W.; Arunachalam, G.; Rahman, I. Regulation of SIRT1 in cellular functions: Role of polyphenols. Arch. Biochem. Biophys. 2010, 501, 79–90. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Sirtuin (SIRT) | Class | Main Cell Localization | Potential Signaling Pathways Impact and Affected Molecules | Functions | Level | Probable Role in AML | Ref. |
|---|---|---|---|---|---|---|---|
| SIRT1 | I | Nucleus Cytoplasm | p53, c-Myc, USP22, FOXO1, FOXP1, STAT5, DOT1L, miR-9 | Deacetylase Deacylase | High | Promoter | [43,44,47,55,56,57,58,60] |
| SIRT2 | I | Cytoplasm Nucleus (during mitosis) | NAMPT, MAPK, VEGF, MRP1, ERK1/2, BCL-2, p53, Caspase-1, AKT/GSK3β/β-catenin, PI3K/AKT/mTOR, pentose-phosphate cycle, p21, cyclin E1, miR-140-5p, miR-145 | Deacetylase Deacylase | High | Promoter | [68,69,73,74,75,76,77,78,79,80] |
| SIRT3 | I | Mitochondria | PTEN, MDM2, p53, cyclophilin D, IDH2, tricarboxylic acid cycle, Fatty acid oxidation, sumoylation, HES1, Notch1 | Deacetylase Decrotonylase | Low | Undetermined/ Bi-directional | [83,84,85,86,89,90] |
| SIRT4 | II | Mitochondria | Glutamine metabolism | Deacetylase Deacylase ADP-rybosil-transferase, | Low (high according to some sources) | Undetermined/ Bi-directional | [94,96,97] |
| SIRT5 | III | Mitochondria | BAX/BCL-2, oxidative phosphorylation | Deacetylase Demalonylase Deglutarylase Desuccinylase | No data | Promoter | [104,105,106,107,110,111,113,114] |
| SIRT6 | IV | Nucleus | NAMPT, NMNAT1, cyclin-CDK | Deacetylase Deacylase ADP-rybosil-transferase | High | Promoter | [140,146,147,148] |
| SIRT7 | IV | Nucleus Nucleolus | NRF1, NPM1, p53 | Deacetylase Dessuccinylase | Low | Suppressor | [147,151,164,165,167,171] |
| Molecule | Mechanism of Action | Targeted Sirtuin (SIRT) | Additional Mechanisms | Reference |
|---|---|---|---|---|
| Selisistat (EX-527) | Inhibitor | SIRT1 | [182] | |
| Nicotinamide-d4 Nicotinamide-13C6 Nicotinamide-15N,13C3 | Inhibitors | SIRT1 |
| [183,184,185] |
| Z26395438 (compound 1) | Inhibitor | SIRT1 | [186] | |
| Antiproliferative agent-17 | Inhibitor | SIRT1 |
| [187] |
| Sirt1/2-IN-3 (compound PS9) | Inhibitor | SIRT1 SIRT2 |
| [188] |
| Sirt1/2-IN-2 (compound hsa55) | Inhibitor | SIRT1 SIRT2 |
| [188] |
| Tenovin-1 | Inhibitor | SIRT1 SIRT2 |
| [5,189,190,191] |
| Tenovin-6 | Inhibitor | SIRT1 SIRT2 |
| [181] |
| 4′-Bromo-resveratrol | Inhibitor | SIRT1 SIRT3 |
| [192] |
| Sirt1/2-IN-4 (compound PS3) | Inhibitor | SIRT1 SIRT2 SIRT3 |
| [188] |
| BZD9Q1 | Inhibitor | SIRT1 SIRT2 SIRT3 |
| [193] |
| SIRT1/2/3-IN-1 (compound 10) | Inhibitor | SIRT1 SIRT2 SIRT3 |
| [194] |
| Resveratrol | Activator | SIRT1 | [195] | |
| SIRT1 activator 1(compound 3) | Activator | SIRT1 | [196] | |
| JFD00244 | Inhibitor | SIRT2 |
| [197,198] |
| SirReal1, SirReal2 | Inhibitors | SIRT2 | [199] | |
| 3-aryl-mercapto-butyrylated peptide derivative | Inhibitor | SIRT2 | [200] | |
| SIRT2-IN-12 (compound 3) (xanthone derivative) | Inhibitor | SIRT2 | [201] | |
| Mz325 | Inhibitor | SIRT2 |
| [202] |
| HSP70/SIRT2-IN-2 (Compounds 1a) | Inhibitor | SIRT2 |
| [203] |
| SIRT2/6-IN-1 (Compound 5) | Inhibitor | SIRT2 SIRT6 |
| [204] |
| YC8-02 | Inhibitor | SIRT3 |
| [86] |
| SIRT4-IN-1 (compound 69) | Inhibitor | SIRT4 | [205] | |
| NRD167 | Inhibitor | SIRT5 |
| [115] |
| 179MC3482 | Inhibitor | SIRT5 | [206] | |
| SIRT5 inhibitor 1 | Inhibitor | SIRT5 | [207] | |
| SIRT5 inhibitor 8 (compound 10) | Inhibitor | SIRT5 |
| [208] |
| SIRT5 inhibitor 9 (compound 14) | Inhibitor | SIRT5 |
| [208] |
| SIRT5 Inhibitor 6 (2,4,5-trisubstituted pyrimidine derivative) | Inhibitor | SIRT5 |
| [209] |
| SIRT5 inhibitor 7 (compound 58) (2,4,5-trisubstituted pyrimidine derivative) | Inhibitor | SIRT5 |
| [209] |
| SIRT6-IN-2 (compound 5) | Inhibitor | SIRT6 |
| [210] |
| UBCS039 | Activator | SIRT6 |
| [193,211] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strzałka, P.; Krawiec, K.; Wiśnik, A.; Jarych, D.; Czemerska, M.; Zawlik, I.; Pluta, A.; Wierzbowska, A. The Role of the Sirtuin Family Histone Deacetylases in Acute Myeloid Leukemia—A Promising Road Ahead. Cancers 2025, 17, 1009. https://doi.org/10.3390/cancers17061009
Strzałka P, Krawiec K, Wiśnik A, Jarych D, Czemerska M, Zawlik I, Pluta A, Wierzbowska A. The Role of the Sirtuin Family Histone Deacetylases in Acute Myeloid Leukemia—A Promising Road Ahead. Cancers. 2025; 17(6):1009. https://doi.org/10.3390/cancers17061009
Chicago/Turabian StyleStrzałka, Piotr, Kinga Krawiec, Aneta Wiśnik, Dariusz Jarych, Magdalena Czemerska, Izabela Zawlik, Agnieszka Pluta, and Agnieszka Wierzbowska. 2025. "The Role of the Sirtuin Family Histone Deacetylases in Acute Myeloid Leukemia—A Promising Road Ahead" Cancers 17, no. 6: 1009. https://doi.org/10.3390/cancers17061009
APA StyleStrzałka, P., Krawiec, K., Wiśnik, A., Jarych, D., Czemerska, M., Zawlik, I., Pluta, A., & Wierzbowska, A. (2025). The Role of the Sirtuin Family Histone Deacetylases in Acute Myeloid Leukemia—A Promising Road Ahead. Cancers, 17(6), 1009. https://doi.org/10.3390/cancers17061009